
The MEK1/2 Pathway as a Therapeutic Target in High-Grade Serous Ovarian Carcinoma


 ,
,  ,
,
Abstract
:Simple Summary
Abstract
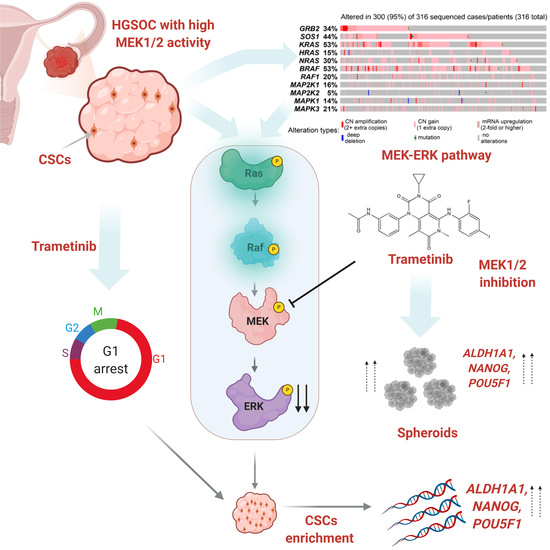
{kind=link}
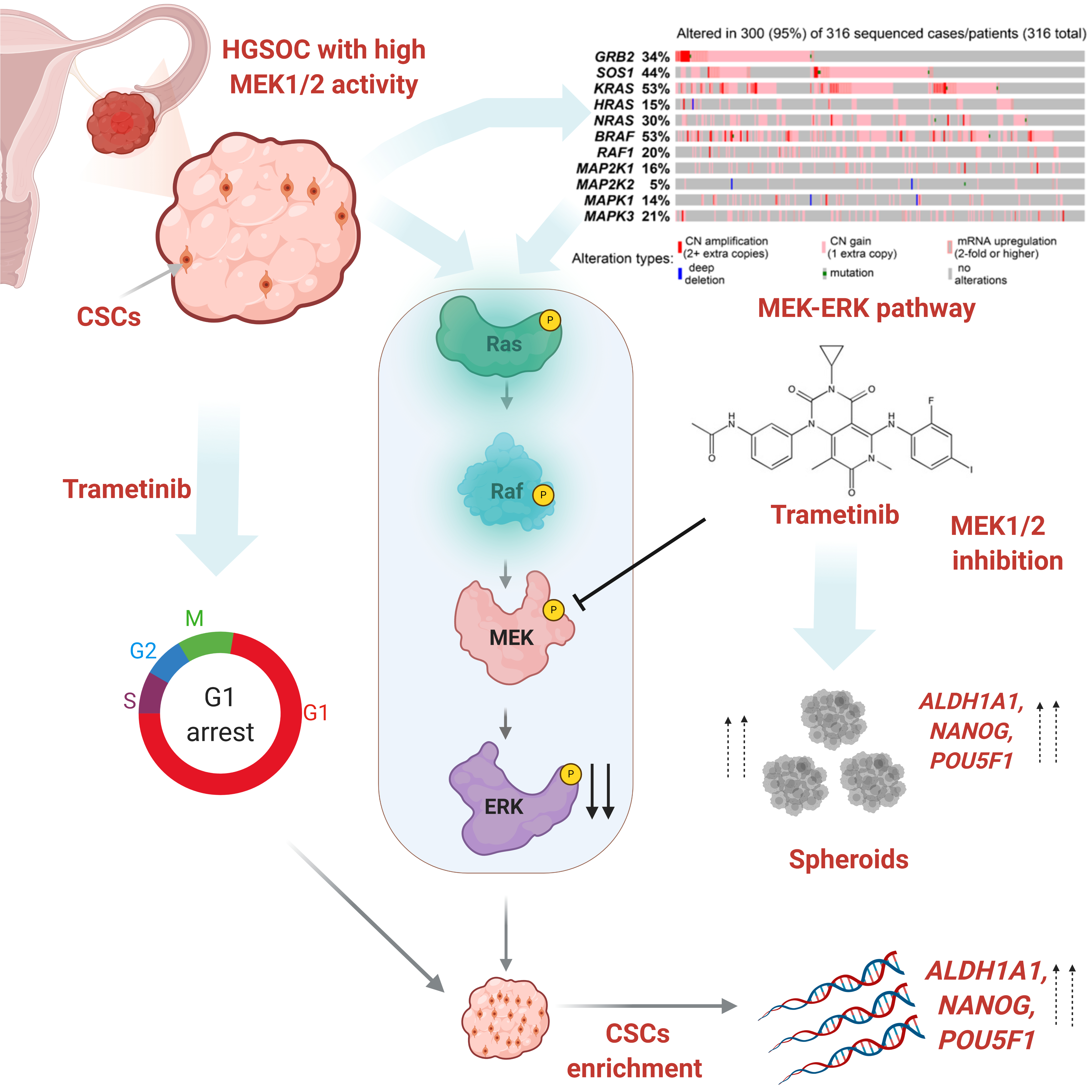
{kind=link}
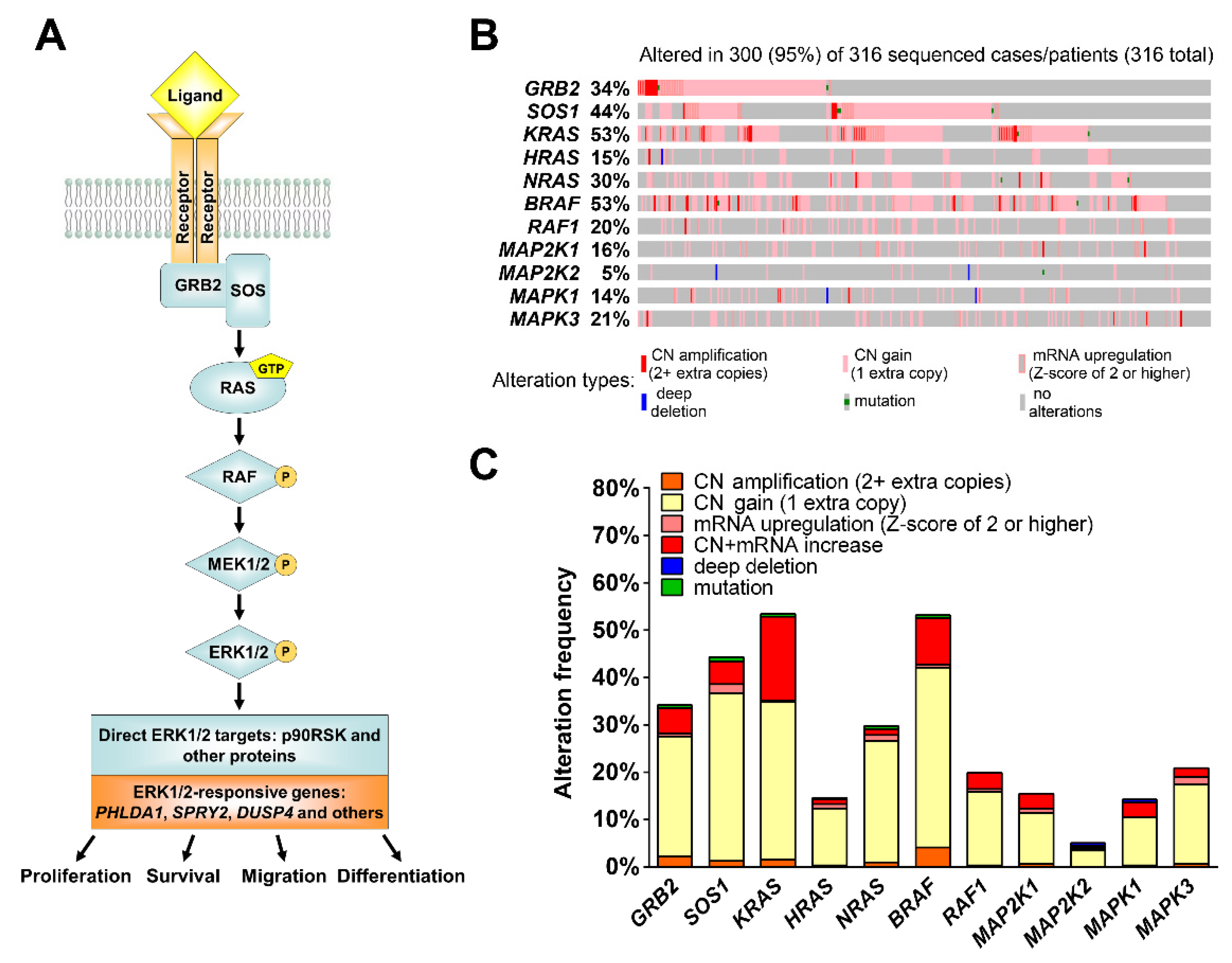
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Clinical Tissue Samples
2.2. Cell Culture
2.3. Drug Treatment of Cultured Cells
2.4. Immunoblotting
2.5. Immunohistochemical Staining of Tissue Slides
2.6. Immunofluorescent Staining of Tissue Slides
2.7. RT-qPCR Analysis of Gene Expression
2.8. Cell Viability Assay
2.9. Real-Time Cytotoxicity Assay
2.10. Average Cell Size Estimation
2.11. Cell Cycle Assay
2.12. Knockdown of Gene Expression with siRNA
2.13. Estimation of CD133+ Cell Fraction
2.14. Estimation of ALDH Activity
2.15. Spheroid Cell Growth Assay
2.16. Animal Studies
2.17. Statistical Analysis
3. Results
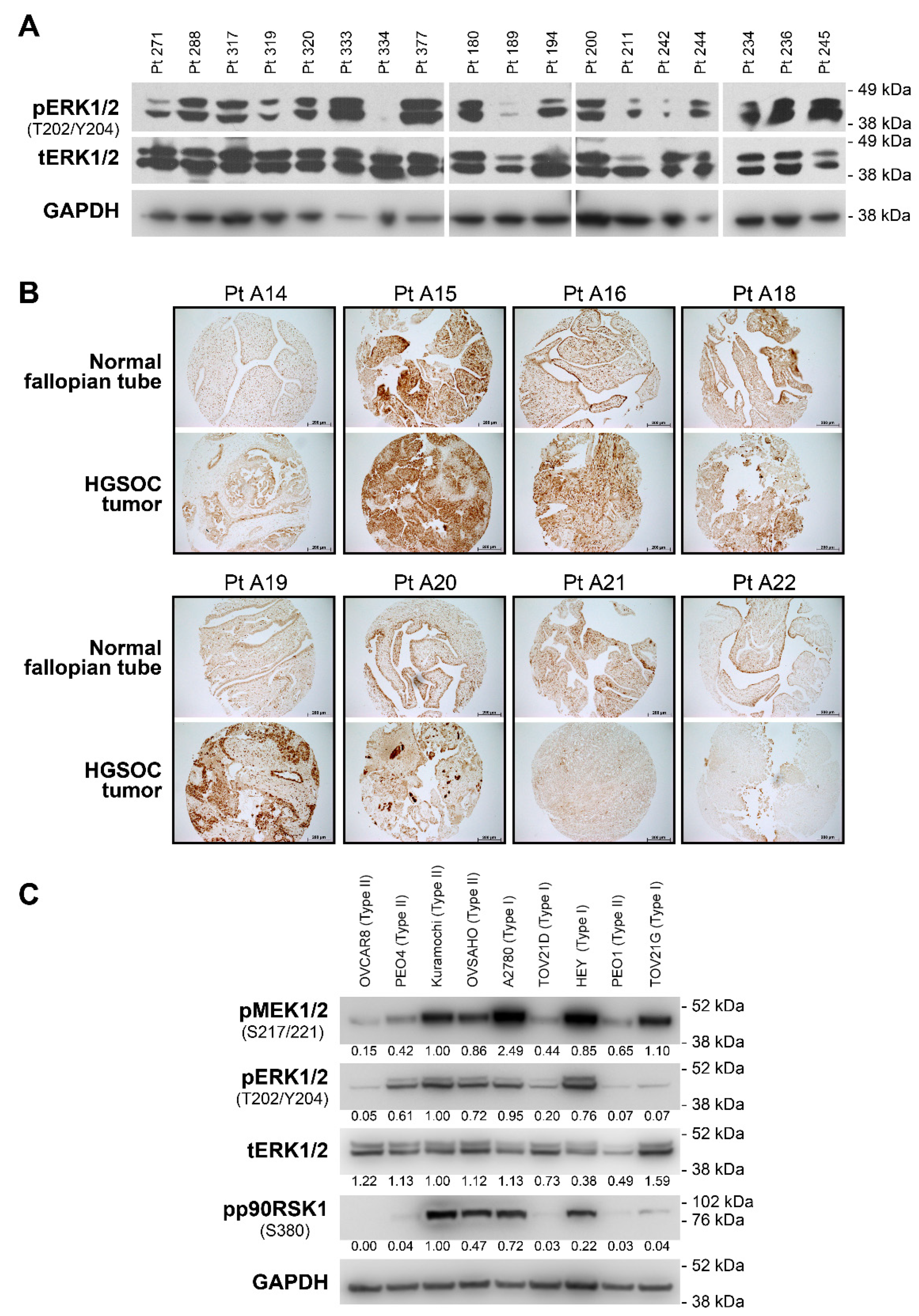
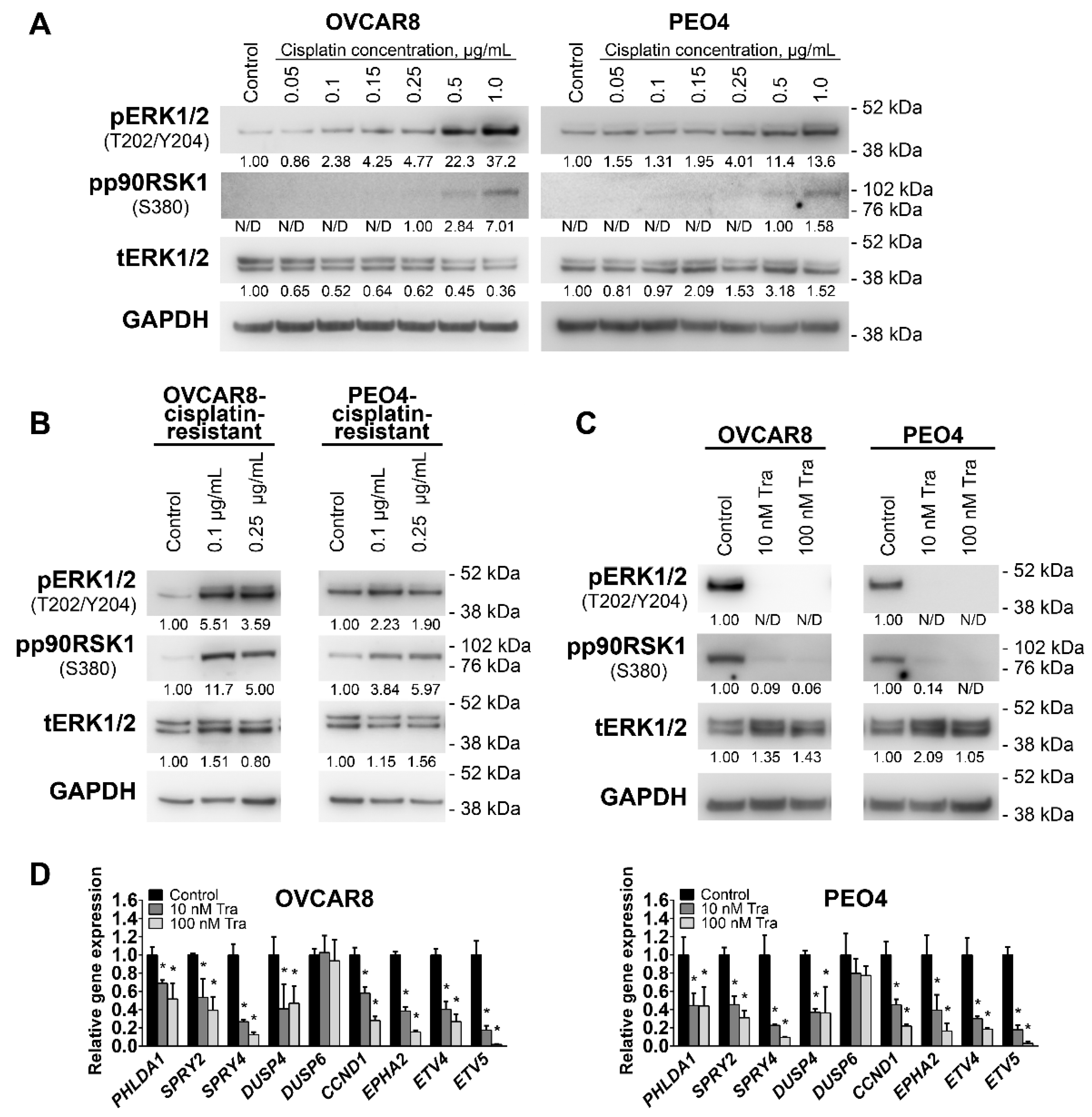
3.1. The MEK1/2 Pathway Is Active in High Grade Ovarian Tumors
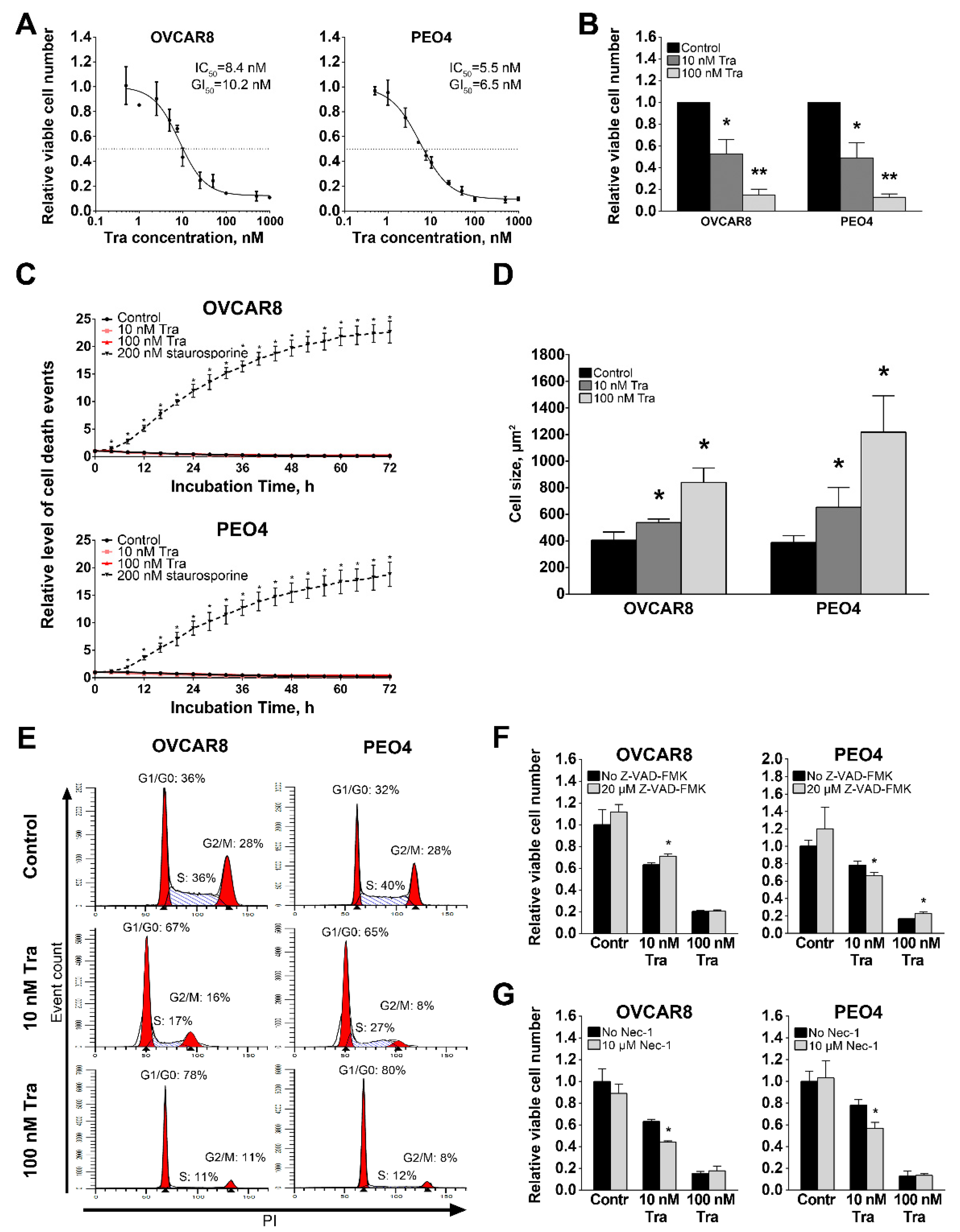
3.2. Inhibition of MEK1/2 Causes Arrest of HGSOC Cell Proliferation without Inducing Cell Death
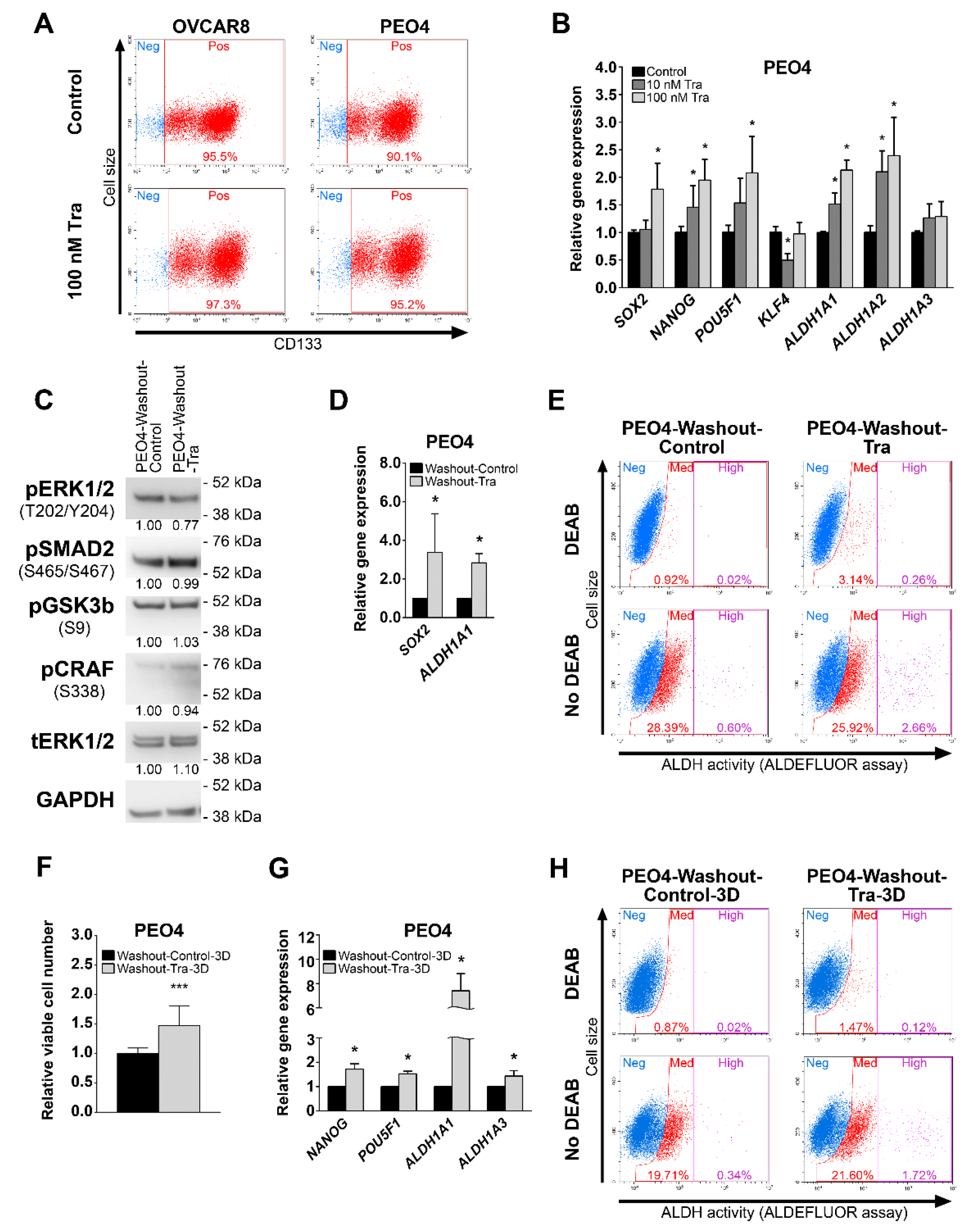
3.3. Trametinib Promotes Stemness of HGSOC Cells
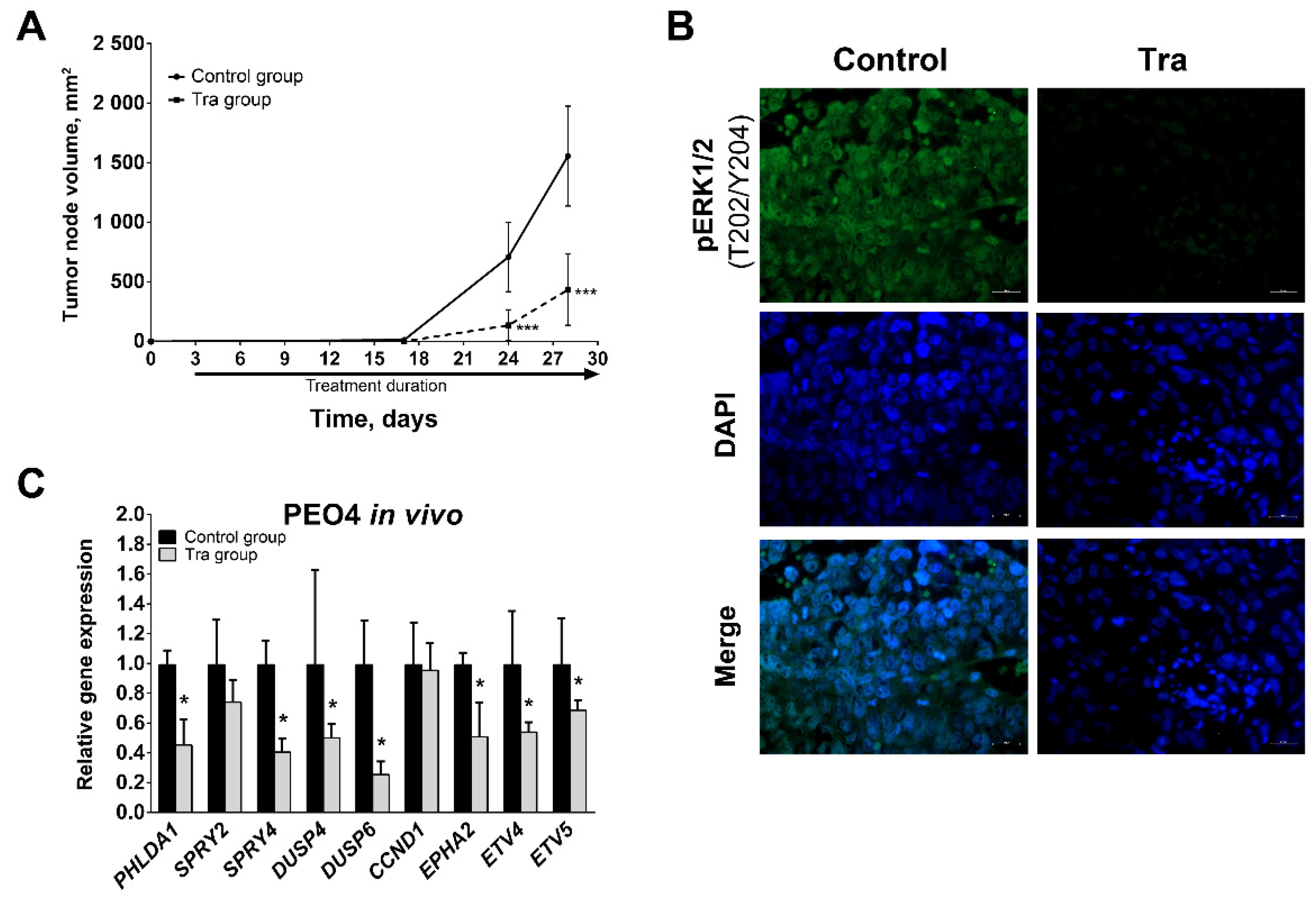
3.4. Effect of MEK1/2 Inhibition In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih, I.M. The dualistic model of ovarian carcinogenesis: Revisited, revised, and expanded. Am. J. Pathol. 2016, 186, 733–747. [Google Scholar] [CrossRef] [Green Version]
- Cooke, S.L.; Brenton, J.D. Evolution of platinum resistance in high-grade serous ovarian cancer. Lancet Oncol. 2011, 12, 1169–1174. [Google Scholar] [CrossRef]
- Vaughan, S.; Coward, J.I.; Bast, R.C., Jr.; Berchuck, A.; Berek, J.S.; Brenton, J.D.; Coukos, G.; Crum, C.C.; Drapkin, R.; Etemadmoghadam, D.; et al. Rethinking ovarian cancer: Recommendations for improving outcomes. Nat. Rev. Cancer 2011, 11, 719–725. [Google Scholar] [CrossRef] [Green Version]
- Morgan, R.J., Jr.; Armstrong, D.K.; Alvarez, R.D.; Bakkum-Gamez, J.N.; Behbakht, K.; Chen, L.M.; Copeland, L.; Crispens, M.A.; DeRosa, M.; Dorigo, O.; et al. Ovarian cancer, version 1.2016, nccn clinical practice guidelines in oncology. J. Natl. Compr. Canc. Netw. 2016, 14, 1134–1163. [Google Scholar] [CrossRef] [PubMed]
- Raja, F.A.; Chopra, N.; Ledermann, J.A. Optimal first-line treatment in ovarian cancer. Ann. Oncol. 2012, 23 (Suppl. 10), x118–x127. [Google Scholar] [CrossRef]
- Davis, A.; Tinker, A.V.; Friedlander, M. “Platinum resistant” ovarian cancer: What is it, who to treat and how to measure benefit? Gynecol. Oncol. 2014, 133, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Cortez, A.J.; Tudrej, P.; Kujawa, K.A.; Lisowska, K.M. Advances in ovarian cancer therapy. Cancer Chemother. Pharmacol. 2018, 81, 17–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christie, E.L.; Bowtell, D.D.L. Acquired chemotherapy resistance in ovarian cancer. Ann. Oncol. 2017, 28, viii13–viii15. [Google Scholar] [CrossRef]
- Agarwal, R.; Kaye, S.B. Ovarian cancer: Strategies for overcoming resistance to chemotherapy. Nat. Rev. Cancer 2003, 3, 502–516. [Google Scholar] [CrossRef] [PubMed]
- Freimund, A.E.; Beach, J.A.; Christie, E.L.; Bowtell, D.D.L. Mechanisms of drug resistance in high-grade serous ovarian cancer. Hematol. Oncol. Clin. N. Am. 2018, 32, 983–996. [Google Scholar] [CrossRef]
- Norouzi-Barough, L.; Sarookhani, M.R.; Sharifi, M.; Moghbelinejad, S.; Jangjoo, S.; Salehi, R. Molecular mechanisms of drug resistance in ovarian cancer. J. Cell. Physiol. 2018, 233, 4546–4562. [Google Scholar] [CrossRef]
- Mittempergher, L. Genomic characterization of high-grade serous ovarian cancer: Dissecting its molecular heterogeneity as a road towards effective therapeutic strategies. Curr. Oncol. Rep. 2016, 18, 44. [Google Scholar] [CrossRef]
- Yin, X.; Jing, Y.; Cai, M.C.; Ma, P.; Zhang, Y.; Xu, C.; Zhang, M.; Di, W.; Zhuang, G. Clonality, heterogeneity, and evolution of synchronous bilateral ovarian cancer. Cancer Res. 2017, 77, 6551–6561. [Google Scholar] [CrossRef] [Green Version]
- Cooke, S.L.; Ng, C.K.; Melnyk, N.; Garcia, M.J.; Hardcastle, T.; Temple, J.; Langdon, S.; Huntsman, D.; Brenton, J.D. Genomic analysis of genetic heterogeneity and evolution in high-grade serous ovarian carcinoma. Oncogene 2010, 29, 4905–4913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chefetz, I.; Alvero, A.B.; Holmberg, J.C.; Lebowitz, N.; Craveiro, V.; Yang-Hartwich, Y.; Yin, G.; Squillace, L.; Gurrea Soteras, M.; Aldo, P.; et al. Tlr2 enhances ovarian cancer stem cell self-renewal and promotes tumor repair and recurrence. Cell Cycle 2013, 12, 511–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mor, G.; Yin, G.; Chefetz, I.; Yang, Y.; Alvero, A. Ovarian cancer stem cells and inflammation. Cancer Biol. Ther. 2011, 11, 708–713. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.H.; Dou, H.T.; Xu, P.; Zhuang, S.C.; Liu, P.S. Tumor recurrence and drug resistance properties of side population cells in high grade ovary cancer. Drug Res. (Stuttg.) 2015, 65, 153–157. [Google Scholar] [CrossRef]
- Kuroda, T.; Hirohashi, Y.; Torigoe, T.; Yasuda, K.; Takahashi, A.; Asanuma, H.; Morita, R.; Mariya, T.; Asano, T.; Mizuuchi, M.; et al. Aldh1-high ovarian cancer stem-like cells can be isolated from serous and clear cell adenocarcinoma cells, and aldh1 high expression is associated with poor prognosis. PLoS ONE 2013, 8, e65158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruscito, I.; Cacsire Castillo-Tong, D.; Vergote, I.; Ignat, I.; Stanske, M.; Vanderstichele, A.; Ganapathi, R.N.; Glajzer, J.; Kulbe, H.; Trillsch, F.; et al. Exploring the clonal evolution of cd133/aldehyde-dehydrogenase-1 (aldh1)-positive cancer stem-like cells from primary to recurrent high-grade serous ovarian cancer (hgsoc). A study of the ovarian cancer therapy-innovative models prolong survival (octips) consortium. Eur. J. Cancer 2017, 79, 214–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chefetz, I.; Grimley, E.; Yang, K.; Hong, L.; Vinogradova, E.V.; Suciu, R.; Kovalenko, I.; Karnak, D.; Morgan, C.A.; Chtcherbinine, M.; et al. A pan-aldh1a inhibitor induces necroptosis in ovarian cancer stem-like cells. Cell Rep. 2019, 26, 3061–3075.e3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chefetz, I.; Holmberg, J.C.; Alvero, A.B.; Visintin, I.; Mor, G. Inhibition of aurora-a kinase induces cell cycle arrest in epithelial ovarian cancer stem cells by affecting nfkb pathway. Cell Cycle 2011, 10, 2206–2214. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, A.; Hong, L.; Chefetz, I. How to win the ovarian cancer stem cell battle: Destroying the roots. Cancer Drug Resist. 2020, 4, 1021–1033. [Google Scholar] [CrossRef]
- O’Connor, M.L.; Xiang, D.; Shigdar, S.; Macdonald, J.; Li, Y.; Wang, T.; Pu, C.; Wang, Z.; Qiao, L.; Duan, W. Cancer stem cells: A contentious hypothesis now moving forward. Cancer Lett. 2014, 344, 180–187. [Google Scholar] [CrossRef]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef]
- Martins-Neves, S.R.; Cleton-Jansen, A.M.; Gomes, C.M.F. Therapy-induced enrichment of cancer stem-like cells in solid human tumors: Where do we stand? Pharmacol. Res. 2018, 137, 193–204. [Google Scholar] [CrossRef]
- Rybak, A.P.; Ingram, A.J.; Tang, D. Propagation of human prostate cancer stem-like cells occurs through egfr-mediated erk activation. PLoS ONE 2013, 8, e61716. [Google Scholar] [CrossRef] [Green Version]
- Ahn, H.J.; Kim, G.; Park, K.S. Ell3 stimulates proliferation, drug resistance, and cancer stem cell properties of breast cancer cells via a mek/erk-dependent signaling pathway. Biochem. Biophys. Res. Commun. 2013, 437, 557–564. [Google Scholar] [CrossRef]
- Wang, Y.; Lin, X.; Fu, X.; Yan, W.; Lin, F.; Kuang, P.; Luo, Y.; Lin, E.; Hong, X.; Wu, G. Long non-coding rna bancr regulates cancer stem cell markers in papillary thyroid cancer via the raf/mek/erk signaling pathway. Oncol. Rep. 2018, 40, 859–866. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.T. Mapk signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Burotto, M.; Chiou, V.L.; Lee, J.M.; Kohn, E.C. The mapk pathway across different malignancies: A new perspective. Cancer 2014, 120, 3446–3456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the raf/mek/erk pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caunt, C.J.; Sale, M.J.; Smith, P.D.; Cook, S.J. Mek1 and mek2 inhibitors and cancer therapy: The long and winding road. Nat. Rev. Cancer 2015, 15, 577–592. [Google Scholar] [CrossRef]
- Mandal, R.; Becker, S.; Strebhardt, K. Stamping out raf and mek1/2 to inhibit the erk1/2 pathway: An emerging threat to anticancer therapy. Oncogene 2016, 35, 2547–2561. [Google Scholar] [CrossRef] [PubMed]
- Singer, G.; Oldt, R., 3rd; Cohen, Y.; Wang, B.G.; Sidransky, D.; Kurman, R.J.; Shih Ie, M. Mutations in braf and kras characterize the development of low-grade ovarian serous carcinoma. J. Natl. Cancer Inst. 2003, 95, 484–486. [Google Scholar] [CrossRef] [Green Version]
- Miller, C.R.; Oliver, K.E.; Farley, J.H. Mek1/2 inhibitors in the treatment of gynecologic malignancies. Gynecol. Oncol. 2014, 133, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Hew, K.E.; Miller, P.C.; El-Ashry, D.; Sun, J.; Besser, A.H.; Ince, T.A.; Gu, M.; Wei, Z.; Zhang, G.; Brafford, P.; et al. Mapk activation predicts poor outcome and the mek inhibitor, selumetinib, reverses antiestrogen resistance in er-positive high-grade serous ovarian cancer. Clin. Cancer Res. 2016, 22, 935–947. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhou, J.Y.; Wu, G.S. Erk-dependent mkp-1-mediated cisplatin resistance in human ovarian cancer cells. Cancer Res. 2007, 67, 11933–11941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wu, G.S. Role of autophagy in cisplatin resistance in ovarian cancer cells. J. Biol. Chem. 2014, 289, 17163–17173. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cbio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cbioportal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Silva, I.A.; Bai, S.; McLean, K.; Yang, K.; Griffith, K.; Thomas, D.; Ginestier, C.; Johnston, C.; Kueck, A.; Reynolds, R.K.; et al. Aldehyde dehydrogenase in combination with cd133 defines angiogenic ovarian cancer stem cells that portend poor patient survival. Cancer Res. 2011, 71, 3991–4001. [Google Scholar] [CrossRef] [Green Version]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time rt-pcr. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; DeZonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. Imagej2: Imagej for the next generation of scientific image data. BMC Bioinf. 2017, 18, 529. [Google Scholar] [CrossRef]
- Bengtsson, M.; Stahlberg, A.; Rorsman, P.; Kubista, M. Gene expression profiling in single cells from the pancreatic islets of langerhans reveals lognormal distribution of mrna levels. Genome Res. 2005, 15, 1388–1392. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Yoon, S.; Kim, D.H. A high nuclear basal level of erk2 phosphorylation contributes to the resistance of cisplatin-resistant human ovarian cancer cells. Gynecol. Oncol. 2007, 104, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Kielbik, M.; Krzyzanowski, D.; Pawlik, B.; Klink, M. Cisplatin-induced erk1/2 activity promotes g1 to s phase progression which leads to chemoresistance of ovarian cancer cells. Oncotarget 2018, 9, 19847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, T.; Tischkowitz, M.; Ameziane, N.; Hodgson, S.V.; Mathew, C.G.; Joenje, H.; Mok, S.C.; D’Andrea, A.D. Disruption of the fanconi anemia-brca pathway in cisplatin-sensitive ovarian tumors. Nat. Med. 2003, 9, 568–574. [Google Scholar] [CrossRef]
- Haley, J.; Tomar, S.; Pulliam, N.; Xiong, S.; Perkins, S.M.; Karpf, A.R.; Mitra, S.; Nephew, K.P.; Mitra, A.K. Functional characterization of a panel of high-grade serous ovarian cancer cell lines as representative experimental models of the disease. Oncotarget 2016, 7, 32810–32820. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.E.; Wei, B.R.; Madigan, J.P.; Hall, M.D.; Simpson, R.M.; Zhuang, Z.; Gottesman, M.M. The protein phosphatase 2a inhibitor lb100 sensitizes ovarian carcinoma cells to cisplatin-mediated cytotoxicity. Mol. Cancer Ther. 2015, 14, 90–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagle, M.C.; Kirouac, D.; Klijn, C.; Liu, B.; Mahajan, S.; Junttila, M.; Moffat, J.; Merchant, M.; Huw, L.; Wongchenko, M.; et al. A transcriptional mapk pathway activity score (mpas) is a clinically relevant biomarker in multiple cancer types. NPJ Precis. Oncol. 2018, 2, 7. [Google Scholar] [CrossRef] [Green Version]
- Khan, I.; Yousif, A.; Chesnokov, M.; Hong, L.; Chefetz, I. A decade of cell death studies: Breathing new life into necroptosis. Pharmacol. Ther. 2020, 107717. [Google Scholar] [CrossRef]
- Foster, R.; Buckanovich, R.J.; Rueda, B.R. Ovarian cancer stem cells: Working towards the root of stemness. Cancer Lett. 2013, 338, 147–157. [Google Scholar] [CrossRef]
- Roy, L.; Cowden Dahl, K.D. Can stemness and chemoresistance be therapeutically targeted via signaling pathways in ovarian cancer? Cancers 2018, 10, 241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. Map kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Zha, J.; Lei, M. Inhibiting erk/mnk/eif4e broadly sensitizes ovarian cancer response to chemotherapy. Clin. Transl. Oncol. 2018, 20, 374–381. [Google Scholar] [CrossRef]
- Gilmartin, A.G.; Bleam, M.R.; Groy, A.; Moss, K.G.; Minthorn, E.A.; Kulkarni, S.G.; Rominger, C.M.; Erskine, S.; Fisher, K.E.; Yang, J.; et al. Gsk1120212 (jtp-74057) is an inhibitor of mek activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin. Cancer Res. 2011, 17, 989–1000. [Google Scholar] [CrossRef] [Green Version]
- Yeh, T.C.; Marsh, V.; Bernat, B.A.; Ballard, J.; Colwell, H.; Evans, R.J.; Parry, J.; Smith, D.; Brandhuber, B.J.; Gross, S.; et al. Biological characterization of arry-142886 (azd6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin. Cancer Res. 2007, 13, 1576–1583. [Google Scholar] [CrossRef] [Green Version]
- Hoeflich, K.P.; Merchant, M.; Orr, C.; Chan, J.; Den Otter, D.; Berry, L.; Kasman, I.; Koeppen, H.; Rice, K.; Yang, N.Y.; et al. Intermittent administration of mek inhibitor gdc-0973 plus pi3k inhibitor gdc-0941 triggers robust apoptosis and tumor growth inhibition. Cancer Res. 2012, 72, 210–219. [Google Scholar] [CrossRef] [Green Version]
- Simpkins, F.; Jang, K.; Yoon, H.; Hew, K.E.; Kim, M.; Azzam, D.J.; Sun, J.; Zhao, D.; Ince, T.A.; Liu, W.; et al. Dual src and mek inhibition decreases ovarian cancer growth and targets tumor initiating stem-like cells. Clin. Cancer Res. 2018, 24, 4874–4886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baba, T.; Convery, P.A.; Matsumura, N.; Whitaker, R.S.; Kondoh, E.; Perry, T.; Huang, Z.; Bentley, R.C.; Mori, S.; Fujii, S.; et al. Epigenetic regulation of cd133 and tumorigenicity of cd133+ ovarian cancer cells. Oncogene 2009, 28, 209–218. [Google Scholar] [CrossRef] [Green Version]
- Curley, M.D.; Therrien, V.A.; Cummings, C.L.; Sergent, P.A.; Koulouris, C.R.; Friel, A.M.; Roberts, D.J.; Seiden, M.V.; Scadden, D.T.; Rueda, B.R.; et al. Cd133 expression defines a tumor initiating cell population in primary human ovarian cancer. Stem Cells 2009, 27, 2875–2883. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Liu, S.; Roh, M.; Vatan, L.; Szeliga, W.; Wei, S.; Banerjee, M.; Mao, Y.; Kotarski, J.; Wicha, M.S.; et al. Expression of aldehyde dehydrogenase and cd133 defines ovarian cancer stem cells. Int. J. Cancer 2012, 130, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.J.; Ingram, P.N.; Yang, K.; Coffman, L.; Iyengar, M.; Bai, S.; Thomas, D.G.; Yoon, E.; Buckanovich, R.J. Identifying an ovarian cancer cell hierarchy regulated by bone morphogenetic protein 2. Proc. Natl. Acad. Sci. USA 2015, 112, E6882–E6888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fremin, C.; Meloche, S. From basic research to clinical development of mek1/2 inhibitors for cancer therapy. J. Hematol. Oncol. 2010, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Meloche, S.; Pouyssegur, J. The erk1/2 mitogen-activated protein kinase pathway as a master regulator of the g1- to s-phase transition. Oncogene 2007, 26, 3227–3239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Adjei, A.A. The clinical development of mek inhibitors. Nat. Rev. Clin. Oncol. 2014, 11, 385–400. [Google Scholar] [CrossRef] [PubMed]
- Cappuccio, S.; Distefano, M.G.; Ghizzoni, V.; Fagotti, A.; Scambia, G. Trametinib response in heavily pretreated high-grade ovarian cancer: One step towards precision medicine. Gynecol. Oncol. Rep. 2020, 32, 100547. [Google Scholar] [CrossRef] [PubMed]
- Thigpen, J.T.; Blessing, J.A.; Ball, H.; Hummel, S.J.; Barrett, R.J. Phase ii trial of paclitaxel in patients with progressive ovarian carcinoma after platinum-based chemotherapy: A gynecologic oncology group study. J. Clin. Oncol. 1994, 12, 1748–1753. [Google Scholar] [CrossRef]
- Jordan, M.A.; Toso, R.J.; Thrower, D.; Wilson, L. Mechanism of mitotic block and inhibition of cell proliferation by taxol at low concentrations. Proc. Natl. Acad. Sci. USA 1993, 90, 9552–9556. [Google Scholar] [CrossRef] [Green Version]
- Takada, M.; Hix, J.M.L.; Corner, S.; Schall, P.Z.; Kiupel, M.; Yuzbasiyan-Gurkan, V. Targeting mek in a translational model of histiocytic sarcoma. Mol. Cancer Ther. 2018, 17, 2439–2450. [Google Scholar] [CrossRef] [Green Version]
- Sumi, T.; Hirai, S.; Yamaguchi, M.; Tanaka, Y.; Tada, M.; Niki, T.; Takahashi, H.; Sakuma, Y. Trametinib downregulates survivin expression in rb1-positive kras-mutant lung adenocarcinoma cells. Biochem. Biophys. Res. Commun. 2018, 501, 253–258. [Google Scholar] [CrossRef]
- Vogel, C.J.; Smit, M.A.; Maddalo, G.; Possik, P.A.; Sparidans, R.W.; van der Burg, S.H.; Verdegaal, E.M.; Heck, A.J.; Samatar, A.A.; Beijnen, J.H.; et al. Cooperative induction of apoptosis in nras mutant melanoma by inhibition of mek and rock. Pigment Cell Melanoma Res. 2015, 28, 307–317. [Google Scholar] [CrossRef]
- Fernandez, M.L.; DiMattia, G.E.; Dawson, A.; Bamford, S.; Anderson, S.; Hennessy, B.T.; Anglesio, M.S.; Shepherd, T.G.; Salamanca, C.; Hoenisch, J.; et al. Differences in mek inhibitor efficacy in molecularly characterized low-grade serous ovarian cancer cell lines. Am. J. Cancer Res. 2016, 6, 2235–2251. [Google Scholar]
- Zhan, T.; Ambrosi, G.; Wandmacher, A.M.; Rauscher, B.; Betge, J.; Rindtorff, N.; Häussler, R.S.; Hinsenkamp, I.; Bamberg, L.; Hessling, B.; et al. Mek inhibitors activate wnt signalling and induce stem cell plasticity in colorectal cancer. Nat. Commun. 2019, 10, 2197. [Google Scholar] [CrossRef]
- Sarvi, S.; Crispin, R.; Lu, Y.; Zeng, L.; Hurley, T.D.; Houston, D.R.; von Kriegsheim, A.; Chen, C.H.; Mochly-Rosen, D.; Ranzani, M.; et al. Aldh1 bio-activates nifuroxazide to eradicate aldh(high) melanoma-initiating cells. Cell Chem. Biol. 2018, 25, 1456–1469.e6. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Guo, X.; Chang, D.Y.; Rosen, D.G.; Mercado-Uribe, I.; Liu, J. Cd133 expression associated with poor prognosis in ovarian cancer. Mod. Pathol. 2012, 25, 456–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Yang, B.; Cao, Q.; Wu, X. Association of vasculogenic mimicry formation and cd133 expression with poor prognosis in ovarian cancer. Gynecol. Obstet. Investig. 2016, 81, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Stemberger-Papic, S.; Vrdoljak-Mozetic, D.; Ostojic, D.V.; Rubesa-Mihaljevic, R.; Krigtofic, I.; Brncic-Fisher, A.; Kragevic, M.; Eminovic, S. Expression of cd133 and cd117 in 64 serous ovarian cancer cases. Coll. Antropol. 2015, 39, 745–753. [Google Scholar] [PubMed]
- Steg, A.D.; Bevis, K.S.; Katre, A.A.; Ziebarth, A.; Dobbin, Z.C.; Alvarez, R.D.; Zhang, K.; Conner, M.; Landen, C.N. Stem cell pathways contribute to clinical chemoresistance in ovarian cancer. Clin. Cancer Res. 2012, 18, 869–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Y.; Wei, X.; Gong, H.; Ni, Y. Aldehyde dehydrogenase serves as a biomarker for worse survival profiles in ovarian cancer patients: An updated meta-analysis. BMC Womens Health 2018, 18, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolcher, A.W.; Bendell, J.C.; Papadopoulos, K.P.; Burris, H.A., 3rd; Patnaik, A.; Jones, S.F.; Rasco, D.; Cox, D.S.; Durante, M.; Bellew, K.M.; et al. A phase ib trial of the oral mek inhibitor trametinib (gsk1120212) in combination with everolimus in patients with advanced solid tumors. Ann. Oncol. 2015, 26, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Ioka, T.; Fukutomi, A.; Morizane, C.; Kasuga, A.; Takahashi, H.; Todaka, A.; Okusaka, T.; Creasy, C.L.; Gorman, S.; et al. Efficacy and safety of trametinib in japanese patients with advanced biliary tract cancers refractory to gemcitabine. Cancer Sci. 2018, 109, 215–224. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chesnokov, M.S.; Khan, I.; Park, Y.; Ezell, J.; Mehta, G.; Yousif, A.; Hong, L.J.; Buckanovich, R.J.; Takahashi, A.; Chefetz, I. The MEK1/2 Pathway as a Therapeutic Target in High-Grade Serous Ovarian Carcinoma. Cancers 2021, 13, 1369. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13061369
Chesnokov MS, Khan I, Park Y, Ezell J, Mehta G, Yousif A, Hong LJ, Buckanovich RJ, Takahashi A, Chefetz I. The MEK1/2 Pathway as a Therapeutic Target in High-Grade Serous Ovarian Carcinoma. Cancers. 2021; 13(6):1369. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13061369
Chicago/Turabian StyleChesnokov, Mikhail S., Imran Khan, Yeonjung Park, Jessica Ezell, Geeta Mehta, Abdelrahman Yousif, Linda J. Hong, Ronald J. Buckanovich, Akimasa Takahashi, and Ilana Chefetz. 2021. "The MEK1/2 Pathway as a Therapeutic Target in High-Grade Serous Ovarian Carcinoma" Cancers 13, no. 6: 1369. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13061369