
Response to FEC Chemotherapy and Oncolytic HSV-1 Is Associated with Macrophage Polarization and Increased Expression of S100A8/A9 in Triple Negative Breast Cancer



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Mouse Experiments
2.3. Chemotherapy Treatments
2.4. Oncolytic Virus Treatments
2.5. Survival Analysis: Kaplan–Meier and ROC Plots
2.6. Clariom S Assay
2.7. Flow Cytometry
2.8. Statistical Analysis
3. Results
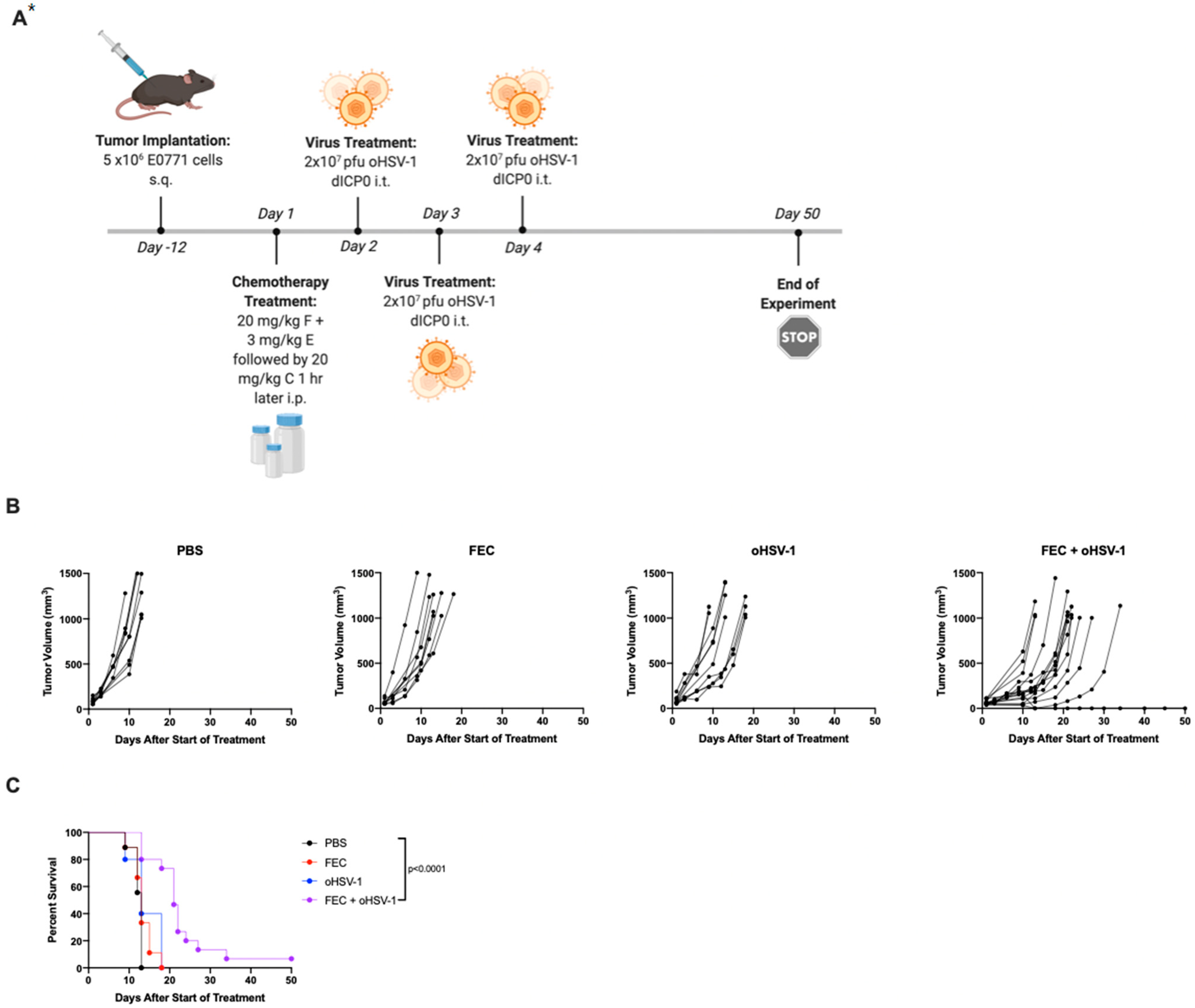
3.1. FEC + oHSV-1 Improves Survival Outcomes
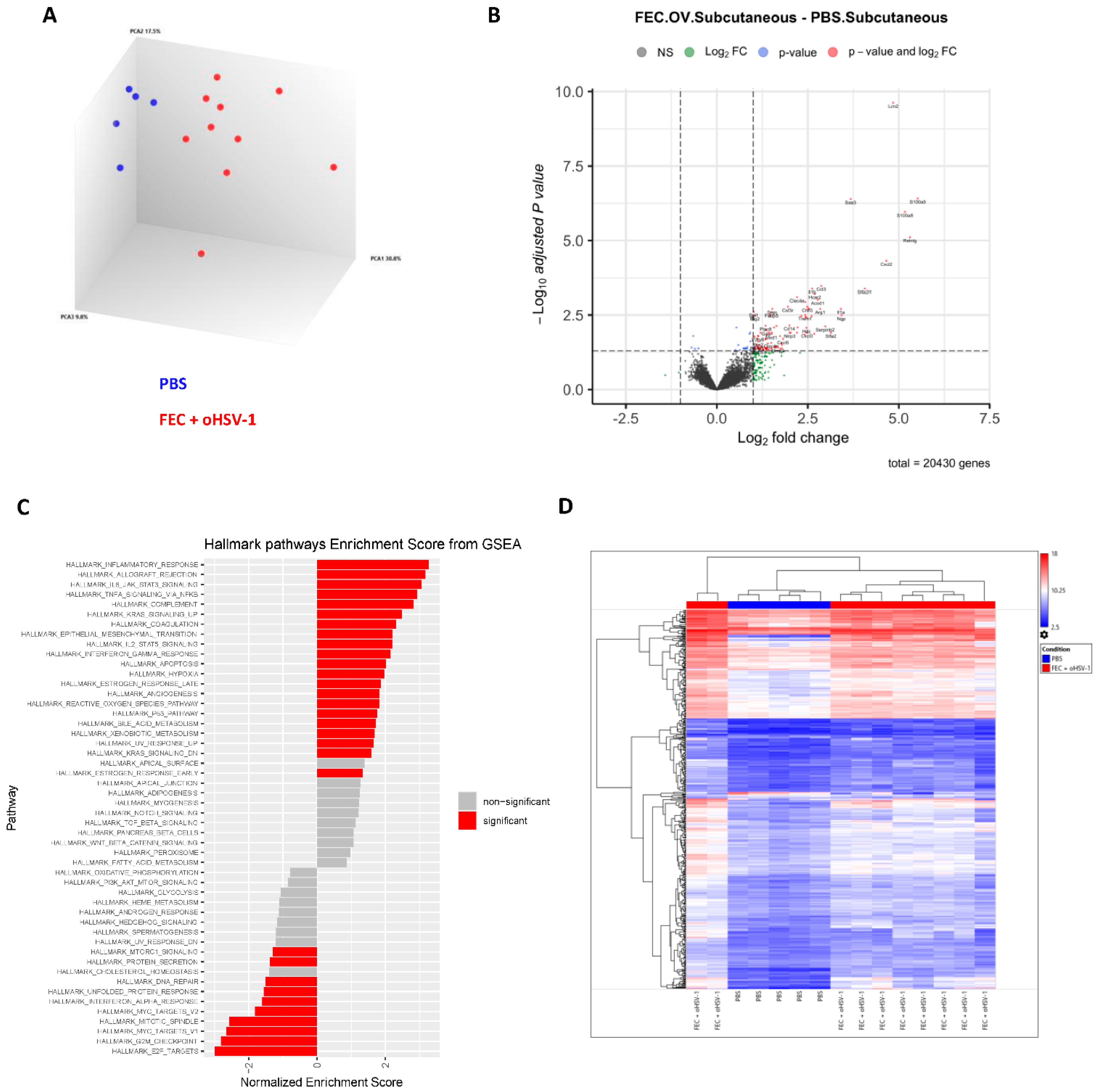
3.2. FEC + oHSV-1 Upregulates Inflammatory Myeloid Cell Pathways
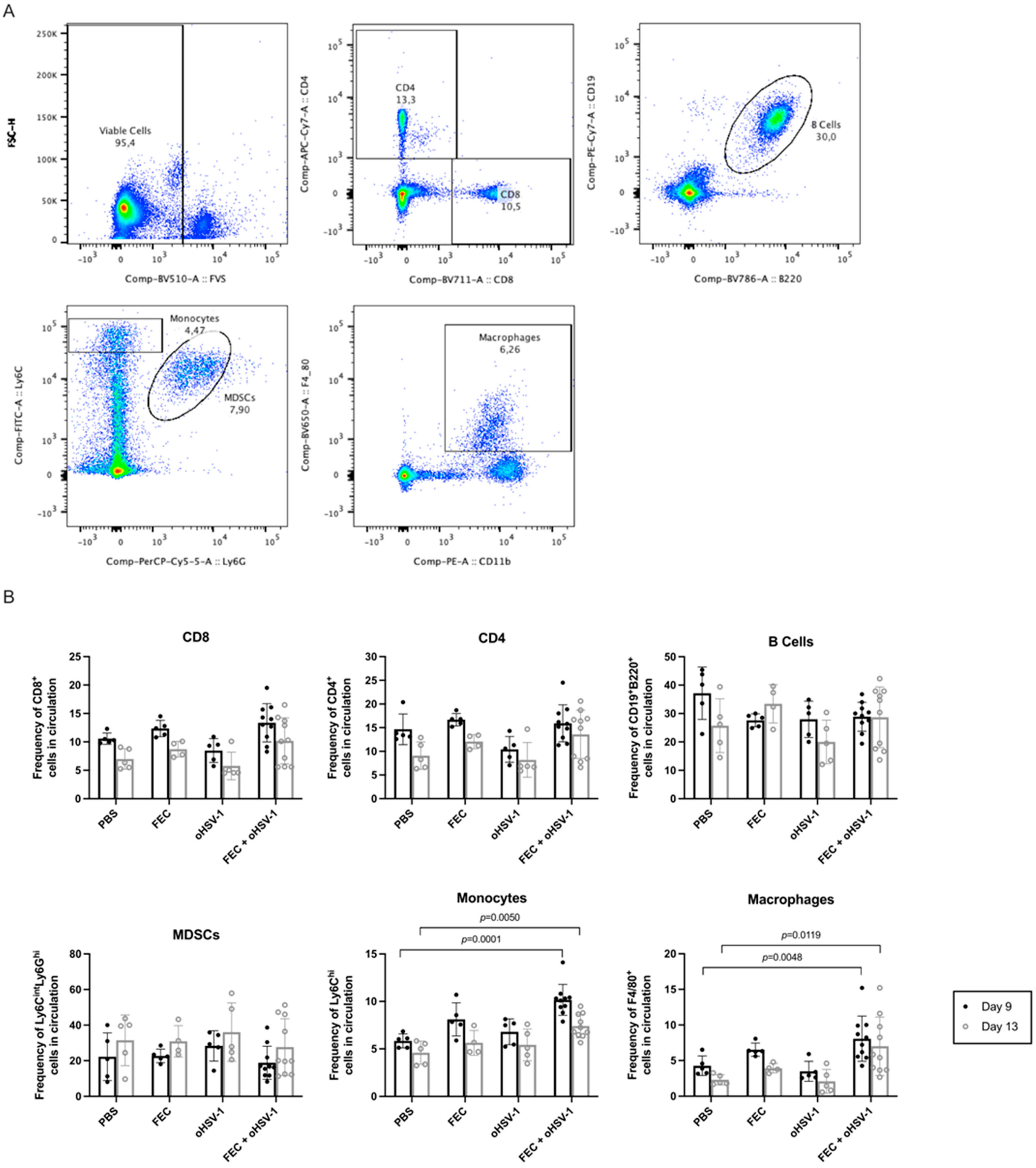
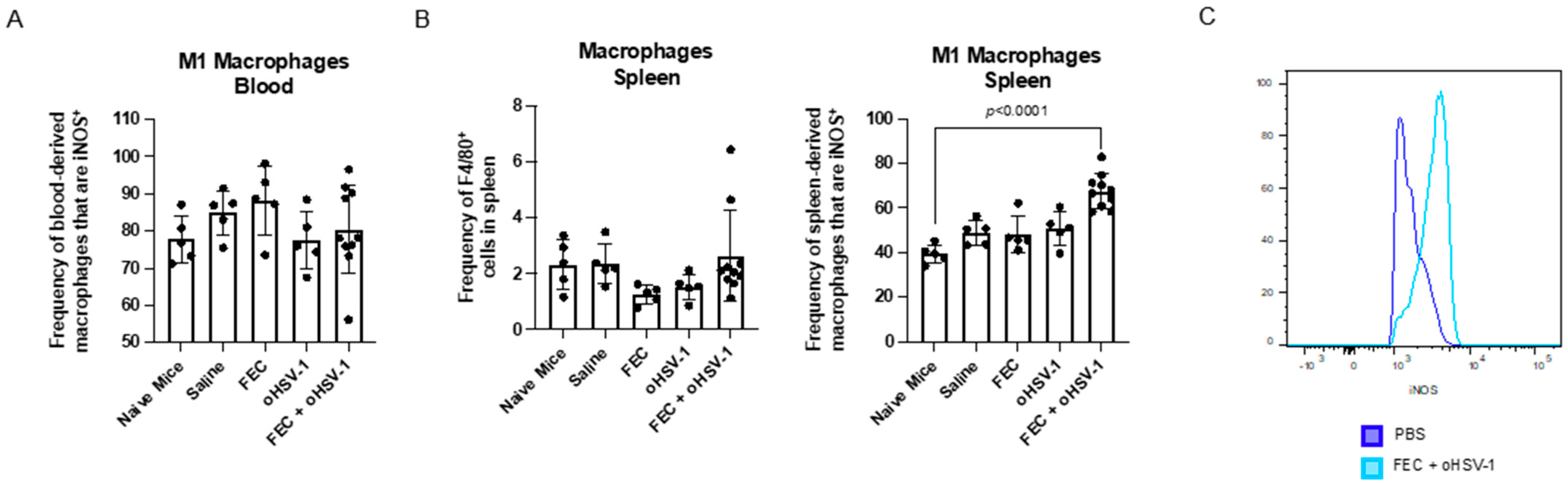
3.3. FEC + oHSV-1 Increases Inflammatory Myeloid Cells in the Peripheral Blood and Spleen
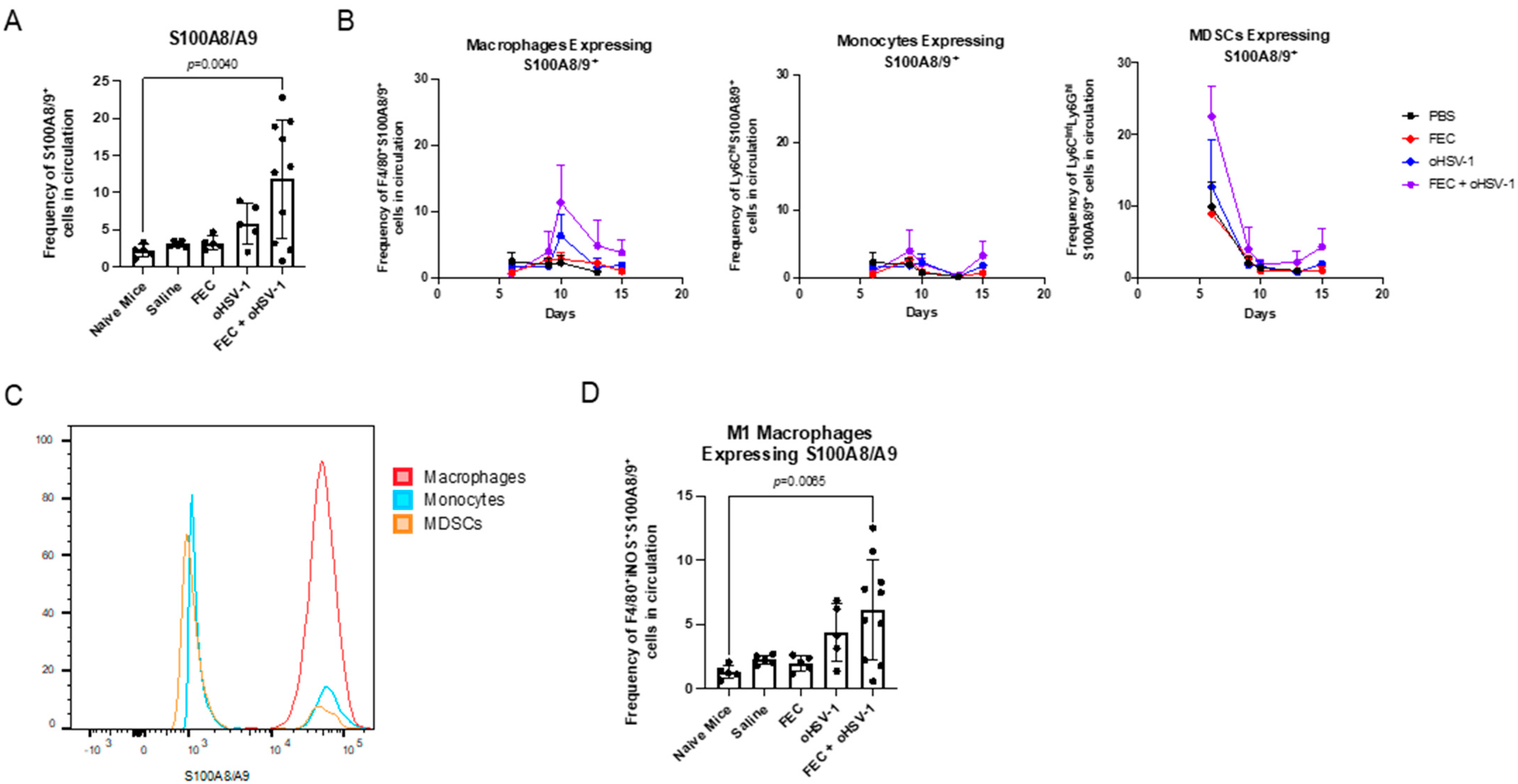
3.4. FEC + oHSV-1 Therapeutic Efficacy Further Potentiated by Upregulation of S100A8/A9
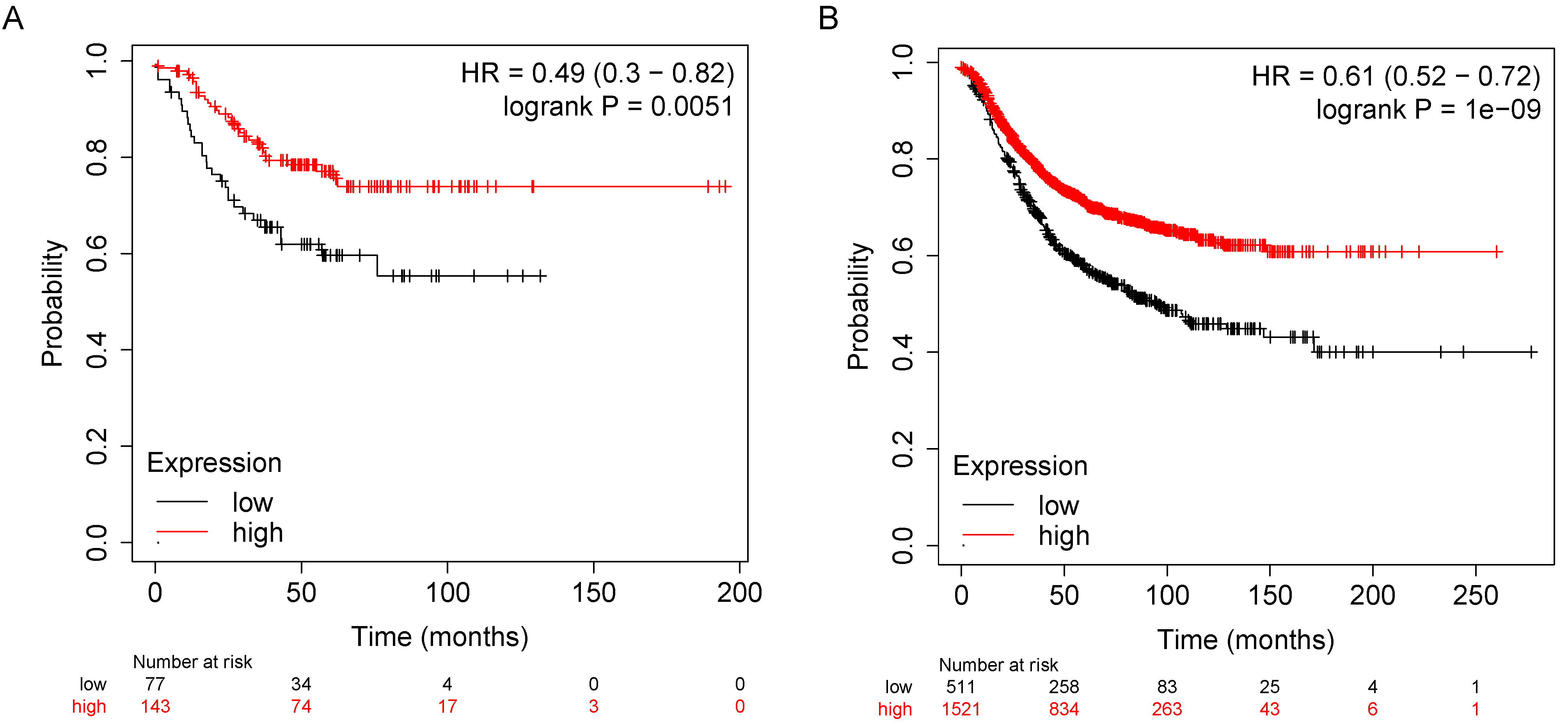
3.5. S100A8 and S100A9 Are Predictive of Response to FEC Therapy in Breast Cancer
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Amodio, G.; Cichy, J.; Conde, P.; Matteoli, G.; Moreau, A.; Ochando, J.; Oral, B.H.; Pekarova, M.; Ryan, E.J.; Roth, J.; et al. Role of myeloid regulatory cells (MRCs) in maintaining tissue homeostasis and promoting tolerance in autoimmunity, inflammatory disease and transplantation. Cancer Immunol. Immunother. 2019, 68, 661–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engblom, C.; Pfirschke, C.; Pittet, M.J. The role of myeloid cells in cancer therapies. Nat. Rev. Cancer 2016, 16, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Vito, A.; El-Sayes, N.; Mossman, K. Hypoxia-Driven Immune Escape in the Tumor Microenvironment. Cells 2020, 9, 992. [Google Scholar] [CrossRef]
- Condamine, T.; Gabrilovich, D.I. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011, 32, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Ueda, Y.; Kondo, M.; Kelsoe, G. Inflammation and the reciprocal production of granulocytes and lymphocytes in bone marrow. J. Exp. Med. 2005, 201, 1771–1780. [Google Scholar] [CrossRef] [Green Version]
- Jordan, K.R.; Kapoor, P.; Spongberg, E.; Tobin, R.P.; Gao, D.; Borges, V.F.; McCarter, M.D. Immunosuppressive myeloid-derived suppressor cells are increased in splenocytes from cancer patients. Cancer Immunol. Immunother. 2017, 66, 503–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bates, J.P.; Derakhshandeh, R.; Jones, L.; Webb, T.J. Mechanisms of immune evasion in breast cancer. BMC Cancer 2018, 18, 556. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Montero, C.M.; Salem, M.L.; Nishimura, M.I.; Garrett-Mayer, E.; Cole, D.J.; Montero, A.J. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol. Immunother. 2009, 58, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Bergenfelz, C.; Larsson, A.M.; Von Stedingk, K.; Gruvberger-Saal, S.; Aaltonen, K.; Jansson, S.; Jernström, H.; Janols, H.; Wullt, M.; Bredberg, A.; et al. Systemic monocytic-MDSCs are generated from monocytes and correlate with disease progression in breast cancer patients. PLoS ONE 2015, 10, e0127028. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef]
- Vito, A.; Salem, O.; El-Sayes, N.; MacFawn, I.; Portillo, A.L.; Milne, K.; Harrington, D.; Ashkar, A.; Wan, Y.; Workenhe, S.; et al. Immune checkpoint blockade in triple negative breast cancer influenced by B cells through myeloid-derived suppressor cells. Commun. Biol. 2021, 4, 859. [Google Scholar] [CrossRef] [PubMed]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage polarization: Different gene signatures in M1(Lps+) vs. Classically and M2(LPS-) vs. Alternatively activated macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef]
- Wong, S.C.; Puaux, A.-L.; Chittezhath, M.; Shalova, I.; Kajiji, T.S.; Wang, X.; Abastado, J.-P.; Lam, K.-P.; Biswas, S.K. Macrophage polarization to a unique phenotype driven by B cells. Eur. J. Immunol. 2010, 40, 2296–2307. [Google Scholar] [CrossRef]
- Pereira, A.; Alvares-Saraiva, A.M.; Konno, F.T.d.C.; Spadacci-Morena, D.D.; Perez, E.C.; Mariano, M.; Lallo, M.A. B-1 cell-mediated modulation of M1 macrophage profile ameliorates microbicidal functions and disrupt the evasion mechanisms of Encephalitozoon cuniculi. PLoS Negl. Trop. Dis. 2019, 13, e0007674. [Google Scholar] [CrossRef] [Green Version]
- Summers, B.C.; Leib, D.A. Herpes Simplex Virus Type 1 Origins of DNA Replication Play No Role in the Regulation of Flanking Promoters. J. Virol. 2002, 76, 7020–7029. [Google Scholar] [CrossRef] [Green Version]
- Györffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1809 patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fekete, J.T.; Győrffy, B. ROCplot.org: Validating predictive biomarkers of chemotherapy/hormonal therapy/anti-HER2 therapy using transcriptomic data of 3,104 breast cancer patients. Int. J. Cancer 2019, 145, 3140–3151. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Jin, D.; Chen, X. Lipocalin 2 is a regulator of macrophage polarization and NF-kB/STAT3 pathway activation. Mol. Endocrinol. 2014, 28, 1616–1628. [Google Scholar] [CrossRef] [Green Version]
- Patterson, D.C.; Ignatowicz, A.P.; Samuels, J.S.; Rayalam, S.; Shashidharamurthy, R. Lipocalin2, an innate immune protein, modulates iNOS and arginase-1 in macrophages to promote the anti-inflammatory function. J. Immunol. 2017, 198, 206–216. [Google Scholar]
- Sanada, Y.; Yamamoto, T.; Satake, R.; Yamashita, A.; Kanai, S.; Kato, N.; Van De Loo, F.A.; Nishimura, F.; Scherer, P.E.; Yanaka, N. Serum Amyloid A3 Gene Expression in Adipocytes is an Indicator of the Interaction with Macrophages. Sci. Rep. 2016, 6, 38697. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhou, H.; Zhu, Z.; Yan, Q.; Wang, L.; Liang, Q.; Ye, R.D. Ex Vivo and In Vitro Effect of Serum Amyloid A in the Induction of Macrophage M2 Markers and Efferocytosis of Apoptotic Neutrophils. J. Immunol. 2015, 194, 4891–4900. [Google Scholar] [CrossRef]
- Torraca, V.; Cui, C.; Boland, R.; Bebelman, J.-P.; van der Sar, A.; Smit, M.; Siderius, M.; Spaink, H.; Meijer, A. The CXCR3-CXCL11 signaling axis mediates macrophage recruitment and dissemination of mycobacterial infection. Co. Biol. 2015, 7, 253–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibaldi, D.; Vilar-Pereira, G.; Pereira, I.R.; Silva, A.A.; Barrios, L.C.; Ramos, I.P.; Mata dos Santos, H.A.; Gazzinelli, R.; Lannes-Vieira, J. CCL3/Macrophage Inflammatory Protein-1α Is Dually Involved in Parasite Persistence and Induction of a TNF- and IFNγ-Enriched Inflammatory Milieu in Trypanosoma cruzi-Induced Chronic Cardiomyopathy. Front. Immunol. 2020, 11, 306. [Google Scholar] [CrossRef]
- Rider, P.; Carmi, Y.; Guttman, O.; Braiman, A.; Cohen, I.; Voronov, E.; White, M.R.; Dinarello, C.A.; Apte, R.N. IL-1α and IL-1β Recruit Different Myeloid Cells and Promote Different Stages of Sterile Inflammation. J. Immunol. 2011, 187, 4835–4843. [Google Scholar] [CrossRef] [Green Version]
- Lv, L.L.; Tang, P.M.K.; Li, C.J.; You, Y.K.; Li, J.; Huang, X.R.; Ni, J.; Feng, M.; Liu, B.C.; Lan, H.Y. The pattern recognition receptor, Mincle, is essential for maintaining the M1 macrophage phenotype in acute renal inflammation. Kidney Int. 2017, 91, 587–602. [Google Scholar] [CrossRef]
- Li, J.; Chen, J.; Zhang, M.; Zhang, C.; Wu, R.; Yang, T.; Qiu, Y.; Liu, J.; Zhu, T.; Zhang, Y.; et al. The mTOR Deficiency in Monocytic Myeloid-Derived Suppressor Cells Protects Mouse Cardiac Allografts by Inducing Allograft Tolerance. Front. Immunol. 2021, 12, 995. [Google Scholar] [CrossRef]
- Rodriguez, R.M.; Suarez-Alvarez, B.; Lopez-Larrea, C. Therapeutic Epigenetic Reprogramming of Trained Immunity in Myeloid Cells. Trends Immunol. 2019, 40, 66–80. [Google Scholar] [CrossRef]
- Chen, S.; Yang, J.; Wei, Y.; Wei, X. Epigenetic regulation of macrophages: From homeostasis maintenance to host defense. Cell. Mol. Immunol. 2020, 17, 36–49. [Google Scholar] [CrossRef] [Green Version]
- Álvarez-Errico, D.; Vento-Tormo, R.; Sieweke, M.; Ballestar, E. Epigenetic control of myeloid cell differentiation, identity and function. Nat. Rev. Immunol. 2015, 15, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Hua, Q.; Zheng, L. Generation of Myeloid Cells in Cancer: The Spleen Matters. Front. Immunol. 2020, 11, 1126. [Google Scholar] [CrossRef]
- Wang, S.; Song, R.; Wang, Z.; Jing, Z.; Wang, S.; Ma, J. S100A8/A9 in inflammation. Front. Immunol. 2018, 9, 1298. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Chitayat, S.; Hashemi, M.; Eshraghi, M.; Chazin, W.J.; Halayko, A.J.; Kerkhoff, C. S100A8/A9: A Janus-faced molecule in cancer therapy and tumorgenesis. Eur. J. Pharmacol. 2009, 625, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Rashedi, I.; Dattilo, B.M.; Eshraghi, M.; Chazin, W.J.; Hashemi, M.; Wesselborg, S.; Kerkhoff, C.; Los, M. S100A8/A9 at low concentration promotes tumor cell growth via RAGE ligation and MAP kinase-dependent pathway. J. Leukoc. Biol. 2008. [Google Scholar] [CrossRef] [PubMed]
- Turovskaya, O.; Foell, D.; Sinha, P.; Vogl, T.; Newlin, R.; Nayak, J.; Nguyen, M.; Olsson, A.; Nawroth, P.P.; Bierhaus, A.; et al. RAGE, carboxylated glycans and S100A8/A9 play essential roles in colitis-associated carcinogenesis. Carcinogenesis 2008, 83, 1484–1492. [Google Scholar] [CrossRef]
- Sieow, J.L.; Gun, S.Y.; Wong, S.C. The sweet surrender: How myeloid cell metabolic plasticity shapes the tumor microenvironment. Front. Cell Dev. Biol. 2018, 6, 168. [Google Scholar] [CrossRef] [Green Version]
- Cha, Y.J.; Koo, J.S. Role of Tumor-Associated Myeloid Cells in Breast Cancer. Cells 2020, 9, 1785. [Google Scholar] [CrossRef]
- Goyette, J.; Geczy, C.L. Inflammation-associated S100 proteins: New mechanisms that regulate function. Amino Acids 2011, 41, 821–842. [Google Scholar] [CrossRef]
- Bagheri, V.; Apostolopoulos, V. Pro-inflammatory S100A9 Protein: A Double-Edged Sword in Cancer? Inflammation 2019, 42, 1137–1138. [Google Scholar] [CrossRef] [PubMed]
- Srikrishna, G. S100A8 and S100A9: New insights into their roles in malignancy. J. Innate Immun. 2011, 4, 31–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trikha, P.; Carson, W.E. Signaling pathways involved in MDSC regulation. Biochim. Biophys. Acta—Rev. Cancer 2014, 1846, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Zhong, X.; Xie, F.; Chen, L.; Liu, Z.; Wang, Q. S100A8 and S100A9 promote endothelial cell activation through the RAGE-mediated mammalian target of rapamycin complex 2 pathway. Mol. Med. Rep. 2020, 22, 5293–5303. [Google Scholar] [CrossRef] [PubMed]
- Ohata, H.; Shiokawa, D.; Obata, Y.; Sato, A.; Sakai, H.; Fukami, M.; Hara, W.; Taniguchi, H.; Ono, M.; Nakagama, H.; et al. NOX1-Dependent mTORC1 Activation via S100A9 Oxidation in Cancer Stem-like Cells Leads to Colon Cancer Progression. Cell Rep. 2019, 28, 1282–1295.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidiropoulos, D.N.; Rafie, C.; Christmas, B.J.; Davis-Marcisak, E.F.; Sharma, G.; Bigelow, E.; Gupta, A.; Yegnasubramanian, S.; Stearns, V.; Connolly, R.M.; et al. Phenotypic shifts of tumor associated macrophages and STAT3 mediated suppression of myeloid derived suppressor cells drive sensitization of HER2 + tumor immunity. bioRxiv 2021. [Google Scholar] [CrossRef]
- Loi, S.; Sirtaine, N.; Piette, F.; Salgado, R.; Viale, G.; Van Eenoo, F.; Rouas, G.; Francis, P.; Crown, J.P.A.; Hitre, E.; et al. Prognostic and predictive value of tumor-infiltrating lymphocytes in a phase III randomized adjuvant breast cancer trial in node-positive breast cancer comparing the addition of docetaxel to doxorubicin with doxorubicin-based chemotherapy: BIG 02-98. J. Clin. Oncol. 2013, 31, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; Gray, R.J.; Demaria, S.; Goldstein, L.; Perez, E.A.; Shulman, L.N.; Martino, S.; Wang, M.; Jones, V.E.; Saphner, T.J.; et al. Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase III randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J. Clin. Oncol. 2014, 32, 2959. [Google Scholar] [CrossRef] [PubMed]
- Denkert, C.; von Minckwitz, G.; Darb-Esfahani, S.; Lederer, B.; Heppner, B.I.; Weber, K.E.; Budczies, J.; Huober, J.; Klauschen, F.; Furlanetto, J.; et al. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: A pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol. 2018, 19, 40–50. [Google Scholar] [CrossRef]
- Oshi, M.; Tokumaru, Y.; Asaoka, M.; Yan, L.; Satyananda, V.; Matsuyama, R.; Matsuhashi, N.; Futamura, M.; Ishikawa, T.; Yoshida, K.; et al. M1 Macrophage and M1/M2 ratio defined by transcriptomic signatures resemble only part of their conventional clinical characteristics in breast cancer. Sci. Rep. 2020, 10, 16554. [Google Scholar] [CrossRef]
- Kwak, T.; Wang, F.; Deng, H.; Condamine, T.; Kumar, V.; Perego, M.; Kossenkov, A.; Montaner, L.J.; Xu, X.; Xu, W.; et al. Distinct Populations of Immune-Suppressive Macrophages Differentiate from Monocytic Myeloid-Derived Suppressor Cells in Cancer. Cell Rep. 2020, 33, 108571. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene. | F + O vs. P(Fold Change) | Function |
|---|---|---|
| LCN2 | 632.83 | Regulator of macrophage polarization via STAT3 activation [19] Modulates iNOS and Arg1 in macrophages to promote the anti-inflammatory function [20] |
| CXCL2 | 308.84 | Secreted by macrophages, regulator of myeloid cell migration |
| SAA3 | 180.26 | Marker of macrophage infiltration [21] Promotes macrophage differentiation [22] |
| CXCL3 | 50.83 | Mediator of macrophage recruitment [23] |
| CCL3 | 37.62 | Macrophage recruitment [24] |
| IL1A | 34.53 | Associated with activated macrophages [13] |
| IL1B | 27.81 | Associated with activated macrophages [13] Macrophage recruitment [25] |
| CLEC4E | 13.73 | Macrophage activation and differentiation [26] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vito, A.; El-Sayes, N.; Salem, O.; Wan, Y.; Mossman, K.L. Response to FEC Chemotherapy and Oncolytic HSV-1 Is Associated with Macrophage Polarization and Increased Expression of S100A8/A9 in Triple Negative Breast Cancer. Cancers 2021, 13, 5590. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13215590
Vito A, El-Sayes N, Salem O, Wan Y, Mossman KL. Response to FEC Chemotherapy and Oncolytic HSV-1 Is Associated with Macrophage Polarization and Increased Expression of S100A8/A9 in Triple Negative Breast Cancer. Cancers. 2021; 13(21):5590. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13215590
Chicago/Turabian StyleVito, Alyssa, Nader El-Sayes, Omar Salem, Yonghong Wan, and Karen L. Mossman. 2021. "Response to FEC Chemotherapy and Oncolytic HSV-1 Is Associated with Macrophage Polarization and Increased Expression of S100A8/A9 in Triple Negative Breast Cancer" Cancers 13, no. 21: 5590. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13215590