
Alternative Splicing, Epigenetic Modifications and Cancer: A Dangerous Triangle, or a Hopeful One?

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction: Defining the Triangle
2. Epigenetic Modifications That Cause Splicing-Related Alterations Involved in Cancer
2.1. DNA Methylation
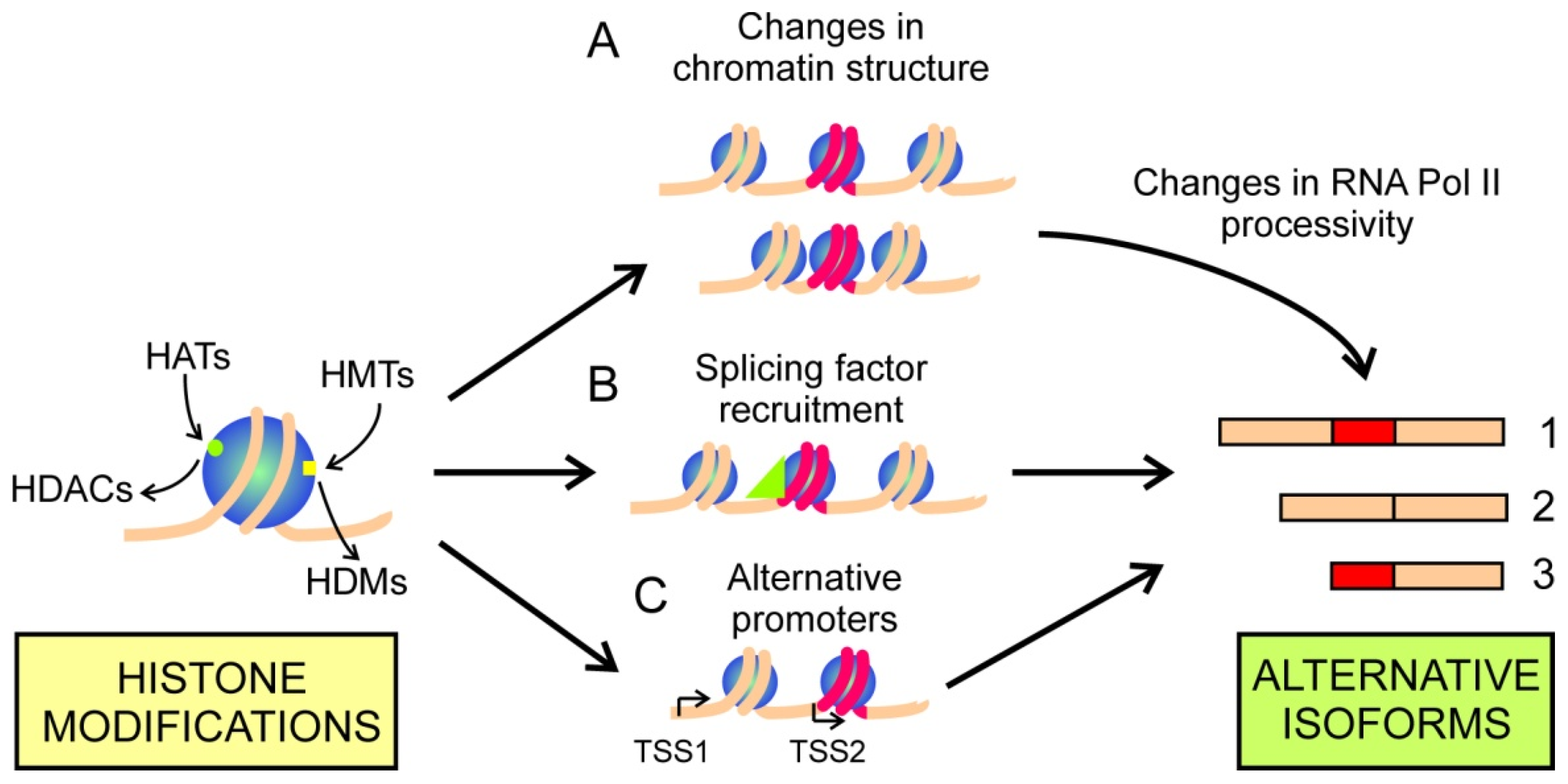
2.2. Histone Modifications
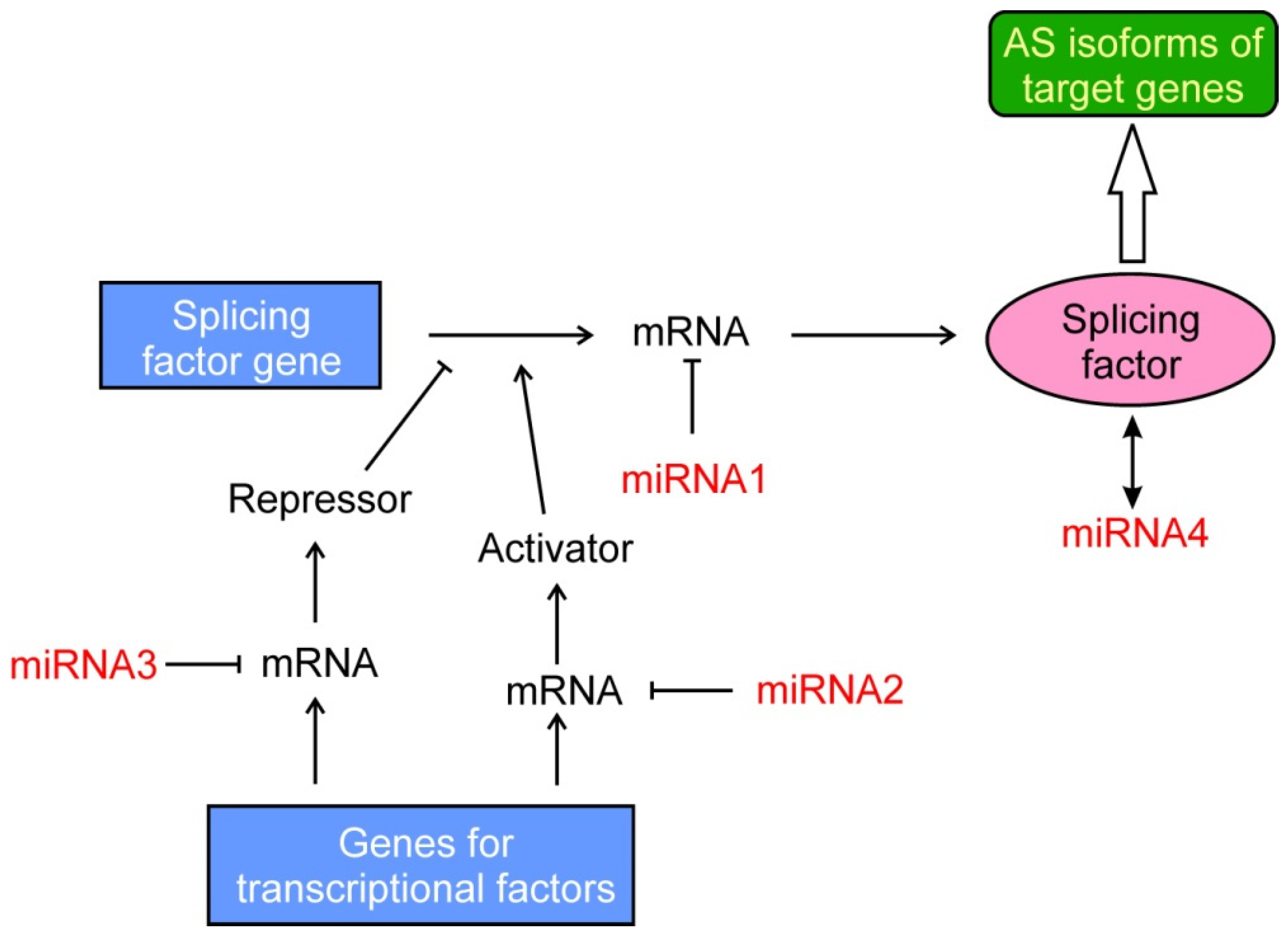
2.3. Binding of ncRNAs
2.4. Remodeling of Chromatin
2.5. Epigenetic Regulations of the Splicing Machinery
3. Alternative Splicing of Epigenetic Writers, Erasers or Readers
4. Other Alternative Splicing Events Causing Cancer
5. The Components of the Splicing Machinery: Active and Passive Roles in AS-Related Cancer Development
6. Diagnostic and Prognostic Value of Splicing-Related Epigenetic Marks
7. Therapeutic Possibilities
8. Future Prospects
9. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Denny, J.C.; Collins, F.S. Precision medicine in 2030—seven ways to transform healthcare. Cell 2021, 184, 1415–1419. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilleron, S.; Sarfati, D.; Janssen-Heijnen, M.; Vignat, J.; Ferlay, J.; Bray, F.; Soerjomataram, I. Global cancer incidence in older adults, 2012 and 2035: A population-based study. Int. J. Cancer 2019, 144, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Paredes, M.; Esteller, M. Cancer epigenetics reaches mainstream oncology. Nat. Med. 2011, 17, 330–339. [Google Scholar] [CrossRef]
- Nebbioso, A.; Tambaro, F.P.; Dell’Aversana, C.; Altucci, L. Cancer epigenetics: Moving forward. PLoS Genet. 2018, 14, e1007362. [Google Scholar] [CrossRef] [Green Version]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Barahona, V.; Joshi, R.S.; Esteller, M. Use of DNA methylation profiling in translational oncology. Semin. Cancer Biol. 2020, 19, S1044. [Google Scholar] [CrossRef]
- Castillo, J.; López-Rodas, G.; Franco, L. Histone post-translational modifications and nucleosome organisation in transcriptional regulation: Some open questions. Adv. Exp. Med. Biol. 2017, 966, 65–92. [Google Scholar]
- Loidl, P. Towards an understanding of the biological function of histone acetylation. FEBS Lett. 1988, 227, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Audia, J.E.; Campbell, R.M. Histone modifications and cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef] [PubMed]
- Werner, R.J.; Kelly, A.D.; Issa, J.-P.J. Epigenetics and Precision Oncology. Cancer J. 2017, 23, 262–269. [Google Scholar] [CrossRef]
- Clapier, C.R.; Cairns, B.R. The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 2009, 78, 273–304. [Google Scholar] [CrossRef] [PubMed]
- Blossey, R.; Schiessel, H. The latest twists in chromatin remodeling. Biophys. J. 2018, 114, 2255–2261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Centore, R.C.; Sandoval, G.J.; Soares, L.M.M.; Kadoch, C.; Chan, H.M. Mammalian SWI/SNF chromatin remodeling complexes: Emerging mechanisms and therapeutic strategies. Trends Genet. 2020, 36, 936–950. [Google Scholar] [CrossRef] [PubMed]
- Sévenet, N.; Sheridan, E.; Amram, D.; Schneider, P.; Handgretinger, R.; Delattre, O. Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. Am. J. Hum. Genet. 1999, 65, 1342–1348. [Google Scholar] [CrossRef] [Green Version]
- Kanai, Y.; Ushijima, S.; Nakanishi, Y.; Sakamoto, M.; Hirohashi, S. Mutation of the DNA methyltransferase (DNMT) 1 gene in human colorectal cancers. Cancer Lett. 2003, 192, 75–82. [Google Scholar] [CrossRef]
- Ozdağ, H.; Teschendorff, A.E.; Ahmed, A.A.; Hyland, S.J.; Blenkiron, C.; Bobrow, L.; Veerakumarasivam, A.; Burtt, G.; Subkhankulova, T.; Arends, M.J.; et al. Differential expression of selected histone modifier genes in human solid cancers. BMC Genom. 2006, 7, 90. [Google Scholar] [CrossRef] [Green Version]
- Ley, T.J.; Ding, L.; Walter, M.J.; McLellan, M.D.; Lamprecht, T.; Larson, D.E.; Kandoth, C.; Payton, J.E.; Baty, J.; Welch, J.; et al. Mutations in Acute Myeloid Leukemia. N. Engl. J. Med. 2010, 363, 2424–2433. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.; Laird, P.W. Interplay between the cancer genome and epigenome. Cell 2013, 153, 38–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357, eaal2380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Qian, J.; Gu, C.; Yang, Y. Alternative splicing and cancer: A systematic review. Signal Transduct. Target. Ther. 2021, 6, 78. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Rio, D.C. Mechanisms and regulation of alternative pre-mRNA splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tellier, M.; Maudlin, I.; Murphy, S. Transcription and splicing: A two-way street. Wiley Interdiscip. Rev. RNA 2020, 11, e1593. [Google Scholar] [CrossRef]
- Chen, L.L. The expanding regulatory mechanisms and cellular functions of circular RNAs. Nat. Rev. Mol. Cell Biol. 2020, 21, 475–490. [Google Scholar] [CrossRef] [PubMed]
- Doshi, J.; Willis, K.; Madurga, A.; Stelzer, C.; Benenson, Y. Multiple alternative promoters and alternative splicing enable universal transcription-based logic computation in mammalian cells. Cell Rep. 2020, 33, 108437. [Google Scholar] [CrossRef]
- Pereira-Castro, I.; Moreira, A. On the function and relevance of alternative 3′-UTRs in gene expression regulation. Wiley Interdiscip. Rev. RNA 2021, 12, e1653. [Google Scholar] [CrossRef]
- Girard, C.; Will, C.L.; Peng, J.; Makarov, E.M.; Kastner, B.; Lemm, I.; Urlaub, H.; Hartmuth, K.; Luhrmann, R. Post-transcriptional spliceosomes are retained in nuclear speckles until splicing completion. Nat. Commun. 2012, 3, 994. [Google Scholar] [CrossRef]
- Narayanan, S.P.; Singh, S.; Shukla, S. A saga of cancer epigenetics: Linking epigenetics to alternative splicing. Biochem. J. 2017, 474, 885–896. [Google Scholar] [CrossRef]
- Neugebauer, K.M. Nascent RNA and the coordination of splicing with transcription. Cold Spring Harb. Perspect. Biol. 2019, 11, a032227. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Sailu, S.; Palchamy, E.; Coumar, M.S.; Baluchamy, S. Identification of a novel leukemic-specific splice variant of DNMT3B and its stability. Med. Oncol. 2017, 34, 145. [Google Scholar] [CrossRef] [PubMed]
- Patounas, O.; Papacharalampous, I.; Eckerich, C.; Markopoulos, G.S.; Kolettas, E.; Fackelmayer, F.O. A novel splicing isoform of protein arginine methyltransferase 1 (PRMT1) that lacks the dimerization arm and correlates with cellular malignancy. J. Cell. Biochem. 2018, 119, 2110–2123. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, H.L.; Huang, X.; Gu, X.; Xue, W.; Xu, Y. Identification and functional characterization of a new splicing variant of EZH2 in the central nervous system. Int. J. Biol. Sci. 2019, 15, 69–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuźbicki, Ł.; Lange, D.; Stanek-Widera, A.; Chwirot, B.W. Prognostic significance of RBP2-H1 variant of JARID1B in melanoma. BMC Cancer 2017, 17, 854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Xie, N.; Chen, R.; Lee, A.R.; Lovnicki, J.; Morrison, E.A.; Fazli, L.; Zhang, Q.; Musselman, C.A.; Wang, Y.; et al. RNA splicing of the BHC80 gene contributes to neuroendocrine prostate cancer progression. Eur. Urol. 2019, 76, 157–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rona, G.B.; Almeida, D.S.G.; Pinheiro, A.S.; Eleutherio, E.C.A. The PWWP domain of the human oncogene WHSC1L1/NSD3 induces a metabolic shift toward fermentation. Oncotarget 2017, 8, 54068–54081. [Google Scholar] [CrossRef] [Green Version]
- Teslow, E.A.; Bao, B.; Dyson, G.; Legendre, C.; Mitrea, C.; Sakr, W.; Carpten, J.D.; Powell, I.; Bollig-Fischer, A. Exogenous IL-6 induces mRNA splice variant MBD2_v2 to promote stemness in TP53 wild-type, African American PCa cells. Mol. Oncol. 2018, 12, 1138–1152. [Google Scholar] [CrossRef] [Green Version]
- Teslow, E.A.; Mitrea, C.; Bao, B.; Mohammad, R.M.; Polin, L.A.; Dyson, G.; Purrington, K.S.; Bollig-Fischer, A. Obesity-induced MBD2_v2 expression promotes tumor-initiating triple-negative breast cancer stem cells. Mol. Oncol. 2019, 13, 894–908. [Google Scholar] [CrossRef]
- Amirkhah, R.; Naderi-Meshkin, H.; Shah, J.S.; Dunne, P.D.; Schmitz, U. The intricate interplay between epigenetic events, alternative splicing and noncoding RNA deregulation in colorectal cancer. Cells 2019, 8, 929. [Google Scholar] [CrossRef] [Green Version]
- Miranda Furtado, C.L.; Dos Santos Luciano, M.C.; Da Silva Santos, R.; Furtado, G.P.; Moraes, M.O.; Pessoa, C. Epidrugs: Targeting epigenetic marks in cancer treatment. Epigenetics 2019, 14, 1164–1176. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chan, Y.T.; Tan, H.Y.; Li, S.; Wang, N.; Feng, Y. Epigenetic regulation in human cancer: The potential role of epi-drug in cancer therapy. Mol. Cancer 2020, 19, 79. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Montiel, N.; Rosas-Murrieta, N.H.; Ruiz, M.A.; Monjaraz-Guzman, E.; Martinez-Contreras, R. Alternative splicing as a target for cancer treatment. Int. J. Mol. Sci. 2018, 19, 545. [Google Scholar] [CrossRef] [Green Version]
- Black, A.J.; Gamarra, J.R.; Giudice, J. More than a messenger: Alternative splicing as a therapeutic target. Biochim. Biophys. Acta-Gene Regul. Mech. 2019, 1862, 194395. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Zhao, J.; Pearson, Z.J.; Boskovic, Z.V.; Wang, J. RNA-targeting splicing modifiers: Drug development and screening assays. Molecules 2021, 26, 2263. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.P.; Li, J.B.; Gao, Y.; Lee, J.-H.; Leproust, E.; Park, I.-H.; Xie, B.; Daley, G.Q.; Church, G.M. Targeted and genome-scale methylomics reveals gene body signatures in human cell lines. Nat. Biotechnol. 2009, 27, 361–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giono, L.E.; Kornblihtt, A.R. Linking transcription, RNA polymerase II elongation and alternative splicing. Biochem. J. 2020, 477, 3091–3104. [Google Scholar] [CrossRef]
- Yearim, A.; Gelfman, S.; Shayevitch, R.; Melcer, S.; Glaich, O.; Mallm, J.-P.; Nissim-Rafinia, M.; Cohen, A.-H.S.; Rippe, K.; Meshorer, E.; et al. HP1 is involved in regulating the global impact of DNA methylation on alternative splicing. Cell Rep. 2015, 10, 1122–1134. [Google Scholar] [CrossRef] [Green Version]
- Shukla, S.; Kavak, E.; Gregory, M.; Imashimizu, M.; Shutinoski, B.; Kashlev, M.; Oberdoerffer, P.; Sandberg, R.; Oberdoerffer, S. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 2011, 479, 74–79. [Google Scholar] [CrossRef]
- Chodavarapu, R.K.; Feng, S.; Bernatavichute, Y.V.; Chen, P.-Y.; Stroud, H.; Yu, Y.; Hetzel, J.A.; Kuo, F.; Kim, J.; Cokus, S.J.; et al. Relationship between nucleosome positioning and DNA methylation. Nature 2010, 466, 388–392. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Useche, I.; Ke, J.; Tian, Y.; Shim, D.; Howell, S.C.; Qiu, X.; Yuan, C. DNA methylation regulated nucleosome dynamics. Sci. Rep. 2013, 3, 2121. [Google Scholar] [CrossRef] [Green Version]
- Dubois, F.; Bergot, E.; Levallet, G. Cancer and RASSF1A/RASSF1C, the Two Faces of Janus. Trends Cancer 2019, 5, 662–665. [Google Scholar] [CrossRef]
- Calanca, N.; Paschoal, A.P.; Munhoz, É.P.; Galindo, L.T.; Barbosa, B.M.; Caldeira, J.R.F.; Oliveira, R.A.; Cavalli, L.R.; Rogatto, S.R.; Rainho, C.A. The long non-coding RNA ANRASSF1 in the regulation of alternative protein-coding transcripts RASSF1A and RASSF1C in human breast cancer cells: Implications to epigenetic therapy. Epigenetics 2019, 14, 741–750. [Google Scholar] [CrossRef]
- Schwenzer, H.; Abdel Mouti, M.; Neubert, P.; Morris, J.; Stockton, J.; Bonham, S.; Fellermeyer, M.; Chettle, J.; Fischer, R.; Beggs, A.D.; et al. LARP1 isoform expression in human cancer cell lines. RNA Biol. 2021, 18, 237–247. [Google Scholar] [CrossRef] [Green Version]
- Mura, M.; Hopkins, T.G.; Michael, T.; Abd-Latip, N.; Weir, J.; Aboagye, E.; Mauri, F.; Jameson, C.; Sturge, J.; Gabra, H.; et al. LARP1 post-transcriptionally regulates mTOR and contributes to cancer progression. Oncogene 2015, 34, 5025–5036. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, T.G.; Mura, M.; Al-Ashtal, H.A.; Lahr, R.M.; Abd-Latip, N.; Sweeney, K.; Lu, H.; Weir, J.; El-Bahrawy, M.; Steel, J.H.; et al. The RNA-binding protein LARP1 is a post-transcriptional regulator of survival and tumorigenesis in ovarian cancer. Nucleic Acids Res. 2016, 44, 1227–1246. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Lin, S.-T.; Mi, Y.-H.; Liu, Y.; Ma, Y.; Sun, H.-M.; Peng, Z.-H.; Fan, J.-W. Overexpression of LARP1 predicts poor prognosis of colorectal cancer and is expected to be a potential therapeutic target. Tumor Biol. 2016, 37, 14585–14594. [Google Scholar] [CrossRef] [Green Version]
- Teles, S.P.; Ferreira, M.; Carvalho, J. Integrated analysis of structural variation and RNA expression of FGFR2 and its splicing modulator ESRP1 highlight the ESRP1amp-FGFR2norm-FGFR2-IIIchigh axis in diffuse gastric cancer. Cancers 2020, 12, 70. [Google Scholar] [CrossRef] [Green Version]
- Ashok, C.; Ahuja, N.; Natua, S.; Mishra, J.; Samaiya, A.; Shukla, S. E2F1 and epigenetic modifiers orchestrate breast cancer progression by regulating oxygen-dependent ESRP1 expression. Oncogenesis 2021, 10, 58. [Google Scholar] [CrossRef]
- Li, S.; Zhang, J.; Huang, S.; He, X. Genome-wide analysis reveals that exon methylation facilitates its selective usage in the human transcriptome. Brief. Bioinform. 2018, 19, 754–764. [Google Scholar] [CrossRef]
- Singh, S.; Narayanan, S.P.; Biswas, K.; Gupta, A.; Ahuja, N.; Yadav, S.; Panday, R.K.; Samaiya, A.; Sharan, S.K.; Shukla, S. Intragenic DNA methylation and BORIS-mediated cancer-specific splicing contribute to the Warburg effect. Proc. Natl. Acad. Sci. USA 2017, 114, 11440–11445. [Google Scholar] [CrossRef] [Green Version]
- Yadav, S.; Bhagat, S.D.; Gupta, A.; Samaiya, A.; Srivastava, A.; Shukla, S. Dietary-phytochemical mediated reversion of cancer-specific splicing inhibits Warburg effect in head and neck cancer. BMC Cancer 2019, 19, 1031. [Google Scholar] [CrossRef]
- Nguyen, P.; Cui, H.; Bisht, K.S.; Sun, L.; Patel, K.; Lee, R.S.; Kugoh, H.; Oshimura, M.; Feinberg, A.P.; Gius, D. CTCFL/BORIS is a methylation-independent DNA-binding protein that preferentially binds to the paternal H19 differentially methylated region. Cancer Res. 2008, 68, 5546–5551. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Huang, Y.; Liu, Z.; Zhao, R.; Liu, Q.; Wei, L.; Yu, X.; Li, B.; Qin, Y. Hypomethylation of BORIS is a promising prognostic biomarker in hepatocellular carcinoma. Gene 2017, 629, 29–34. [Google Scholar] [CrossRef]
- Batsché, E.; Yi, J.; Mauger, O.; Kornobis, E.; Hopkins, B.; Hanmer-Lloyd, C.; Muchardt, C. CD44 alternative splicing senses intragenic DNA methylation in tumors via direct and indirect mechanisms. Nucleic Acids Res. 2021, 49, 6213–6237. [Google Scholar] [CrossRef]
- Wong, J.J.L.; Gao, D.; Nguyen, T.V.; Kwok, C.T.; Van Geldermalsen, M.; Middleton, R.; Pinello, N.; Thoeng, A.; Nagarajah, R.; Holst, J.; et al. Intron retention is regulated by altered MeCP2-mediated splicing factor recruitment. Nat. Commun. 2017, 8, 15134. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.; Lee, D.; Lee, J.; Park, D.; Kim, Y.J.; Park, W.Y.; Hong, D.; Park, P.J.; Lee, E. Intron retention is a widespread mechanism of tumor-suppressor inactivation. Nat. Genet. 2015, 47, 1242–1248. [Google Scholar] [CrossRef]
- Mendizabal, I.; Zeng, J.; Keller, T.E.; Yi, S.V. Body-hypomethylated human genes harbor extensive intragenic transcriptional activity and are prone to cancer-associated dysregulation. Nucleic Acids Res. 2017, 45, 4390–4400. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, U.; Shah, J.S.; Dhungel, B.P.; Monteuuis, G.; Luu, P.L.; Petrova, V.; Metierre, C.; Nair, S.S.; Bailey, C.G.; Saunders, V.A.; et al. Widespread aberrant alternative splicing despite molecular remission in chronic myeloid leukemia patients. Cancers 2020, 12, 3738. [Google Scholar] [CrossRef]
- Nogués, G.; Kadener, S.; Cramer, P.; Bentley, D.; Kornblihtt, A.R. Transcriptional activators differ in their abilities to control alternative splicing. J. Biol. Chem. 2002, 277, 43110–43114. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Chang, S.; Lu, Y.; Wang, J.; Si, Y.; Zhang, L.; Cheng, S.; Jiang, W.G. Enhanced osteopontin splicing regulated by RUNX2 is HDAC-dependent and induces invasive phenotypes in NSCLC cells. Cancer Cell Int. 2019, 19, 306. [Google Scholar] [CrossRef]
- Zardo, G. The role of H3K4 trimethylation in CpG islands hypermethylation in cancer. Biomolecules 2021, 11, 143. [Google Scholar] [CrossRef] [PubMed]
- Enroth, S.; Bornelöv, S.; Wadelius, C.; Komorowski, J. Combinations of histone modifications mark exon inclusion levels. PLoS ONE 2012, 7, e29911. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; Greene, C.S.; Heller, E.A. Specific histone modifications associate with alternative exon selection during mammalian development. Nucleic Acids Res. 2020, 48, 4709–4724. [Google Scholar] [CrossRef] [Green Version]
- Steinmann, S.; Kunze, P.; Hampel, C.; Eckstein, M.; Bertram Bramsen, J.; Muenzner, J.K.; Carlé, B.; Ndreshkjana, B.; Kemenes, S.; Gasparini, P.; et al. DAPK1 loss triggers tumor invasion in colorectal tumor cells. Cell Death Dis. 2019, 10, 895. [Google Scholar] [CrossRef]
- Song, Z.; Li, Z.; Han, W.; Zhu, C.; Lou, N.; Li, X.; Luo, G.; Peng, S.; Li, G.; Zhao, Y.; et al. Low DAPK1 expression correlates with poor prognosis and sunitinib resistance in clear cell renal cell carcinoma. Aging 2021, 13, 1842–1858. [Google Scholar] [CrossRef]
- Brocks, D.; Schmidt, C.R.; Daskalakis, M.; Jang, H.S.; Shah, N.M.; Li, D.; Li, J.; Zhang, B.; Hou, Y.; Laudato, S.; et al. DNMT and HDAC inhibitors induce cryptic transcription start sites encoded in long terminal repeats. Nat. Genet. 2017, 49, 1052–1060. [Google Scholar] [CrossRef]
- Guo, T.; Sakai, A.; Afsari, B.; Considine, M.; Danilova, L.; Favorov, A.V.; Yegnasubramanian, S.; Kelley, D.Z.; Flam, E.; Ha, P.K.; et al. A novel functional splice variant of AKT3 defined by analysis of alternative splice expression in HPV-positive oropharyngeal cancers. Cancer Res. 2017, 77, 5248–5258. [Google Scholar] [CrossRef] [Green Version]
- Guo, T.; Zambo, K.D.A.; Zamuner, F.T.; Ou, T.; Hopkins, C.; Kelley, D.Z.; Wulf, H.A.; Winkler, E.; Erbe, R.; Danilova, L.; et al. Chromatin structure regulates cancer-specific alternative splicing events in primary HPV-related oropharyngeal squamous cell carcinoma. Epigenetics 2020, 15, 959–971. [Google Scholar] [CrossRef]
- Hu, R.; Dunn, T.A.; Wei, S.; Isharwal, S.; Veltri, R.W.; Humphreys, E.; Han, M.; Partin, A.W.; Vessella, R.L.; Isaacs, W.B.; et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009, 69, 16–22. [Google Scholar] [CrossRef] [Green Version]
- Hörnberg, E.; Ylitalo, E.B.; Crnalic, S.; Antti, H.; Stattin, P.; Widmark, A.; Bergh, A.; Wikström, P. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS ONE 2011, 6, e19059. [Google Scholar] [CrossRef] [Green Version]
- Fan, L.; Zhang, F.; Xu, S.; Cui, X.; Hussain, A.; Fazli, L.; Gleave, M.; Dong, X.; Qi, J. Histone demethylase JMJD1A promotes alternative splicing of AR variant 7 (AR-V7) in prostate cancer cells. Proc. Natl. Acad. Sci. USA 2018, 115, E4584–E4593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cato, L.; de Tribolet-Hardy, J.; Lee, I.; Rottenberg, J.T.; Coleman, I.; Melchers, D.; Houtman, R.; Xiao, T.; Li, W.; Uo, T.; et al. ARv7 represses tumor-suppressor genes in castration-resistant prostate cancer. Cancer Cell 2019, 35, 401–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paschalis, A.; Welti, J.; Neeb, A.J.; Yuan, W.; Figueiredo, I.; Pereira, R.; Ferreira, A.; Riisnaes, R.; Rodrigues, D.N.; Jiménez-Vacas, J.M.; et al. JMJD6 Is a druggable oxygenase that regulates AR-V7 expression in prostate cancer. Cancer Res. 2021, 81, 1087–1100. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.-X.; Li, X.-W.; Shi, B.-Y.; Wang, F.; Xu, Z.-R.; Meng, H.-L.; Su, Y.-Y.; Wang, J.-M.; Xiao, N.; He, Q.; et al. Effect of histone modifications on hMLH1 alternative splicing in gastric cancer. Tumor Biol. 2017, 39, 1010428317697546. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Liu, Q.; Garza, N.; Kornblau, S.; Jin, V.X. Integrative analysis reveals functional and regulatory roles of H3K79me2 in mediating alternative splicing. Genome Med. 2018, 10, 30. [Google Scholar] [CrossRef]
- González-Rodríguez, P.; Engskog-Vlachos, P.; Zhang, H.; Murgoci, A.N.; Zerdes, I.; Joseph, B. SETD2 mutation in renal clear cell carcinoma suppress autophagy via regulation of ATG12. Cell Death Dis. 2020, 11, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, J.M.; Hacker, K.E.; Singh, D.; Brannon, A.R.; Parker, J.S.; Weiser, M.; Ho, T.H.; Kuan, P.F.; Jonasch, E.; Furey, T.S.; et al. Variation in chromatin accessibility in human kidney cancer links H3K36 methyltransferase loss with widespread RNA processing defects. Genome Res. 2014, 24, 241–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, H.; Li, N.; Fu, D.; Ren, J.; Hui, J.; Peng, J.; Liu, Y.; Qiu, T.; Jiang, M.; Pan, Q.; et al. Histone methyltransferase SETD2 modulates alternative splicing to inhibit intestinal tumorigenesis. J. Clin. Investig. 2017, 127, 3375–3391. [Google Scholar] [CrossRef] [PubMed]
- Li, X.W.; Shi, B.Y.; Yang, Q.L.; Wu, J.; Wu, H.M.; Wang, Y.F.; Wu, Z.J.; Fan, Y.M.; Wang, Y.P. Epigenetic regulation of CDH1 exon 8 alternative splicing in gastric cancer. BMC Cancer 2015, 15, 954. [Google Scholar] [CrossRef] [Green Version]
- Dhamodharan, S.; Rose, M.M.; Chakkarappan, S.R.; Umadharshini, K.V.; Arulmurugan, R.; Subbiah, S.; Inoue, I.; Munirajan, A.K. Genetic variant rs10251977 (G>A) in EGFR-AS1 modulates the expression of EGFR isoforms A and D. Sci. Rep. 2021, 11, 8808. [Google Scholar] [CrossRef] [PubMed]
- Sharif, J.; Muto, M.; Takebayashi, S.I.; Suetake, I.; Iwamatsu, A.; Endo, T.A.; Shinga, J.; Mizutani-Koseki, Y.; Toyoda, T.; Okamura, K.; et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 2007, 450, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Karagianni, P.; Amazit, L.; Qin, J.; Wong, J. ICBP90, a novel methyl K9 H3 binding protein linking protein ubiquitination with heterochromatin formation. Mol. Cell. Biol. 2008, 28, 705–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajakumara, E.; Wang, Z.; Ma, H.; Hu, L.; Chen, H.; Lin, Y.; Guo, R.; Wu, F.; Li, H.; Lan, F.; et al. PHD finger recognition of unmodified histone H3R2 links UHRF1 to regulation of euchromatic gene expression. Mol. Cell 2011, 43, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Zhang, L.; Xiao, Y.; Li, W.; Hu, Z.; Zhang, R.; Li, J.; Wu, F.; Xi, Y.; Zou, Q.; et al. UHRF1 regulates alternative splicing by interacting with splicing factors and U snRNAs in a H3R2me involved manner. Hum. Mol. Genet. 2021, 30, 2110–2122. [Google Scholar] [CrossRef]
- Lau, K.S.; Haigis, K.M. Non-redundancy within the RAS oncogene family: Insights into mutational disparities in cancer. Mol Cells 2009, 28, 315–320. [Google Scholar] [CrossRef] [Green Version]
- Riffo-Campos, Á.L.; Gimeno-Valiente, F.; Rodríguez, F.M.; Cervantes, A.; López-Rodas, G.; Franco, L.; Castillo, J. Role of epigenetic factors in the selection of the alternative splicing isoforms of human KRAS in colorectal cancer cell lines. Oncotarget 2018, 9, 20578–20589. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.Z.; Xue, M.Z.; Shen, H.J.; Li, X.G.; Ma, D.; Gong, Y.; Liu, Y.R.; Qiao, F.; Xie, H.Y.; Lian, B.; et al. PHF5A epigenetically inhibits apoptosis to promote breast cancer progression. Cancer Res. 2018, 78, 3190–3206. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Yang, X.; Liu, C.; Li, X.; Zhang, B.; Wang, B.; Zhang, Y.; Song, C.; Zhang, T.; Liu, M.; et al. Acetylation of PHF5A Modulates Stress Responses and Colorectal Carcinogenesis through Alternative Splicing-Mediated Upregulation of KDM3A. Mol. Cell 2019, 74, 1250–1263. [Google Scholar] [CrossRef]
- Saxena, A.; Carninci, P. Long non-coding RNA modifies chromatin: Epigenetic silencing by long non-coding RNAs. BioEssays 2011, 33, 830–839. [Google Scholar] [CrossRef] [Green Version]
- Kishore, S.; Stamm, S. The snoRNA HBII-52 regulates alternative splicing of the serotonin receptor 2C. Science 2006, 311, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Nimura, K. Regulation of RNA splicing: Aberrant splicing regulation and therapeutic targets in cancer. Cells 2021, 10, 923. [Google Scholar] [CrossRef] [PubMed]
- Saul, M.J.; Baumann, I.; Bruno, A.; Emmerich, A.C.; Wellstein, J.; Ottinger, S.M.; Contursi, A.; Dovizio, M.; Donnini, S.; Tacconelli, S.; et al. miR-574-5p as RNA decoy for CUGBP1 stimulates human lung tumor growth by mPGES-1 induction. FASEB J. 2019, 33, 6933–6947. [Google Scholar] [CrossRef] [PubMed]
- Emmerich, A.C.; Wellstein, J.; Ossipova, E.; Baumann, I.; Lengqvist, J.; Kultima, K.; Jakobsson, P.J.; Steinhilber, D.; Saul, M.J. Proteomics-based characterization of miR-574-5p decoy to CUGBP1 suggests specificity for mPGES-1 regulation in human lung cancer cells. Front. Pharmacol. 2020, 11, 196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero-Barrios, N.; Legascue, M.F.; Benhamed, M.; Ariel, F.; Crespi, M. Splicing regulation by long noncoding RNAs. Nucleic Acids Res. 2018, 46, 2169–2184. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Zheng, J.; Hao, W.; Zeng, H.; Zhang, Z.; Shao, G. lncRNA PCAT6 facilitates cell proliferation and invasion via regulating the miR-326/hnRNPA2B1 axis in liver cancer. Oncol. Lett. 2021, 21, 471. [Google Scholar] [CrossRef]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar] [CrossRef] [Green Version]
- Paronetto, M.P.; Dimauro, I.; Grazioli, E.; Palombo, R.; Guidotti, F.; Fantini, C.; Sgrò, P.; De Francesco, D.; Di Luigi, L.; Capranica, L.; et al. Exercise-mediated downregulation of MALAT1 expression and implications in primary and secondary cancer prevention. Free Radic. Biol. Med. 2020, 160, 28–39. [Google Scholar] [CrossRef]
- Han, L.; Lai, H.; Yang, Y.; Hu, J.; Li, Z.; Ma, B.; Xu, W.; Liu, W.; Wei, W.; Li, D.; et al. A 5′-tRNA halve, tiRNA-Gly promotes cell proliferation and migration via binding to RBM17 and inducing alternative splicing in papillary thyroid cancer. J. Exp. Clin. Cancer Res. 2021, 40, 222. [Google Scholar] [CrossRef]
- Naftelberg, S.; Schor, I.E.; Ast, G.; Kornblihtt, A.R. Regulation of alternative splicing through coupling with transcription and chromatin structure. Annu. Rev. Biochem. 2015, 84, 165–198. [Google Scholar] [CrossRef]
- Jancewicz, I.; Siedlecki, J.A.; Sarnowski, T.J.; Sarnowska, E. BRM: The core ATPase subunit of SWI/SNF chromatin-remodelling complex - A tumour suppressor or tumour-promoting factor? Epigenet. Chromatin 2019, 12, 68. [Google Scholar] [CrossRef] [PubMed]
- Giles, K.A.; Gould, C.M.; Achinger-Kawecka, J.; Page, S.G.; Kafer, G.R.; Rogers, S.; Luu, P.L.; Cesare, A.J.; Clark, S.J.; Taberlay, P.C. BRG1 knockdown inhibits proliferation through multiple cellular pathways in prostate cancer. Clin. Epigenet. 2021, 13, 37. [Google Scholar] [CrossRef] [PubMed]
- Batsché, E.; Yaniv, M.; Muchardt, C. The human SWI/SNF subunit Brm is a regulator of alternative splicing. Nat. Struct. Mol. Biol. 2006, 13, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Sun, Y.; Ma, L.; Wang, C.; Wu, J.M.; Bi, A.; Liao, D.J. Complex alternative splicing of the Smarca2 gene suggests the importance of Smarca2-B variants. J. Cancer 2011, 2, 386–400. [Google Scholar] [CrossRef] [Green Version]
- Selvanathan, S.P.; Graham, G.T.; Grego, A.R.; Baker, T.M.; Hogg, J.R.; Simpson, M.; Batish, M.; Crompton, B.; Stegmaier, K.; Tomazou, E.M.; et al. EWS-FLI1 modulated alternative splicing of ARID1A reveals novel oncogenic function through the BAF complex. Nucleic Acids Res. 2019, 47, 9619–9636. [Google Scholar] [CrossRef]
- Li, D.Q.; Nair, S.S.; Ohshiro, K.; Kumar, A.; Nair, V.S.; Pakala, S.B.; Reddy, S.D.N.; Gajula, R.P.; Eswaran, J.; Aravind, L.; et al. MORC2 signaling integrates phosphorylation-dependent, ATPase-coupled chromatin remodeling during the DNA damage response. Cell Rep. 2012, 2, 1657–1669. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.L.; Cao, J.L.; Xie, H.Y.; Sun, R.; Yang, L.F.; Shao, Z.M.; Li, D.Q. Cancer-associated MORC2-mutant M276I regulates an hnRNPM-mediated CD44 splicing switch to promote invasion and metastasis in triple-negative breast cancer. Cancer Res. 2018, 78, 5780–5792. [Google Scholar] [CrossRef] [Green Version]
- Chew, G.L.; Bleakley, M.; Bradley, R.K.; Malik, H.S.; Henikoff, S.; Molaro, A.; Sarthy, J. Short H2A histone variants are expressed in cancer. Nat. Commun. 2021, 12, 490. [Google Scholar] [CrossRef]
- Boutz, P.L.; Chawla, G.; Stoilov, P.; Black, D.L. MicroRNAs regulate the expression of the alternative splicing factor nPTB during muscle development. Genes Dev. 2007, 21, 71–84. [Google Scholar] [CrossRef] [Green Version]
- Makeyev, E.V.; Zhang, J.; Carrasco, M.A.; Maniatis, T. The MicroRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol. Cell 2007, 27, 435–448. [Google Scholar] [CrossRef] [Green Version]
- Montero-Conde, C.; Graña-Castro, O.; Martín-Serrano, G.; Martínez-Montes, Á.M.; Zarzuela, E.; Muñoz, J.; Torres-Perez, R.; Pita, G.; Cordero-Barreal, A.; Leandro-García, L.J.; et al. Hsa-miR-139-5p is a prognostic thyroid cancer marker involved in HNRNPF-mediated alternative splicing. Int. J. Cancer 2020, 146, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wu, P.; Yang, Z.; Deng, S.; Ni, L.; Zhang, Y.; Jin, L.; Pan, Y. miR-193a-5p promotes pancreatic cancer cell migration and invasion through SRSF6-mediated alternative Splicing of OGDHL and ECM1. SSRN Electron. J. 2020, 10, 38–59. [Google Scholar] [CrossRef]
- Taniguchi, K.; Uchiyama, K.; Akao, Y. PTBP1-targeting microRNAs regulate cancer-specific energy metabolism through the modulation of PKM1/M2 splicing. Cancer Sci. 2021, 112, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Yang, P.; Amin, S.; Li, Z. A novel miR-206/hnRNPA1/PKM2 axis reshapes the Warburg effect to suppress colon cancer growth. Biochem. Biophys. Res. Commun. 2020, 531, 465–471. [Google Scholar] [CrossRef]
- Wu, H.; Cui, M.; Li, C.; Li, H.; Dai, Y.; Cui, K.; Li, Z. Kaempferol reverses aerobic glycolysis via miR-339-5p-mediated PKM alternative splicing in colon cancer cells. J. Agric. Food Chem. 2021, 69, 3060–3068. [Google Scholar] [CrossRef]
- Pillman, K.A.; Phillips, C.A.; Roslan, S.; Toubia, J.; Dredge, B.K.; Bert, A.G.; Lumb, R.; Neumann, D.P.; Li, X.; Conn, S.J.; et al. miR-200/375 control epithelial plasticity-associated alternative splicing by repressing the RNA -binding protein Quaking. EMBO J. 2018, 37, e99016. [Google Scholar] [CrossRef]
- Wang, J.Z.; Fu, X.; Fang, Z.; Liu, H.; Zong, F.Y.; Zhu, H.; Yu, Y.F.; Zhang, X.Y.; Wang, S.F.; Huang, Y.; et al. QKI-5 regulates the alternative splicing of cytoskeletal gene ADD3 in lung cancer. J. Mol. Cell Biol. 2021, 13, 347–360. [Google Scholar] [CrossRef]
- Huang, Y.Q.; Ling, X.H.; Yuan, R.Q.; Chen, Z.Y.; Yang, S.B.; Huang, H.X.; Zhong, W.D.; Qiu, S.P. miR-30c suppresses prostate cancer survival by targeting the ASF/SF2 splicing factor oncoprotein. Mol. Med. Rep. 2017, 16, 2431–2438. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Li, W.; Pang, Y.; Zhou, Z.; Liu, S.; Cheng, K.; Qin, Q.; Jia, Y.; Liu, S. SF3B4 is regulated by microRNA-133b and promotes cell proliferation and metastasis in hepatocellular carcinoma. EBioMedicine 2018, 38, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; He, X.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Up-regulated miR-133a orchestrates epithelial-mesenchymal transition of airway epithelial cells. Sci. Rep. 2018, 8, 15543. [Google Scholar] [CrossRef]
- Deng, G.; Zhou, X.; Chen, L.; Yao, Y.; Li, J.; Zhang, Y.; Luo, C.; Sun, L.; Tang, J. High expression of ESRP1 regulated by circ-0005585 promotes cell colonization in ovarian cancer. Cancer Cell Int. 2020, 20, 174. [Google Scholar] [CrossRef] [PubMed]
- Muench, D.E.; Ferchen, K.; Velu, C.S.; Pradhan, K.; Chetal, K.; Chen, X.; Weirauch, M.T.; Colmenares, C.; Verma, A.; Salomonis, N.; et al. SKI controls MDS-associated chronic TGF-β signaling, aberrant splicing, and stem cell fitness. Blood 2018, 132, E24–E34. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, T.; Ladd, A.N. The importance of CELF control: Molecular and biological roles of the CUG-BP, Elav-like family of RNA-binding proteins. Wiley Interdiscip. Rev. RNA 2012, 3, 104–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piqué, L.; Martinez de Paz, A.; Piñeyro, D.; Martínez-Cardús, A.; Castro de Moura, M.; Llinàs-Arias, P.; Setien, F.; Gomez-Miragaya, J.; Gonzalez-Suarez, E.; Sigurdsson, S.; et al. Epigenetic inactivation of the splicing RNA-binding protein CELF2 in human breast cancer. Oncogene 2019, 38, 7106–7112. [Google Scholar] [CrossRef]
- Gimeno-Valiente, F.; Riffo-Campos, Á.L.; Torres, L.; Tarazona, N.; Gambardella, V.; Cervantes, A.; López-Rodas, G.; Franco, L.; Castillo, J. Epigenetic mechanisms are involved in the oncogenic properties of ZNF518B in colorectal cancer. Cancers 2021, 13, 1433. [Google Scholar] [CrossRef]
- Chen, K.; Xiao, H.; Zeng, J.; Yu, G.; Zhou, H.; Huang, C.; Yao, W.; Xiao, W.; Hu, J.; Guan, W.; et al. Alternative splicing of EZH2 pre-mRNA by SF3B3 contributes to the tumorigenic potential of renal cancer. Clin. Cancer Res. 2017, 23, 3428–3441. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Sun, L.; Cai, Y.; Shen, S.; Zhang, T.; Wang, N.; Wu, G.; Ma, W.; Li, S.T.; Suo, C.; et al. Hypoxia-induced suppression of alternative splicing of MBD2 promotes breast cancer metastasis via activation of FZD1. Cancer Res. 2021, 81, 1265–1278. [Google Scholar] [CrossRef]
- Sun, J.; Meng, D.; Yu, T.; Li, F.; Zhang, G.; Tian, X.; Zhao, N.; Li, G.; Li, L.; Wang, H.; et al. N-terminal truncated carboxypeptidase E represses E-cadherin expression in lung cancer by stabilizing the Snail-HDAC complex. Am. J. Cancer Res. 2020, 10, 925–938. [Google Scholar]
- Chang, S.; Huang, J.; Niu, H.; Wang, J.; Si, Y.; Bai, Z.; Cheng, S.; Ding, W. Epigenetic regulation of osteopontin splicing isoform c defines its role as a microenvironmental factor to promote the survival of colon cancer cells from 5-FU treatment. Cancer Cell Int. 2020, 20, 452. [Google Scholar] [CrossRef]
- Briones-Orta, M.A.; Avendaño-Vázquez, S.E.; Aparicio-Bautista, D.I.; Coombes, J.D.; Weber, G.F.; Syn, W.K. Osteopontin splice variants and polymorphisms in cancer progression and prognosis. Biochim. Biophys. Acta-Rev. Cancer 2017, 1868, 93–108. [Google Scholar] [CrossRef]
- Shuai, T.; Khan, M.R.; Zhang, X.D.; Li, J.; Thorne, R.F.; Wu, M.; Shao, F. lncRNA TRMP-S directs dual mechanisms to regulate p27-mediated cellular senescence. Mol. Ther.-Nucleic Acids 2021, 24, 971–985. [Google Scholar] [CrossRef] [PubMed]
- Dery, K.J.; Silver, C.; Yang, L.; Shively, J.E. Interferon regulatory factor 1 and a variant of heterogeneous nuclear ribonucleoprotein L coordinately silence the gene for adhesion protein CEACAM1. J. Biol. Chem. 2018, 293, 9277–9291. [Google Scholar] [CrossRef] [Green Version]
- Sueoka, E.; Goto, Y.; Sueoka, N.; Kai, Y.; Kozu, T.; Fujiki, H. Heterogeneous nuclear ribonucleoprotein B1 as a new marker of early detection for human lung cancers. Cancer Res. 1999, 59, 1404–1407. [Google Scholar] [PubMed]
- Makhafola, T.J.; Mbele, M.; Yacqub-Usman, K.; Hendren, A.; Haigh, D.B.; Blackley, Z.; Meyer, M.; Mongan, N.P.; Bates, D.O.; Dlamini, Z. Apoptosis in cancer cells is induced by alternative splicing of hnRNPA2/B1 through splicing of Bcl-x, a mechanism that can be stimulated by an extract of the South African medicinal plant, Cotyledon orbiculata. Front. Oncol. 2020, 10, 547392. [Google Scholar] [CrossRef]
- Weigert, O. The different flavors and splices of MCL. Blood 2020, 136, 526–527. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Chen, R. Understanding aberrant RNA splicing to facilitate cancer diagnosis and therapy. Oncogene 2020, 39, 2231–2242. [Google Scholar] [CrossRef]
- Wang, J.; Wang, X.; Bhat, A.; Chen, Y.; Xu, K.; Mo, Y.Y.; Yi, S.S.; Zhou, Y. Comprehensive network analysis reveals alternative splicing-related lncRNAs in hepatocellular carcinoma. Front. Genet. 2020, 11, 659. [Google Scholar] [CrossRef]
- Giuliani, V.; Miller, M.A.; Liu, C.Y.; Hartono, S.R.; Class, C.A.; Bristow, C.A.; Suzuki, E.; Sanz, L.A.; Gao, G.; Gay, J.P.; et al. PRMT1-dependent regulation of RNA metabolism and DNA damage response sustains pancreatic ductal adenocarcinoma. Nat. Commun. 2021, 12, 4626. [Google Scholar] [CrossRef]
- Fedoriw, A.; Rajapurkar, S.R.; O’Brien, S.; Gerhart, S.V.; Mitchell, L.H.; Adams, N.D.; Rioux, N.; Lingaraj, T.; Ribich, S.A.; Pappalardi, M.B.; et al. Anti-tumor activity of the type I PRMT inhibitor, GSK3368715, synergizes with PRMT5 inhibition through MTAP loss. Cancer Cell 2019, 36, 100–114.e25. [Google Scholar] [CrossRef]
- Radzisheuskaya, A.; Shliaha, P.V.; Grinev, V.; Lorenzini, E.; Kovalchuk, S.; Shlyueva, D.; Gorshkov, V.; Hendrickson, R.C.; Helin, K. PRMT5 methylome profiling uncovers a direct link to splicing regulation in acute myeloid leukemia. Nat. Struct. Mol. Biol. 2019, 26, 999–1012. [Google Scholar] [CrossRef]
- Li, W.-J.; He, Y.-H.; Yang, J.-J.; Hu, G.-S.; Lin, Y.-A.; Ran, T.; Peng, B.-L.; Xie, B.-L.; Huang, M.-F.; Gao, X.; et al. Profiling PRMT methylome reveals roles of hnRNPA1 arginine methylation in RNA splicing and cell growth. Nat. Commun. 2021, 12, 1946. [Google Scholar] [CrossRef] [PubMed]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–221. [Google Scholar] [CrossRef] [PubMed]
- Suñé-Pou, M.; Limeres, M.J.; Moreno-Castro, C.; Hernández-Munain, C.; Suñé-Negre, J.M.; Cuestas, M.L.; Suñé, C. Innovative therapeutic and delivery approaches using nanotechnology to correct splicing defects underlying disease. Front. Genet. 2020, 11, 731. [Google Scholar] [CrossRef]
- Roda, D.; Castillo, J.; Telechea-Fernández, M.; Gil, A.; López-Rodas, G.; Franco, L.; González-Rodríguez, P.; Roselló, S.; Pérez-Fidalgo, J.A.; García-Trevijano, E.R.; et al. EGF-induced acetylation of heterogeneous nuclear ribonucleoproteins is dependent on KRAS mutational status in colorectal cancer cells. PLoS ONE 2015, 10, e0130543. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Epigenetic Partner | AS Event | Type of Cancer | Potential Role | Go to Section | Ref. |
|---|---|---|---|---|---|
| JARID | RBP2-H1 splice variant | MM | prognostic marker | 3 | [35] |
| miR-574-5p | PGES-1 3′UTR isoform | NSCLC | stratification marker | 2.3 | [103] |
| TET3, DNMT3A/B | ESRP1 | BC | prognostic marker | 2.1 | [59] |
| miRNA-133b | miRNA targets SF3B4 | HCC | prognostic marker | 2.5 | [129] |
| PCAT | downregulation of hnRNPA2B1 | HCC | prognostic marker | 2.3 | [106] |
| miR-30c | miRNA targets SF2 | PCa | prognostic marker | 2.5 | [128] |
| JMJD6 | AS of AR giving AR-V7 | CRPC | prognostic marker | 2.2 | [84] |
| miR-193a-5p | miRNA targets SRSF6 | PCa | prognostic marker | 2.5 | [122] |
| miR-23a/b miR-15a/15b/16 | ESRP1 expression controlled by the miRNAs | EOC | prognostic marker | 2.5 | [131] |
| miR-139-5p | miRNA targets hnRNPF | THCA | prognostic marker | 2.5 | [121] |
| KDM3A | PHF5Aac increases level of KDM3A | CRC | prognostic markers | 2.2 | [99] |
| DNAme | BORIS hypomethylation | HCC | prognostic marker | 2.1 | [64] |
| DNAme | exon methylation | LC | prognostic marker | 2.1 | [60] |
| PHF5A | FASTK intron retention | BC | prognostic marker | 2.2 | [98] |
| DNAme | CELF2me aberrant AS | BC | prognostic marker | 2.5 | [134] |
| DNAme | FGFR2-IIIc | GC | prognostic marker | 2.1 | [58] |
| SETD2 | short ATG12 at the expense of canonical ATG12 | RCC | prognostic marker | 2.2 | [87] |
| HDAC1/HDAC3/ EZH2 | increased CPEΔN | LC | prognostic marker | 4 | [138] |
| CDKN2B-AS1 UBE2SP1 | related to AS according to informatics analysis | HCC | prognostic markers | 2.3 | [147] |
| SETD2 | involved in AS of several genes | CRC | predictive of TNM status | 2.2 | [89] |
| Therapeutic Target | AS Event Involved | Cancer Characteristics | Molecular Mechanisms | Possible Drug | Refs. |
|---|---|---|---|---|---|
| miR-193a-5p | 2 aberrant isoforms which favor EMT | overexpressed in PDAC | miRNA targets SRSF6 | antisense oligonucleotides | [122] |
| miR-206 | switch PKM2 to PKM1 | downregulated in CRC | miRNA targets hnRNPA1 | mimic | [124] |
| miR-339-5p | switch PKM2 to PKM1 | downregulated in CRC | miRNA targets hnRNPA1 | kaempferol (activates synthesis of the miRNA) | [125] |
| miR-574-5p | aberrant production of microsomal prostaglandin E synthase-1 (mPGES1) isoform | NSCLC | miRNA is a decoy to CUG binding protein (CUGBP1), preventing its binding to 3′ UTR of mPGES1 | antisense oligonucleotides; inhibitors of prostaglandin E2 synthesis | [103,104] |
| hypermethylated PKM exon 10 | inclusion of exon 10 and skipping of exon 9 resulting in PKM2 | squamous cell carcinoma of the buccal mucosa | (inhibits DNMT3B and reduces exon 10 methylation) | curcumin | [62] |
| KDM3A | acetylation of PHF5A reduces intron retention in KDM3A | upregulated in CRC | increased level of KDM3A mRNA activates Wnt pathway | KDM3Ai | [99] |
| PRMTs | Many changes in splicing events | PDAC, myeloid leukaemia, melanoma and other cancers | multiple proteins involved in AS are substrates of PRMTs | type I PRMTi type II PRMTi | [148,149,150,151] |
| hnRNPA2B1 | switch hnRNPB1 to hnRNPA2 | CRC and oesophageal cancer cell lines | switch Bcl-xL to Bcl-xS, caspase activation | extracts of Cotyledon orbiculata | [144] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gimeno-Valiente, F.; López-Rodas, G.; Castillo, J.; Franco, L. Alternative Splicing, Epigenetic Modifications and Cancer: A Dangerous Triangle, or a Hopeful One? Cancers 2022, 14, 560. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14030560
Gimeno-Valiente F, López-Rodas G, Castillo J, Franco L. Alternative Splicing, Epigenetic Modifications and Cancer: A Dangerous Triangle, or a Hopeful One? Cancers. 2022; 14(3):560. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14030560
Chicago/Turabian StyleGimeno-Valiente, Francisco, Gerardo López-Rodas, Josefa Castillo, and Luis Franco. 2022. "Alternative Splicing, Epigenetic Modifications and Cancer: A Dangerous Triangle, or a Hopeful One?" Cancers 14, no. 3: 560. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14030560