
Assessment of a Size-Based Method for Enriching Circulating Tumour Cells in Colorectal Cancer


 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Approval
2.2. Cell Culture
2.3. Immunostaining to Assess MetaCell Recovery Rates and WBC Depletion Rates
2.4. Gene Expression Analysis for Assessing Recovery and WBC Depletion Rates for MetaCell
2.5. Patient Sampling and Blood Processing
2.6. Statistical Analysis
3. Results
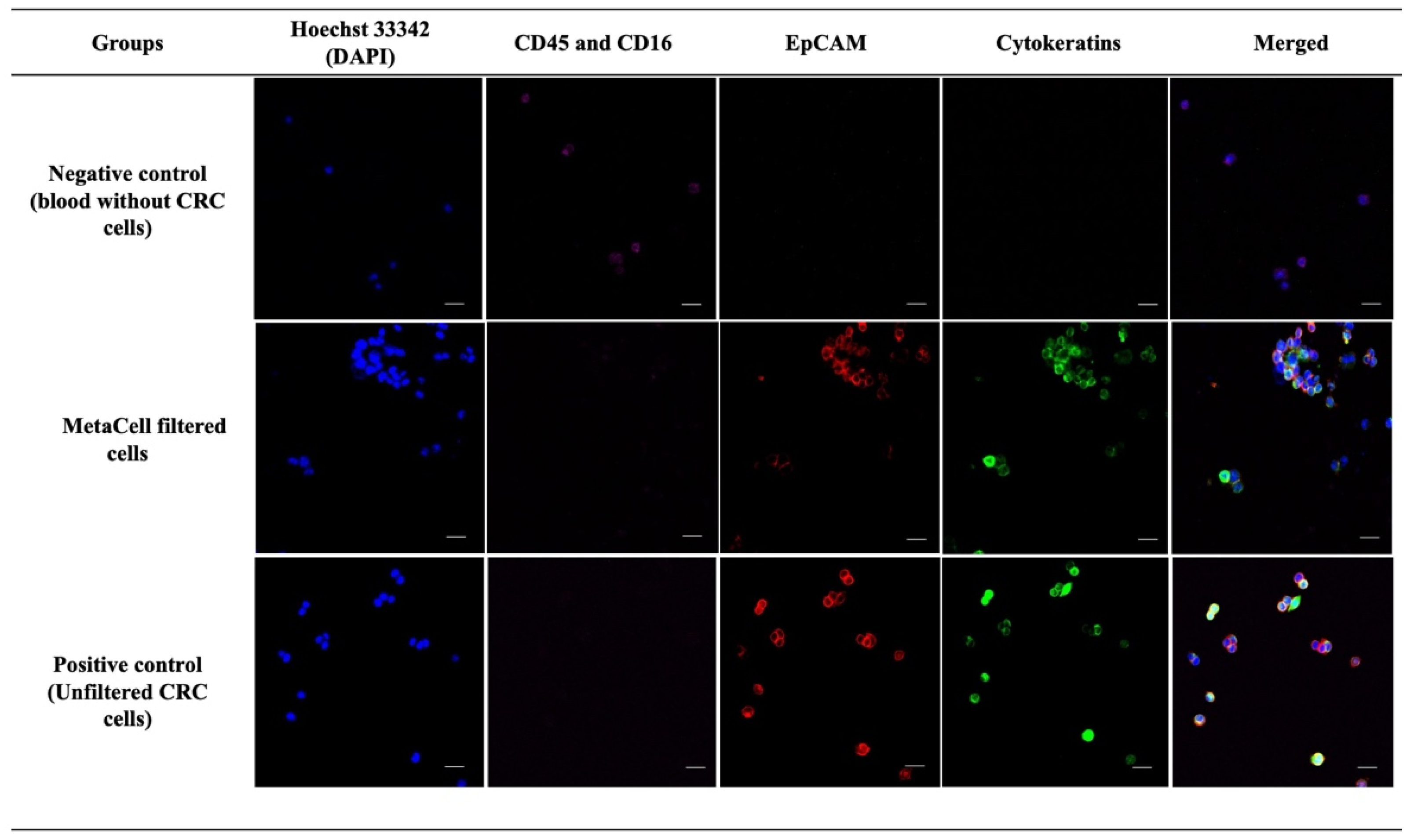
3.1. Qualitative Analysis Demonstrates MetaCell’s Ability for Capturing CRC Cells from Blood Spiked with High CRC Cell Numbers
3.2. Gene Expression Analysis Confirmed MetaCells’s Ability to Highly Enrich for CRC Cells
3.3. Quantitative Assessment Demonstrates That MetaCell Recovery Rates Were Not Affected by Low Cell Numbers
3.4. Gene Expression Analysis Revealed Different CRC Cell Numbers Did Not Have a Significant Effect on MetaCell Recovery Rates
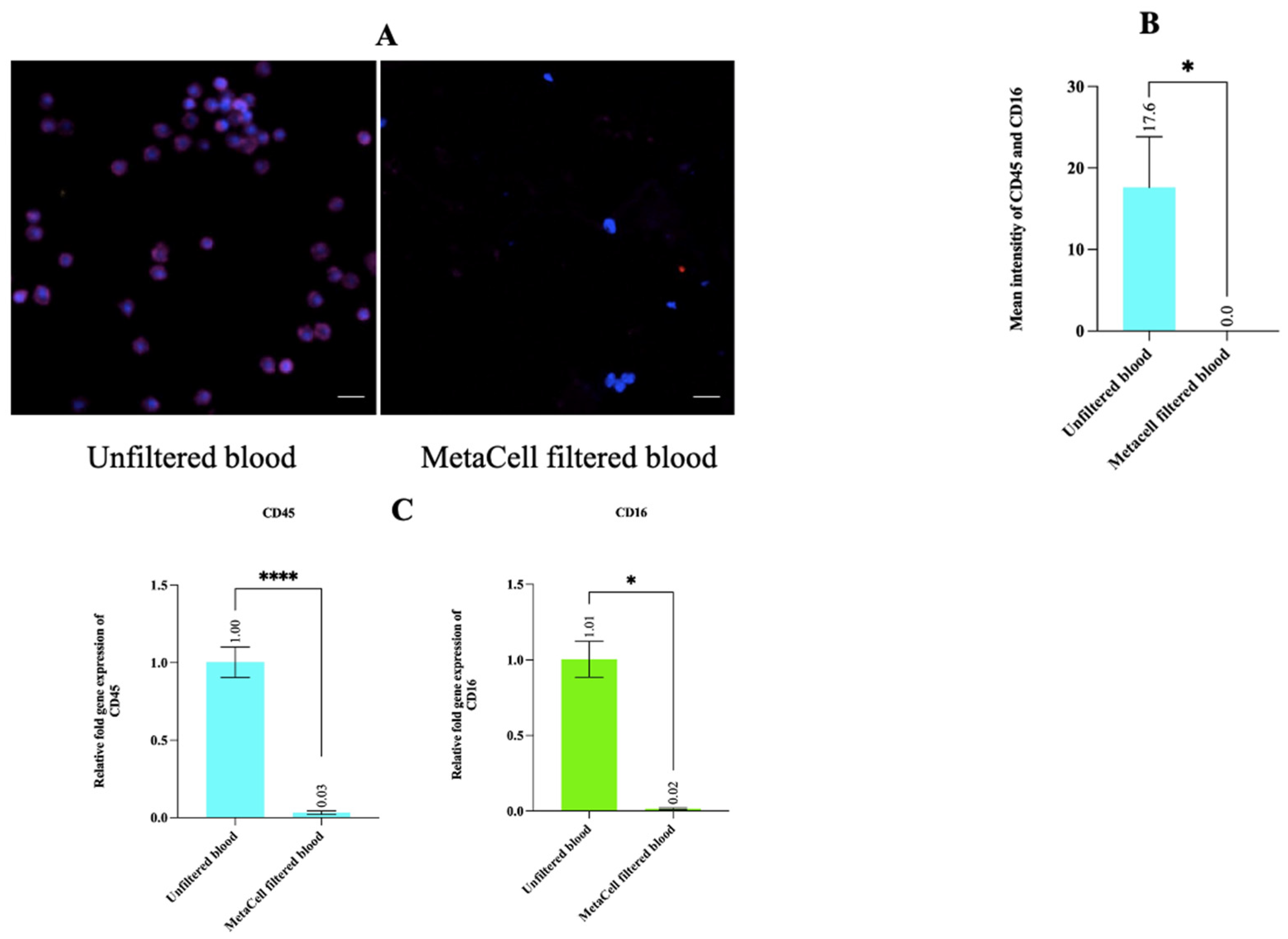
3.5. Estimating WBC Depletion Rate Revealed That MetaCell Significantly Depleted the WBC Population
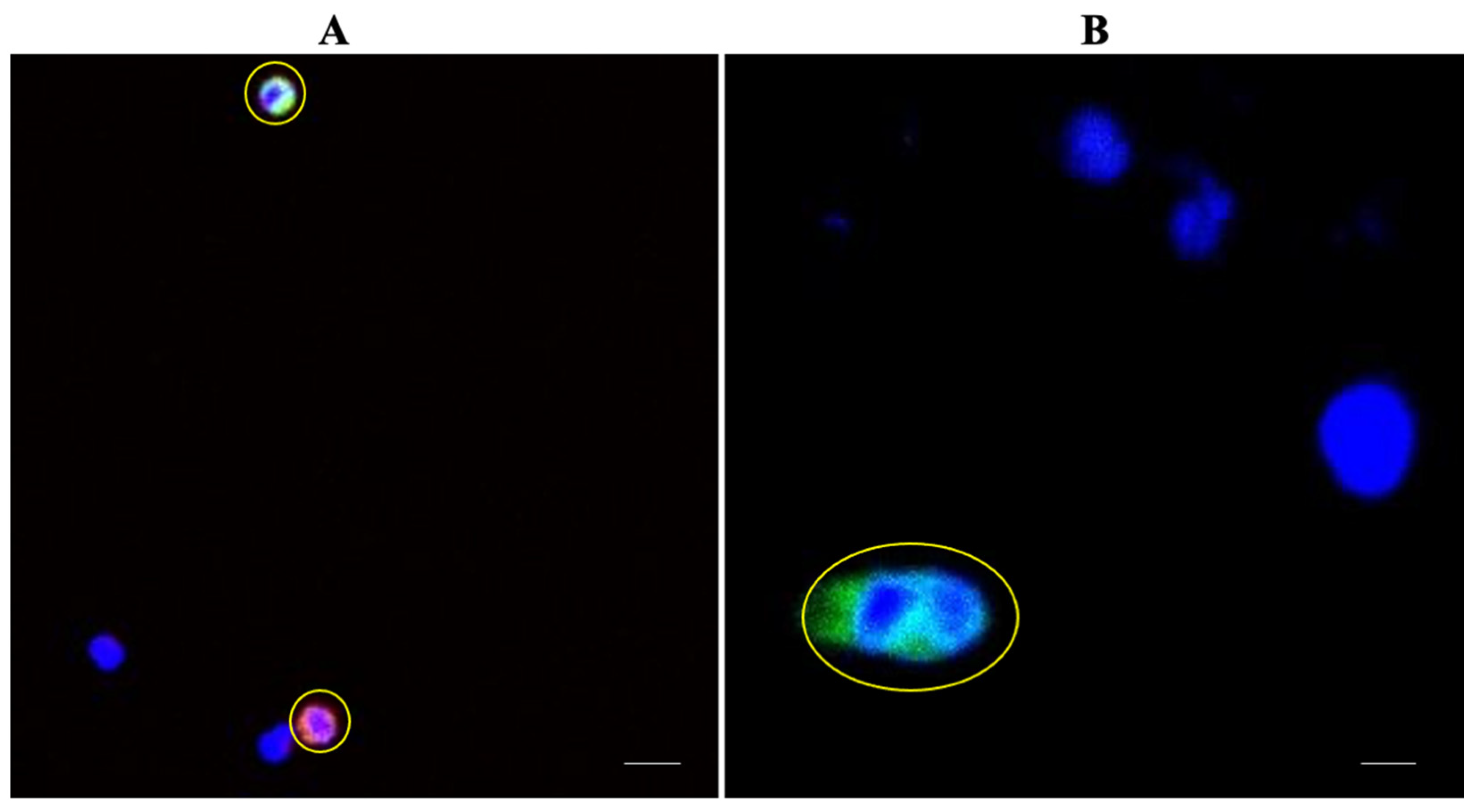
3.6. Patient Characteristics and CTC Detection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Seyfried, T.N.; Huysentruyt, L.C. On the origin of cancer metastasis. Crit. Rev. Oncog. 2013, 18, 43. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Guo, B.-C.; Sun, L.-R.; Wang, J.-W.; Fu, X.-H.; Zhang, S.-Z.; Poston, G.; Ding, K.-F. TNM staging of colorectal cancer should be reconsidered by T stage weighting. World J. Gastroenterol. 2014, 20, 5104. [Google Scholar] [CrossRef] [PubMed]
- Uraoka, T.; Hosoe, N.; Yahagi, N. Colonoscopy: Is it as effective as an advanced diagnostic tool for colorectal cancer screening? Expert Rev. Gastroenterol. Hepatol. 2015, 9, 129–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasantharajan, S.S.; Eccles, M.R.; Rodger, E.J.; Pattison, S.; McCall, J.L.; Gray, E.S.; Calapre, L.; Chatterjee, A. The epigenetic landscape of circulating tumour cells. Biochim. Biophys. Acta 2021, 1875, 188514. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, T. A case of cancer in which cells similar to those in the tumours were seen in the blood after death. Aust. Med. J. 1869, 14, 146. [Google Scholar]
- Lozar, T.; Gersak, K.; Cemazar, M.; Kuhar, C.G.; Jesenko, T. The biology and clinical potential of circulating tumor cells. Radiol. Oncol. 2019, 53, 131–147. [Google Scholar] [CrossRef] [Green Version]
- Paterlini-Bréchot, P. Organ-specific markers in circulating tumor cell screening: An early indicator of metastasis-capable malignancy. Future Oncol. 2011, 7, 849–871. [Google Scholar] [CrossRef]
- Miller, M.C.; Doyle, G.V.; Terstappen, L.W. Significance of circulating tumor cells detected by the CellSearch system in patients with metastatic breast colorectal and prostate cancer. J. Oncol. 2010, 2010, 617421. [Google Scholar] [CrossRef]
- Torino, F.; Bonmassar, E.; Bonmassar, L.; De Vecchis, L.; Barnabei, A.; Zuppi, C.; Capoluongo, E.; Aquino, A. Circulating tumor cells in colorectal cancer patients. Cancer Treat. Rev. 2013, 39, 759–772. [Google Scholar] [CrossRef]
- Kilgour, E.; Rothwell, D.G.; Brady, G.; Dive, C. Liquid Biopsy-Based Biomarkers of Treatment Response and Resistance. Cancer Cell 2020, 37, 485–495. [Google Scholar] [CrossRef]
- Kim, T.H.; Wang, Y.; Oliver, C.R.; Thamm, D.H.; Cooling, L.; Paoletti, C.; Smith, K.J.; Nagrath, S.; Hayes, D.F. A temporary indwelling intravascular aphaeretic system for in vivo enrichment of circulating tumor cells. Nat. Commun. 2019, 10, 1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paoletti, C.; Hayes, D.F. Circulating tumor cells. In Novel Biomarkers in the Continuum of Breast Cancer; Springer: Berlin/Heidelberg, Germany, 2016; pp. 235–258. [Google Scholar]
- Alix-Panabières, C.; Pantel, K. Technologies for detection of circulating tumor cells: Facts and vision. Lab Chip 2014, 14, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Vasseur, A.; Kiavue, N.; Bidard, F.C.; Pierga, J.Y.; Cabel, L. Clinical utility of circulating tumor cells: An update. Mol. Oncol. 2021, 15, 1647–1666. [Google Scholar] [CrossRef] [PubMed]
- Andreopoulou, E.; Yang, L.Y.; Rangel, K.; Reuben, J.; Hsu, L.; Krishnamurthy, S.; Valero, V.; Fritsche, H.; Cristofanilli, M. Comparison of assay methods for detection of circulating tumor cells in metastatic breast cancer: Adna Gen Adna Test Breast Cancer Select/Detect™ versus Veridex CellSearch™ system. Int. J. Cancer 2012, 130, 1590–1597. [Google Scholar] [CrossRef]
- Mazard, T.; Cayrefourcq, L.; Perriard, F.; Senellart, H.; Linot, B.; De La Fouchardière, C.; Terrebonne, E.; François, E.; Obled, S.; Guimbaud, R. Clinical Relevance of Viable Circulating Tumor Cells in Patients with Metastatic Colorectal Cancer: The COLOSPOT Prospective Study. Cancers 2021, 13, 2966. [Google Scholar] [CrossRef]
- Murlidhar, V.; Rivera-Báez, L.; Nagrath, S. Affinity Versus Label-Free Isolation of Circulating Tumor Cells: Who Wins? Small 2016, 12, 4450–4463. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, N.; Gheldof, A.; Tatari, M.; Christofori, G. EMT as the ultimate survival mechanism of cancer cells. In Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2012; Volume 3, pp. 194–207. [Google Scholar]
- Dianat-Moghadam, H.; Azizi, M.; Eslami-S, Z.; Cortés-Hernández, L.E.; Heidarifard, M.; Nouri, M.; Alix-Panabières, C. The role of circulating tumor cells in the metastatic cascade: Biology, technical challenges, and clinical relevance. Cancers 2020, 12, 867. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, M.M.; Ramani, V.C.; Jeffrey, S.S. Circulating tumor cell technologies. Mol. Oncol. 2016, 10, 374–394. [Google Scholar] [CrossRef] [Green Version]
- Zavridou, M.; Mastoraki, S.; Strati, A.; Koutsodontis, G.; Klinakis, A.; Psyrri, A.; Lianidou, E. Direct comparison of size-dependent versus EpCAM-dependent CTC enrichment at the gene expression and DNA methylation level in head and neck squamous cell carcinoma. Sci. Rep. 2020, 10, 6551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francescangeli, F.; Magri, V.; De Angelis, M.L.; De Renzi, G.; Gandini, O.; Zeuner, A.; Gazzaniga, P.; Nicolazzo, C. Sequential Isolation and Characterization of Single CTCs and Large CTC Clusters in Metastatic Colorectal Cancer Patients. Cancers 2021, 13, 6362. [Google Scholar] [CrossRef] [PubMed]
- Kolostova, K.; Zhang, Y.; Hoffman, R.M.; Bobek, V. Morphological and gene-expression characterization of viable heterogeneous circulating tumor cells size-captured and cultured from triple-negative breast cancer mouse models. Int. J. Clin. Exp. Med. 2016, 9, 7772–7779. [Google Scholar]
- Kolostova, K.; Spicka, J.; Matkowski, R.; Bobek, V. Isolation, primary culture, morphological and molecular characterization of circulating tumor cells in gynecological cancers. Am. J. Transl. Res. 2015, 7, 1203. [Google Scholar] [PubMed]
- Kolostova, K.; Zhang, Y.; Hoffman, R.M.; Bobek, V. In vitro culture and characterization of human lung cancer circulating tumor cells isolated by size exclusion from an orthotopic nude-mouse model expressing fluorescent protein. J. Fluoresc. 2014, 24, 1531–1536. [Google Scholar] [CrossRef] [Green Version]
- Bobek, V.; Matkowski, R.; Gürlich, R.; Grabowski, K.; Szelachowska, J.; Lischke, R.; Schützner, J.; Harustiak, T.; Pazdro, A.; Rzechonek, A. Cultivation of circulating tumor cells in esophageal cancer. Folia Histochem. Cytobiol. 2014, 52, 171–177. [Google Scholar] [CrossRef] [Green Version]
- Kolostova, K.; Matkowski, R.; Gürlich, R.; Grabowski, K.; Soter, K.; Lischke, R.; Schützner, J.; Bobek, V. Detection and cultivation of circulating tumor cells in gastric cancer. Cytotechnology 2016, 68, 1095–1102. [Google Scholar] [CrossRef] [Green Version]
- Kolostova, K.; Cegan, M.; Bobek, V. Circulating tumour cells in patients with urothelial tumours: Enrichment and in vitro culture. Can. Urol. Assoc. J. 2014, 8, E715. [Google Scholar]
- Lemma, S.; Perrone, A.M.; De Iaco, P.; Gasparre, G.; Kurelac, I. Current methodologies to detect circulating tumor cells: A focus on ovarian cancer. Am. J. Cancer Res. 2021, 11, 4111. [Google Scholar]
- Acheampong, E.; Abed, A.; Morici, M.; Spencer, I.; Beasley, A.B.; Bowyer, S.; Asante, D.-B.; Lomma, C.; Lin, W.; Millward, M. Evaluation of PD-L1 expression on circulating tumour cells in small-cell lung cancer. Transl. Lung Cancer Res. 2022, 11, 440. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-ΔΔC(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Weiser, M.R. AJCC 8th edition: Colorectal cancer. Ann. Surg. Oncol. 2018, 25, 1454–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macrae, F.A.; Bendell, J.; Tanabe, K.; Savarese, D.; Grover, S. Clinical Presentation, Diagnosis, and Staging of Colorectal Cancer. 2016. Available online: https://www.uptodate.com/contents/clinical-presentation-diagnosis-and-staging-of-colorectal-cancer (accessed on 22 October 2018).
- Welinder, C.; Jansson, B.; Lindell, G.; Wenner, J. Cytokeratin 20 improves the detection of circulating tumor cells in patients with colorectal cancer. Cancer Lett. 2015, 358, 43–46. [Google Scholar] [CrossRef]
- Tamminga, M.; Oomens, L.; Hiltermann, T.J.N.; Andree, K.C.; Tibbe, A.; Broekmaat, J.; Schuuring, E.; Terstappen, L.W.; Groen, H.J. Microsieves for the detection of circulating tumor cells in leukapheresis product in non-small cell lung cancer patients. Transl. Lung Cancer Res. 2020, 9, 1093. [Google Scholar] [CrossRef]
- Alvarez Cubero, M.; Lorente, J.; Robles-Fernandez, I.; Rodriguez-Martinez, A.; Puche, J.; Serrano, M. Circulating tumor cells: Markers and methodologies for enrichment and detection. In Circulating Tumor Cells; Springer: Berlin/Heidelberg, Germany, 2017; pp. 283–303. [Google Scholar]
- Riethdorf, S.; Fritsche, H.; Muller, V.; Rau, T.; Schindlbeck, C.; Rack, B.; Janni, W.; Coith, C.; Beck, K.; Jänicke, F.; et al. Detection of circulating tumor cells in peripheral blood of patients with metastatic breast cancer: A validation study of the CellSearch system. Clin. Cancer Res. 2007, 13, 920–928. [Google Scholar] [CrossRef] [Green Version]
- Warkiani, M.E.; Khoo, B.L.; Wu, L.; Tay, A.K.P.; Bhagat, A.A.S.; Han, J.; Lim, C.T. Ultra-fast, label-free isolation of circulating tumor cells from blood using spiral microfluidics. Nat. Protoc. 2016, 11, 134–148. [Google Scholar] [CrossRef]
- Harouaka, R.A.; Nisic, M.; Zheng, S.-Y. Circulating tumor cell enrichment based on physical properties. J. Lab. Autom. 2013, 18, 455–468. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Wang, S.; Fang, Y.; Zheng, L.; Zhi, X.; Cheng, B.; Chen, Y.; Zhang, C.; Shi, D.; Song, H. Feasibility of a novel one-stop ISET device to capture CTCs and its clinical application. Oncotarget 2017, 8, 3029. [Google Scholar] [CrossRef] [Green Version]
- Mendelaar, P.A.; Kraan, J.; Van, M.; Zeune, L.L.; Terstappen, L.W.; Oomen-de Hoop, E.; Martens, J.W.; Sleijfer, S. Defining the dimensions of circulating tumor cells in a large series of breast, prostate, colon, and bladder cancer patients. Mol. Oncol. 2021, 15, 116–125. [Google Scholar] [CrossRef]
- Marrinucci, D.; Bethel, K.; Lazar, D.; Fisher, J.; Huynh, E.; Clark, P.; Bruce, R.; Nieva, J.; Kuhn, P. Cytomorphology of circulating colorectal tumor cells: A small case series. J. Oncol. 2010, 2010, 861341. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Rodríguez, A.J.; Molina-Vallejo, M.P.; Aznar-Peralta, I.; González Puga, C.; Cañas García, I.; González, E.; Lorente, J.A.; Serrano, M.J.; Garrido-Navas, M.C. Deep Phenotypic Characterisation of CTCs by Combination of Microfluidic Isolation (IsoFlux) and Imaging Flow Cytometry (ImageStream). Cancers 2021, 13, 6386. [Google Scholar] [CrossRef] [PubMed]
- Vilhav, C.; Engström, C.; Naredi, P.; Novotny, A.; Bourghardt-Fagman, J.; Iresjö, B.M.; Asting, A.G.; Lundholm, K. Fractional uptake of circulating tumor cells into liver-lung compartments during curative resection of periampullary cancer. Oncol. Lett. 2018, 16, 6331–6338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendricks, A.; Dall, K.; Brandt, B.; Geisen, R.; Röder, C.; Schafmayer, C.; Becker, T.; Hinz, S.; Sebens, S. Longitudinal Analysis of Circulating Tumor Cells in Colorectal Cancer Patients by a Cytological and Molecular Approach: Feasibility and Clinical Application. Front. Oncol. 2021, 11, 2038. [Google Scholar] [CrossRef] [PubMed]
- Born, J.; Hendricks, A.; Hauser, C.; Egberts, J.-H.; Becker, T.; Röder, C.; Sebens, S. Detection of Marker Associated with CTC in Colorectal Cancer in Mononuclear Cells of Patients with Benign Inflammatory Intestinal Diseases. Cancers 2021, 14, 47. [Google Scholar] [CrossRef]
- Pecot, C.V.; Bischoff, F.Z.; Mayer, J.A.; Wong, K.L.; Pham, T.; Bottsford-Miller, J.; Stone, R.L.; Lin, Y.G.; Jaladurgam, P.; Roh, J.W. A Novel Platform for Detection of CK+ and CK− CTCsDetection of CK+ and CK− CTCs. Cancer Discov. 2011, 1, 580–586. [Google Scholar] [CrossRef] [Green Version]
- Holch, J.W.; Demmer, M.; Lamersdorf, C.; Michl, M.; Schulz, C.; von Einem, J.C.; Modest, D.P.; Heinemann, V. Pattern and dynamics of distant metastases in metastatic colorectal cancer. Visc. Med. 2017, 33, 70–75. [Google Scholar] [CrossRef]
- Hugen, N.; Nagtegaal, I.D. Distinct metastatic patterns in colorectal cancer patients based on primary tumour location. Eur. J. Cancer 2017, 75, 3–4. [Google Scholar] [CrossRef]
- Yoshino, T.; Arnold, D.; Taniguchi, H.; Pentheroudakis, G.; Yamazaki, K.; Xu, R.-H.; Kim, T.; Ismail, F.; Tan, I.; Yeh, K.-H. Pan-Asian adapted ESMO consensus guidelines for the management of patients with metastatic colorectal cancer: A JSMO–ESMO initiative endorsed by CSCO, KACO, MOS, SSO and TOS. Ann. Oncol. 2018, 29, 44–70. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Cervantes, A.; Adam, R.; Sobrero, A.; Van Krieken, J.; Aderka, D.; Aguilar, E.A.; Bardelli, A.; Benson, A.; Bodoky, G. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. 2016, 27, 1386–1422. [Google Scholar] [CrossRef]
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.A.; Kang, G.H.; Widschwendter, M.; Weener, D.; Buchanan, D. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat. Genet. 2006, 38, 787–793. [Google Scholar] [CrossRef]
- Toh, J.W.; Lim, S.H.; MacKenzie, S.; de Souza, P.; Bokey, L.; Chapuis, P.; Spring, K.J. Association between microsatellite instability status and peri-operative release of circulating tumour cells in colorectal cancer. Cells 2020, 9, 425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, H.-Y.; Lu, L.-S.; Cho, W.; Wu, S.-Y.; Chang, Y.-C.; Lin, C.-P.; Yang, C.-Y.; Lin, C.-H.; Jiang, J.-K.; Tseng, F.-G. Enumerating circulating tumor cells with a Self-assembled cell array (SACA) Chip: A feasibility study in patients with colorectal cancer. Cancers 2019, 11, 56. [Google Scholar] [CrossRef] [Green Version]
- Fakih, M.G.; Padmanabhan, A. CEA monitoring in colorectal cancer. What you should know. Oncology 2006, 20, 579–587. [Google Scholar] [PubMed]
- Gkountela, S.; Castro-Giner, F.; Szczerba, B.M.; Vetter, M.; Landin, J.; Scherrer, R.; Krol, I.; Scheidmann, M.C.; Beisel, C.; Stirnimann, C.U. Circulating tumor cell clustering shapes DNA methylation to enable metastasis seeding. Cell 2019, 176, 98–112.e114. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Number of Patients (%) |
|---|---|
| Age (years) | |
| 40–70 | 11 (58.8) |
| 71–80 | 6 (35.2) |
| Gender | |
| Male | 7 (41.2) |
| Female | 10 (58.8) |
| Ethnicity | |
| New Zealand European | 15 (88.2) |
| Other European | 2 (11.8) |
| Clinical Stage (AJCC 8th edition) | |
| Early Stage (stage I and II) | 7 (41.2) |
| Late Stage (stage III and IV) | 10 (58.8) |
| Tumour Site | |
| Left sided CRC | 10 (58.8) |
| Right sided CRC | 7 (41.2) |
| Metastasis | |
| No distant metastases | 11 (64.7) |
| Distant metastases present | 6 (35.3) |
| MMR (IHC) | |
| MMR proficient | 11 (64.7) |
| MMR deficient | 6 (35.3) |
| BRAF (IHC) | |
| BRAF V600E −ve | 5 (29.4) |
| BRAF V600E +ve | 7 (41.2) |
| ND | 5 (29.4) |
| Clinical or Pathological Variables | n | % CTC +ve Patients (n) | % CTC −ve Patients (n) | p-Value | % cCTC +ve Patients (n) | % cCTC −ve Patients (n) | p-Value |
|---|---|---|---|---|---|---|---|
| Age (years) | |||||||
| 40–70 | 11 | 54.5 (6) | 45.5 (5) | >0.99 | 18.2 (2) | 81.8 (9) | 0.58 |
| 71–85 | 6 | 50 (3) | 50 (3) | 33.3 (2) | 66.7 (4) | ||
| Gender | |||||||
| Male | 7 | 71.4 (5) | 28.6 (2) | 0.33 | 42.8 (3) | 57.2 (4) | 0.25 |
| Female | 10 | 40 (4) | 60 (6) | 10 (1) | 90 (9) | ||
| Clinical stage | |||||||
| Early stage (I & II) | 7 | 71.4 (5) | 28.6 (2) | 0.33 | 14.3 (1) | 85.7 (6) | 0.60 |
| Late stage (III & IV) | 10 | 40 (4) | 60 (6) | 30 (3) | 70 (7) | ||
| Tumour site | |||||||
| Left side | 10 | 60 (6) | 40 (4) | 0.63 | 30 (3) | 70 (7) | 0.60 |
| Right side | 7 | 42.8 (3) | 57.2 (4) | 14.3 (1) | 85.7 (6) | ||
| Metastasis | |||||||
| No distant metastases | 11 | 54.5 (6) | 45.5 (5) | >0.99 | 18.2 (2) | 81.8 (9) | 0.58 |
| Distant metastases present | 6 | 50 (3) | 50 (3) | 33.3 (2) | 66.7 (4) | ||
| MMR (IHC) | |||||||
| MMR proficient | 11 | 63.6 (7) | 36.4 (4) | 0.33 | 27.2 (3) | 72.8 (8) | >0.99 |
| MMR deficient | 6 | 33.3 (2) | 66.7 (4) | 16.6 (1) | 83.4 (5) | ||
| BRAF V600E (IHC) | |||||||
| BRAF −ve | 5 | 40 (2) | 60 (3) | >0.99 | 40 (2) | 60 (3) | 0.52 |
| BRAF +ve | 7 | 42.8 (3) | 57.2 (4) | 14.3 (1) | 85.7 (6) | ||
| ND | 5 | 80 (4) | 20 (1) | 20 (1) | 80 (4) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasantharajan, S.S.; Barnett, E.; Gray, E.S.; McCall, J.L.; Rodger, E.J.; Eccles, M.R.; Munro, F.; Pattison, S.; Chatterjee, A. Assessment of a Size-Based Method for Enriching Circulating Tumour Cells in Colorectal Cancer. Cancers 2022, 14, 3446. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14143446
Vasantharajan SS, Barnett E, Gray ES, McCall JL, Rodger EJ, Eccles MR, Munro F, Pattison S, Chatterjee A. Assessment of a Size-Based Method for Enriching Circulating Tumour Cells in Colorectal Cancer. Cancers. 2022; 14(14):3446. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14143446
Chicago/Turabian StyleVasantharajan, Sai Shyam, Edward Barnett, Elin S. Gray, John L. McCall, Euan J. Rodger, Michael R. Eccles, Fran Munro, Sharon Pattison, and Aniruddha Chatterjee. 2022. "Assessment of a Size-Based Method for Enriching Circulating Tumour Cells in Colorectal Cancer" Cancers 14, no. 14: 3446. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14143446