
Machine Learning-Based Genome-Wide Salivary DNA Methylation Analysis for Identification of Noninvasive Biomarkers in Oral Cancer Diagnosis
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Eligibility Criteria
2.2. Oral Rinse Collection and DNA Extraction
2.3. Reduced-Representation Bisulfite Sequencing (RRBS)
2.4. Sequence Alignment and Differential Methylation Analysis
2.5. Machine Learning Models
2.6. Computation
3. Results
3.1. Sequencing Quality and Descriptive Analysis
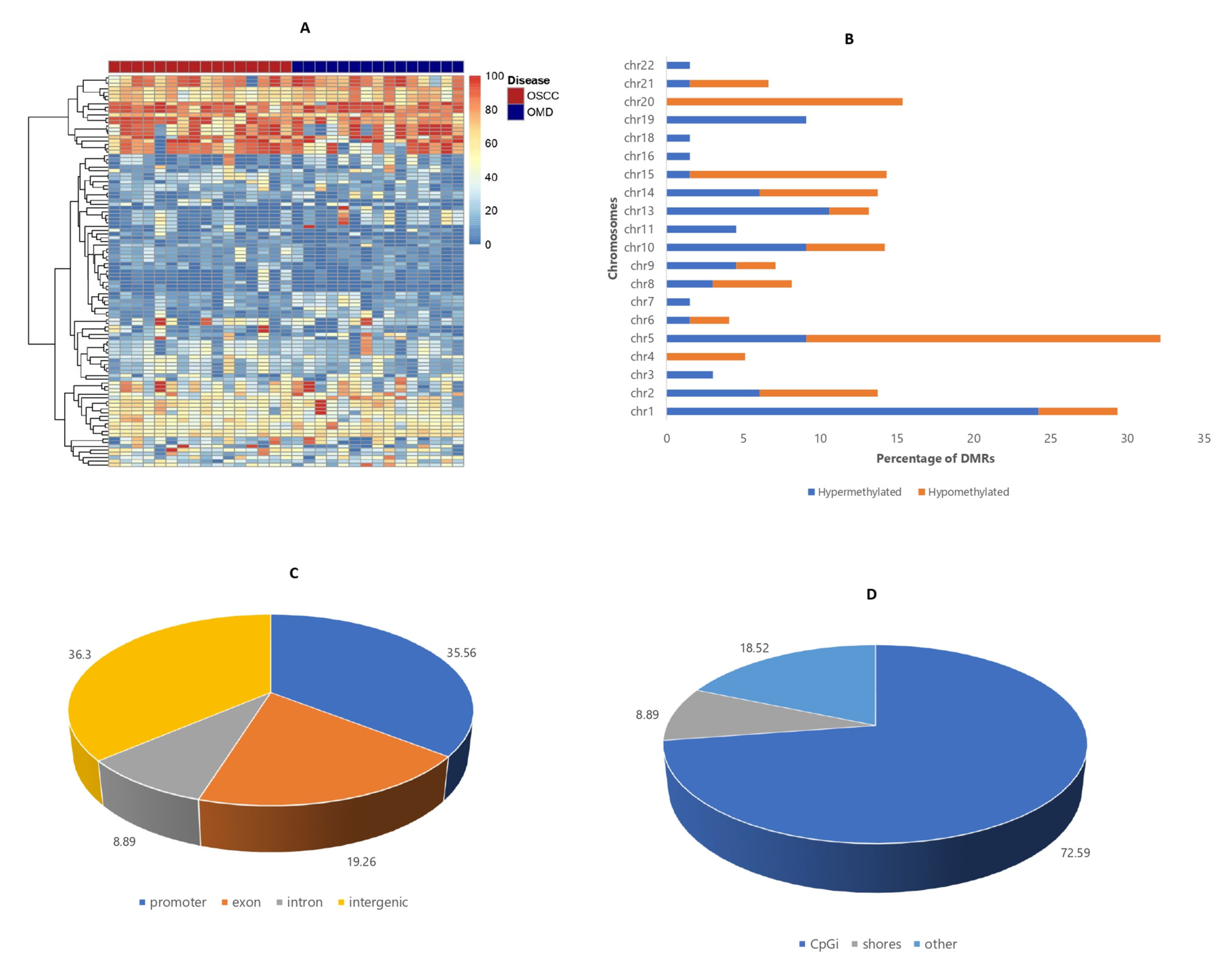
3.2. Base-Pair Resolution Analysis
3.3. Machine-Learning-Based Optimal DMC Selection and Disease Prediction
3.4. Differentially Methylated Region (DMR) Analysis
3.5. Machine-Learning-Based Optimal DMR Selection and Disease Prediction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Adeoye, J.; Thomson, P. Strategies to improve diagnosis and risk assessment for oral cancer patients. Fac. Dent. J. 2020, 11, 122–127. [Google Scholar] [CrossRef]
- Awan, K. Oral Cancer: Early Detection is Crucial. J. Int. Oral Health 2014, 6, i–ii. [Google Scholar]
- Baykul, T.; Yilmaz, H.H.; Aydin, U.; Aydin, M.A.; Aksoy, M.; Yildirim, D. Early diagnosis of oral cancer. J. Int. Med. Res. 2010, 38, 737–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciubba, J.J. Oral cancer. The importance of early diagnosis and treatment. Am. J. Clin. Dermatol. 2001, 2, 239–251. [Google Scholar] [CrossRef]
- Lee, J.J.; Hung, H.C.; Cheng, S.J.; Chiang, C.P.; Liu, B.Y.; Yu, C.H.; Jeng, J.H.; Chang, H.H.; Kok, S.H. Factors associated with underdiagnosis from incisional biopsy of oral leukoplakic lesions. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2007, 104, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, R.; Gupta, D.K. Exciting new advances in oral cancer diagnosis: Avenues to early detection. Head Neck Oncol. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Adeoye, J.; Brennan, P.A.; Thomson, P. “Search less, verify more”-Reviewing salivary biomarkers in oral cancer detection. J. Oral Pathol. Med. 2020, 49, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Adeoye, J.; Alade, A.A.; Zhu, W.Y.; Wang, W.; Choi, S.W.; Thomson, P. Efficacy of hypermethylated DNA biomarkers in saliva and oral swabs for oral cancer diagnosis: Systematic review and meta-analysis. Oral Dis. 2022, 28, 541–558. [Google Scholar] [CrossRef]
- Aro, K.; Wei, F.; Wong, D.T.; Tu, M. Saliva Liquid Biopsy for Point-of-Care Applications. Front. Public Health 2017, 5, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y.; Zhao, H. Liquid biopsy in tumors: Opportunities and challenges. Ann. Transl. Med. 2018, 6, S89. [Google Scholar] [CrossRef]
- Banavar, G.; Ogundijo, O.; Toma, R.; Rajagopal, S.; Lim, Y.K.; Tang, K.; Camacho, F.; Torres, P.J.; Gline, S.; Parks, M.; et al. The salivary metatranscriptome as an accurate diagnostic indicator of oral cancer. NPJ Genom. Med. 2021, 6, 105. [Google Scholar] [CrossRef]
- Sodnom-Ish, B.; Eo, M.Y.; Myoung, H.; Lee, J.H.; Kim, S.M. Next generation sequencing-based salivary biomarkers in oral squamous cell carcinoma. J. Korean Assoc. Oral Maxillofac. Surg. 2022, 48, 3–12. [Google Scholar] [CrossRef]
- Rapado-González, Ó.; López-Cedrún, J.L.; López-López, R.; Rodríguez-Ces, A.M.; Suárez-Cunqueiro, M.M. Saliva Gene Promoter Hypermethylation as a Biomarker in Oral Cancer. J. Clin. Med. 2021, 10, 1931. [Google Scholar] [CrossRef]
- Liyanage, C.; Wathupola, A.; Muraleetharan, S.; Perera, K.; Punyadeera, C.; Udagama, P. Promoter Hypermethylation of Tumor-Suppressor Genes p16 (INK4a), RASSF1A, TIMP3, and PCQAP/MED15 in Salivary DNA as a Quadruple Biomarker Panel for Early Detection of Oral and Oropharyngeal Cancers. Biomolecules 2019, 9, 148. [Google Scholar] [CrossRef] [Green Version]
- Khurshid, Z.; Zafar, M.S.; Khan, R.S.; Najeeb, S.; Slowey, P.D.; Rehman, I.U. Chapter Two—Role of Salivary Biomarkers in Oral Cancer Detection. In Advances in Clinical Chemistry; Makowski, G.S., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 86, pp. 23–70. [Google Scholar]
- Rapado-González, Ó.; Martínez-Reglero, C.; Salgado-Barreira, Á.; Muinelo-Romay, L.; Muinelo-Lorenzo, J.; López-López, R.; Díaz-Lagares, Á.; Suárez-Cunqueiro, M.M. Salivary DNA Methylation as an Epigenetic Biomarker for Head and Neck Cancer. Part I: A Diagnostic Accuracy Meta-Analysis. J. Pers. Med. 2021, 11, 568. [Google Scholar] [CrossRef]
- Piyarathne, N.S.; Rasnayake, R.; Angammana, R.; Chandrasekera, P.; Ramachandra, S.; Weerasekera, M.; Yasawardene, S.; Abu-Eid, R.; Jayasinghe, J.A.P.; Gupta, E. Diagnostic salivary biomarkers in oral cancer and oral potentially malignant disorders and their relationships to risk factors—A systematic review. Expert Rev. Mol. Diagn. 2021, 21, 789–807. [Google Scholar] [CrossRef]
- Adeoye, J.; Wan, C.C.J.; Thomson, P. Mock clinical testing in the validation of fluid-phase biomarkers for head and neck carcinoma diagnosis: Scoping review. Head Neck 2021, 43, 691–704. [Google Scholar] [CrossRef]
- Kourou, K.; Exarchos, T.P.; Exarchos, K.P.; Karamouzis, M.V.; Fotiadis, D.I. Machine learning applications in cancer prognosis and prediction. Comput. Struct. Biotechnol. J. 2015, 13, 8–17. [Google Scholar] [CrossRef] [Green Version]
- Tran, K.A.; Kondrashova, O.; Bradley, A.; Williams, E.D.; Pearson, J.V.; Waddell, N. Deep learning in cancer diagnosis, prognosis and treatment selection. Genome Med. 2021, 13, 152. [Google Scholar] [CrossRef]
- López-Cortés, X.A.; Matamala, F.; Venegas, B.; Rivera, C. Machine-Learning Applications in Oral Cancer: A Systematic Review. Appl. Sci. 2022, 12, 5715. [Google Scholar] [CrossRef]
- Al-Rawi, N.; Sultan, A.; Rajai, B.; Shuaeeb, H.; Alnajjar, M.; Alketbi, M.; Mohammad, Y.; Shetty, S.R.; Mashrah, M.A. The Effectiveness of Artificial Intelligence in Detection of Oral Cancer. Int. Dent. J. 2022, 72, 436–447. [Google Scholar] [CrossRef]
- Adeoye, J.; Koohi-Moghadam, M.; Lo, A.W.I.; Tsang, R.K.; Chow, V.L.Y.; Zheng, L.W.; Choi, S.W.; Thomson, P.; Su, Y.X. Deep Learning Predicts the Malignant-Transformation-Free Survival of Oral Potentially Malignant Disorders. Cancers 2021, 13, 6054. [Google Scholar] [CrossRef]
- Adeoye, J.; Sakeen Alkandari, A.; Tan, J.Y.; Wang, W.; Zhu, W.Y.; Thomson, P.; Zheng, L.W.; Choi, S.W.; Su, Y.X. Performance of a simplified scoring system for risk stratification in oral cancer and oral potentially malignant disorders screening. J. Oral Pathol. Med. 2022, 51, 464–473. [Google Scholar] [CrossRef]
- Montero, P.H.; Patel, S.G. Cancer of the oral cavity. Surg. Oncol. Clin. N. Am. 2015, 24, 491–508. [Google Scholar] [CrossRef] [Green Version]
- Conway, D.I.; Purkayastha, M.; Chestnutt, I.G. The changing epidemiology of oral cancer: Definitions, trends, and risk factors. Br. Dent. J. 2018, 225, 867–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warnakulasuriya, S.; Kujan, O.; Aguirre-Urizar, J.M.; Bagan, J.V.; González-Moles, M.; Kerr, A.R.; Lodi, G.; Mello, F.W.; Monteiro, L.; Ogden, G.R.; et al. Oral potentially malignant disorders: A consensus report from an international seminar on nomenclature and classification, convened by the WHO Collaborating Centre for Oral Cancer. Oral Dis. 2021, 27, 1862–1880. [Google Scholar] [CrossRef]
- Adeoye, J.; Wan, C.C.J.; Thomson, P. An appraisal of pivotal evaluation designs in validating noninvasive biomarkers for head and neck cancer detection. Acta Oncol. 2020, 59, 1500–1502. [Google Scholar] [CrossRef]
- Pepe, M.S.; Etzioni, R.; Feng, Z.; Potter, J.D.; Thompson, M.L.; Thornquist, M.; Winget, M.; Yasui, Y. Phases of Biomarker Development for Early Detection of Cancer. JNCI J. Natl. Cancer Inst. 2001, 93, 1054–1061. [Google Scholar] [CrossRef] [Green Version]
- Pepe, M.S.; Feng, Z.; Janes, H.; Bossuyt, P.M.; Potter, J.D. Pivotal evaluation of the accuracy of a biomarker used for classification or prediction: Standards for study design. J. Natl. Cancer Inst. 2008, 100, 1432–1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.-Q.; Tuominen, L.K.; Tsai, C.-J. SLIM: A sliding linear model for estimating the proportion of true null hypotheses in datasets with dependence structures. Bioinformatics 2010, 27, 225–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Tadist, K.; Najah, S.; Nikolov, N.S.; Mrabti, F.; Zahi, A. Feature selection methods and genomic big data: A systematic review. J. Big Data 2019, 6, 79. [Google Scholar] [CrossRef] [Green Version]
- Collins, G.S.; Reitsma, J.B.; Altman, D.G.; Moons, K.G. Transparent reporting of a multivariable prediction model for individual prognosis or diagnosis (TRIPOD): The TRIPOD statement. Br. J. Cancer 2015, 112, 251–259. [Google Scholar] [CrossRef] [Green Version]
- Irimie, A.I.; Ciocan, C.; Gulei, D.; Mehterov, N.; Atanasov, A.G.; Dudea, D.; Berindan-Neagoe, I. Current insights into oral cancer epigenetics. Int. J. Mol. Sci. 2018, 19, 670. [Google Scholar] [CrossRef] [Green Version]
- Jithesh, P.V.; Risk, J.M.; Schache, A.G.; Dhanda, J.; Lane, B.; Liloglou, T.; Shaw, R.J. The epigenetic landscape of oral squamous cell carcinoma. Br. J. Cancer 2013, 108, 370–379. [Google Scholar] [CrossRef]
- Viet, C.T.; Schmidt, B.L. Methylation array analysis of preoperative and postoperative saliva DNA in oral cancer patients. Cancer Epidemiol. Biomark. Prev. 2008, 17, 3603–3611. [Google Scholar] [CrossRef] [Green Version]
- Viet, C.T.; Zhang, X.; Xu, K.; Yu, G.; Asam, K.; Thomas, C.M.; Callahan, N.F.; Doan, C.; Walker, P.C.; Nguyen, K.; et al. Brush swab as a noninvasive surrogate for tissue biopsies in epigenomic profiling of oral cancer. Biomark. Res. 2021, 9, 90. [Google Scholar] [CrossRef]
- Huang, J.; Soupir, A.C.; Schlick, B.D.; Teng, M.; Sahin, I.H.; Permuth, J.B.; Siegel, E.M.; Manley, B.J.; Pellini, B.; Wang, L. Cancer Detection and Classification by CpG Island Hypermethylation Signatures in Plasma Cell-Free DNA. Cancers 2021, 13, 5611. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Chen, G.; Shi, T. Integrative analysis identifies potential DNA methylation biomarkers for pan-cancer diagnosis and prognosis. Epigenetics 2019, 14, 67–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, R.; Song, J.; Xiao, X.; Xie, Z.; Zhao, Z.; Zhang, W.; Miao, S.; Tang, Y.; Ran, L. Identifying CpG methylation signature as a promising biomarker for recurrence and immunotherapy in non-small-cell lung carcinoma. Aging 2020, 12, 14649–14676. [Google Scholar] [CrossRef] [PubMed]
- Freijeiro-González, L.; Febrero-Bande, M.; González-Manteiga, W. A critical review of LASSO and its derivatives for variable selection under dependence among covariates. Int. Stat. Rev. 2022, 90, 118–145. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Ser. B 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Lechner, M.; Liu, J.; Masterson, L.; Fenton, T.R. HPV-associated oropharyngeal cancer: Epidemiology, molecular biology and clinical management. Nat. Rev. Clin. Oncol. 2022, 19, 306–327. [Google Scholar] [CrossRef]
- Woods, R., Sr.; O’Regan, E.M.; Kennedy, S.; Martin, C.; O’Leary, J.J.; Timon, C. Role of human papillomavirus in oropharyngeal squamous cell carcinoma: A review. World J. Clin. Cases 2014, 2, 172–193. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | OSCC (%) | OPMD (%) | Total | p-Value | |
|---|---|---|---|---|---|
| Age | Median (IQR) | 65 (57–72) | 65.5 (53.8–72.8) | 65 (57–72) | 0.986 a |
| Sex | Female | 9 (52.9) | 8 (50.0) | 17 (51.5) | 0.866 b |
| Male | 8 (47.1) | 8 (50.0) | 16 (48.5) | ||
| Site affected | Buccal | 3 (17.6) | 12 (75.0) | 15 (45.5) | 0.011 b |
| Palate | 1 (5.9) | 1 (6.3) | 2 (6.1) | ||
| Tongue | 8 (47.1) | 3 (18.8) | 11 (33.3) | ||
| Gingiva | 5 (29.4) | 0 | 5 (15.2) | ||
| Risk habit category | NSND | 11 (64.7) | 7 (43.8) | 18 (54.5) | 0.227 b |
| SD | 6 (35.3) | 9 (56.3) | 15 (45.5) | ||
| Charlson comorbidity index | Median (IQR) | 1 (0–2.5) | 0 | 0 (0–1) | 0.046 a |
| Family history of cancer | Yes | 4 (23.5) | 4 (25.0) | 8 (24.2) | 0.922 b |
| No | 13 (76.5) | 12 (75.0) | 25 (75.8) | ||
| Hypertension | Yes | 5 (29.4) | 2 (12.5) | 7 (21.2) | 0.235 b |
| No | 12 (70.6) | 14 (87.5) | 26 (78.8) | ||
| Tumor stage | Stage I/II | 6 (35.6) | |||
| Stage III/IV | 11 (64.7) | ||||
| Tumor grade | Well differentiated | 5 (29.4) | |||
| Moderately differentiated | 9 (52.9) | ||||
| Poorly differentiated | 3 (17.6) | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adeoye, J.; Wan, C.C.J.; Zheng, L.-W.; Thomson, P.; Choi, S.-W.; Su, Y.-X. Machine Learning-Based Genome-Wide Salivary DNA Methylation Analysis for Identification of Noninvasive Biomarkers in Oral Cancer Diagnosis. Cancers 2022, 14, 4935. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14194935
Adeoye J, Wan CCJ, Zheng L-W, Thomson P, Choi S-W, Su Y-X. Machine Learning-Based Genome-Wide Salivary DNA Methylation Analysis for Identification of Noninvasive Biomarkers in Oral Cancer Diagnosis. Cancers. 2022; 14(19):4935. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14194935
Chicago/Turabian StyleAdeoye, John, Chi Ching Joan Wan, Li-Wu Zheng, Peter Thomson, Siu-Wai Choi, and Yu-Xiong Su. 2022. "Machine Learning-Based Genome-Wide Salivary DNA Methylation Analysis for Identification of Noninvasive Biomarkers in Oral Cancer Diagnosis" Cancers 14, no. 19: 4935. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14194935