
Combination of Resminostat with Ruxolitinib Exerts Antitumor Effects in the Chick Embryo Chorioallantoic Membrane Model for Cutaneous T Cell Lymphoma

 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
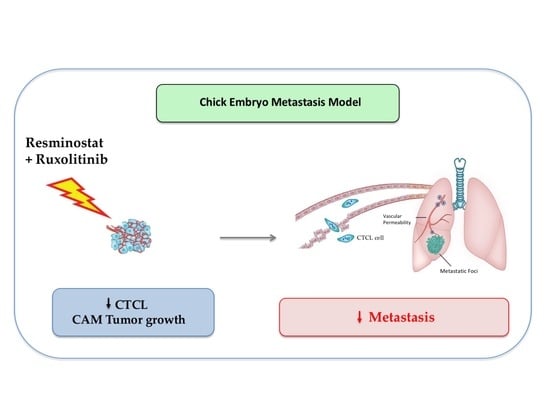
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culturing
2.2. Drugs Tested
2.3. Chick Embryo CAM Model: Xenografted Tumours and Spontaneous Metastasis
2.4. Quantitative Detection of Human Tumor Cell Metastasis
2.5. Experimental Metastasis Chick Embryo Model
2.6. Live Cell Imaging
2.7. Proliferation Analyses
2.8. Apoptosis Assays
2.9. Migration Assays
2.10. Invasion Assays
2.11. 3D Tumour Spheroid Invasion Assay
2.12. Immunohistochemistry
2.13. Western Blotting
2.14. Statistical Analysis
3. Results
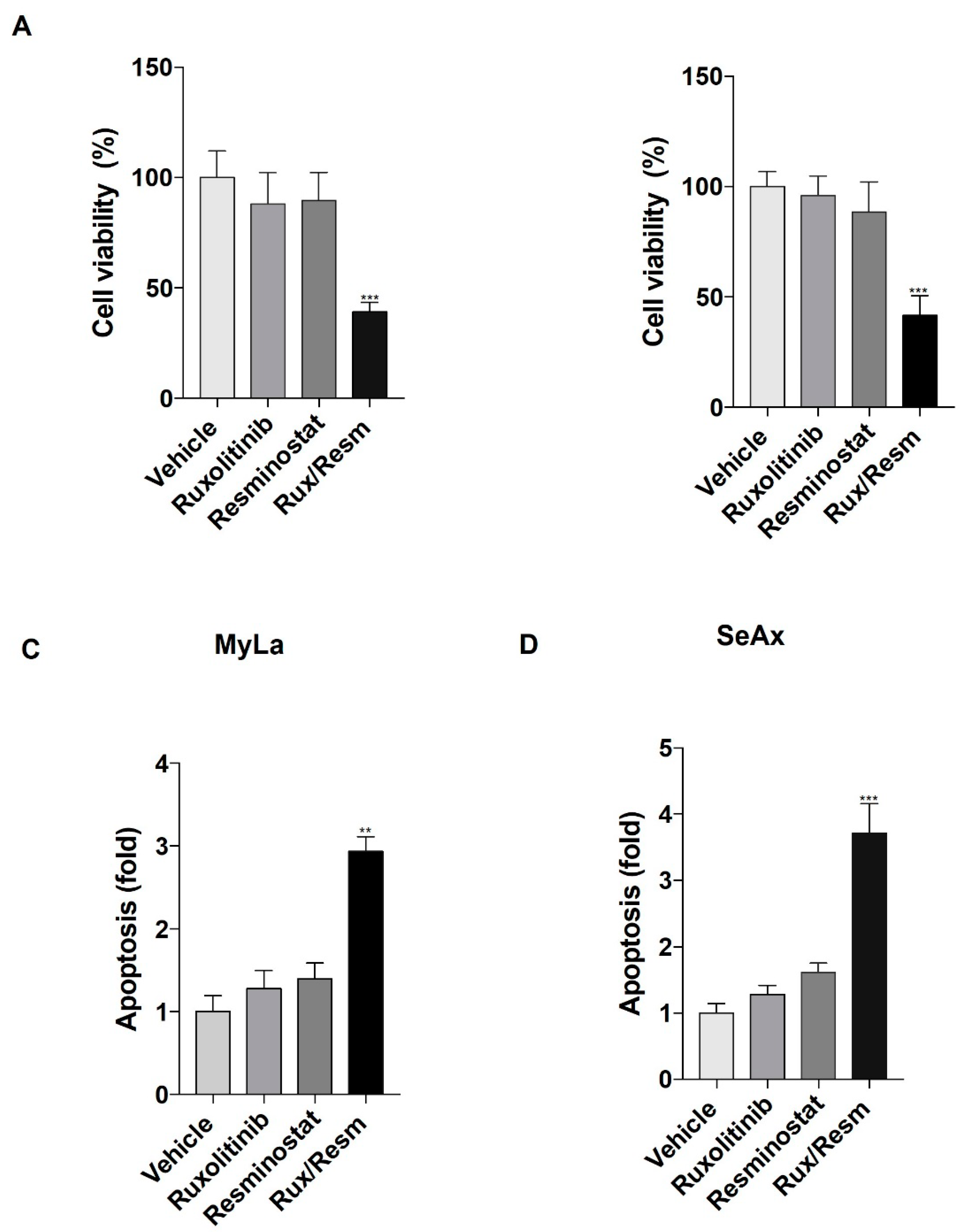
3.1. Resminostat, Ruxolitinib, and Their Combination Inhibit Cell Proliferation and Induce Apoptosis in CTCL Cells
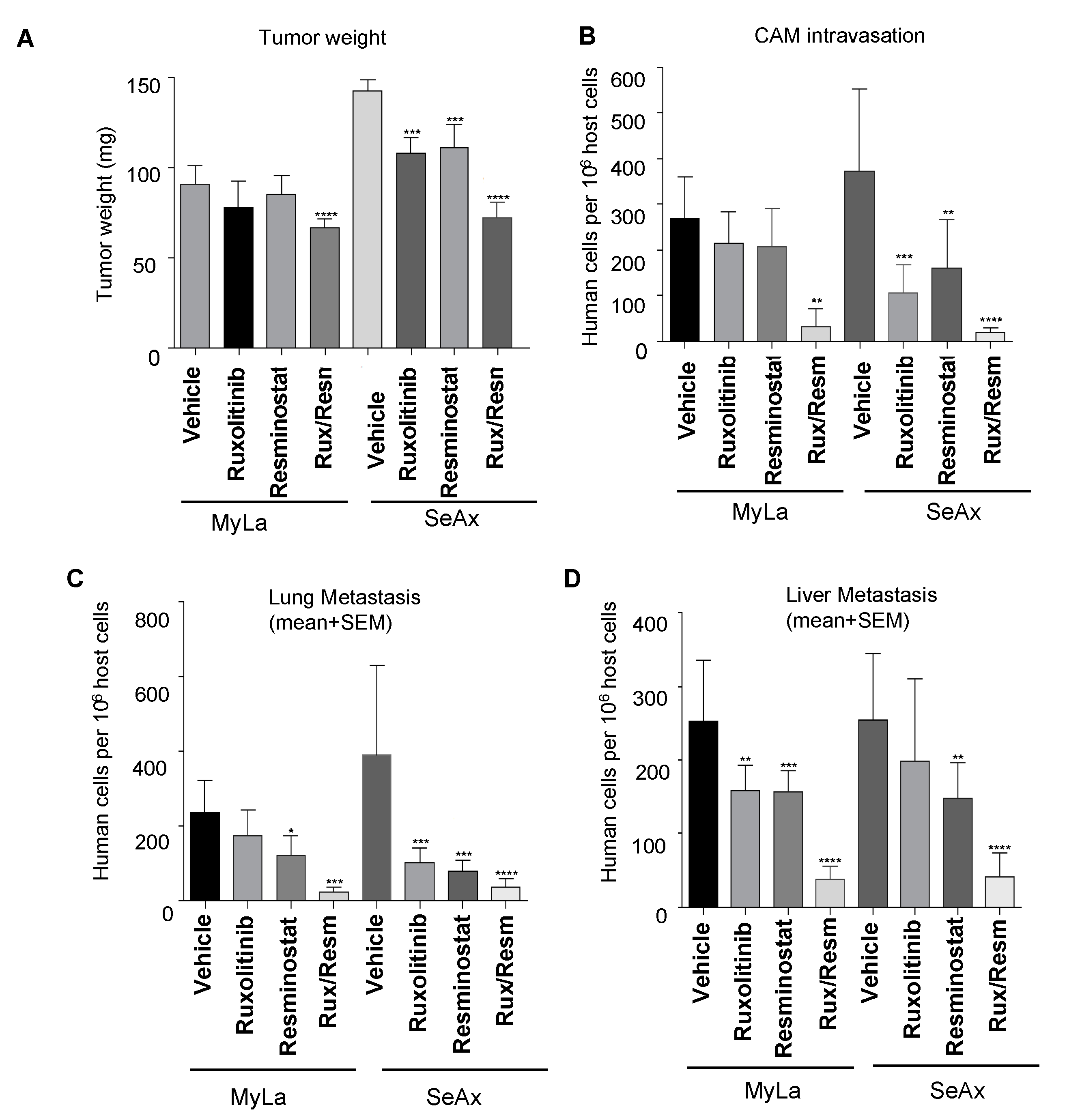
3.2. Combined Treatment of Resminostat and Ruxolitinib Impairs CTCL Tumorigenesis and Metastasis in Spontaneous Metastasis CAM Assay
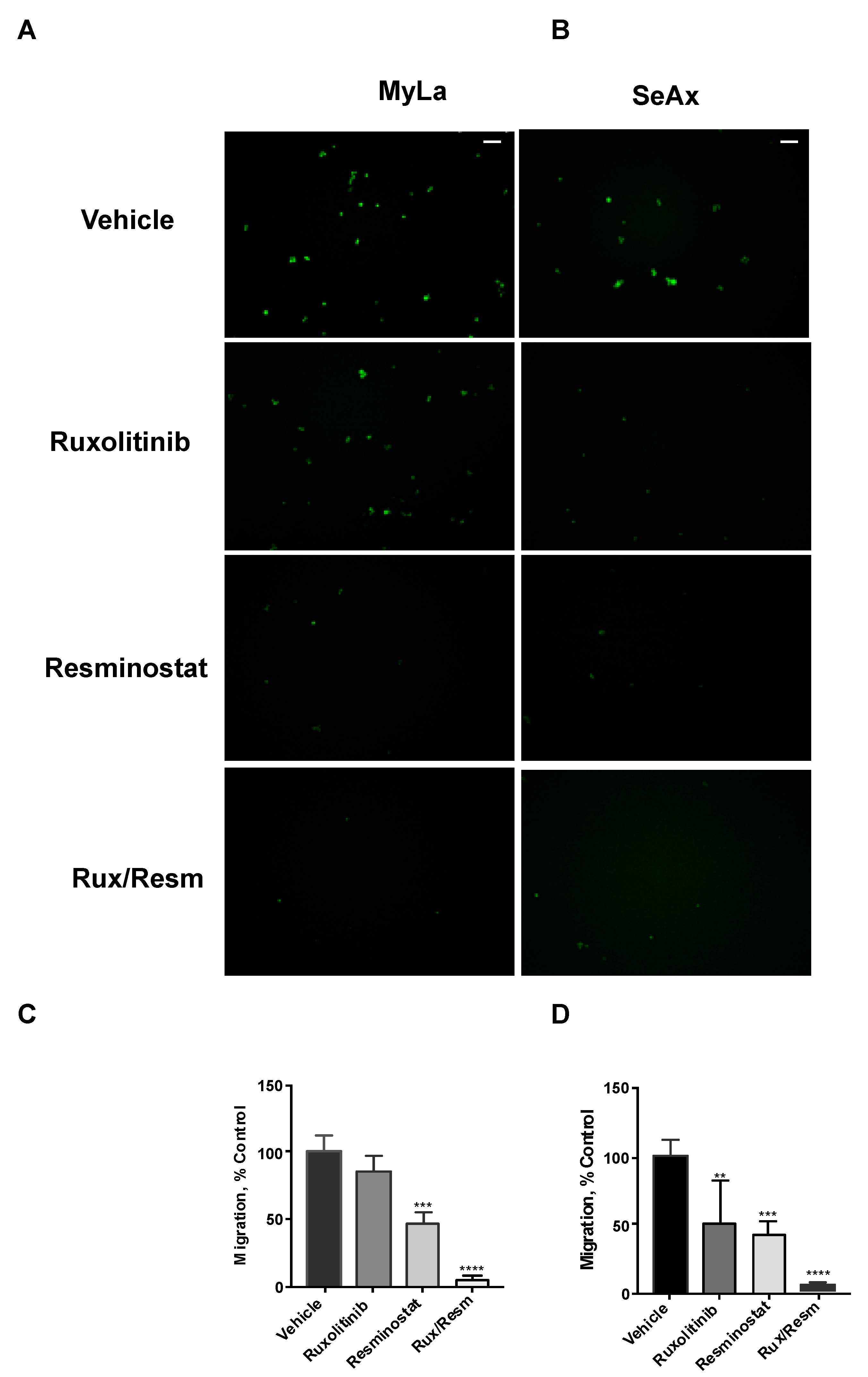
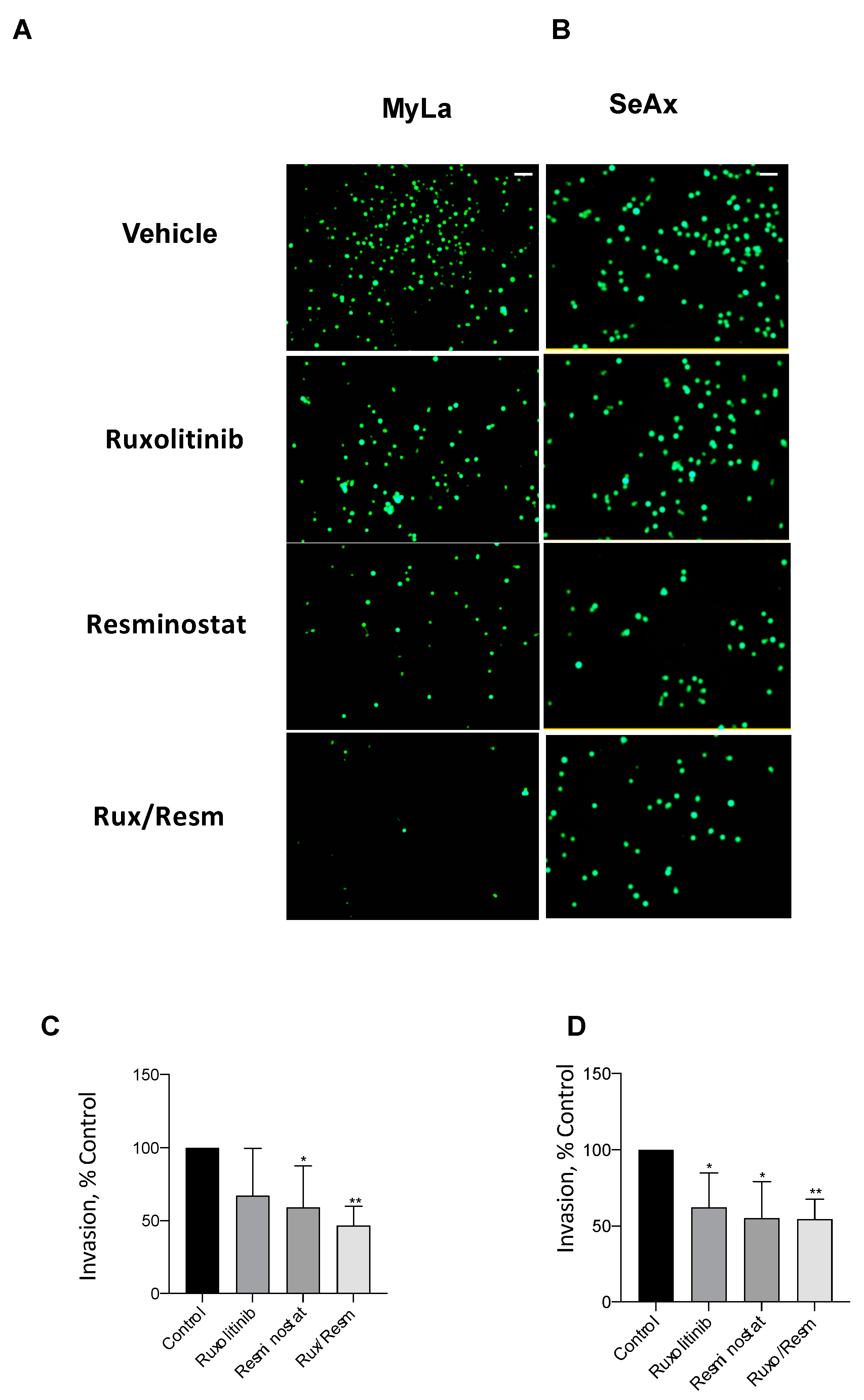
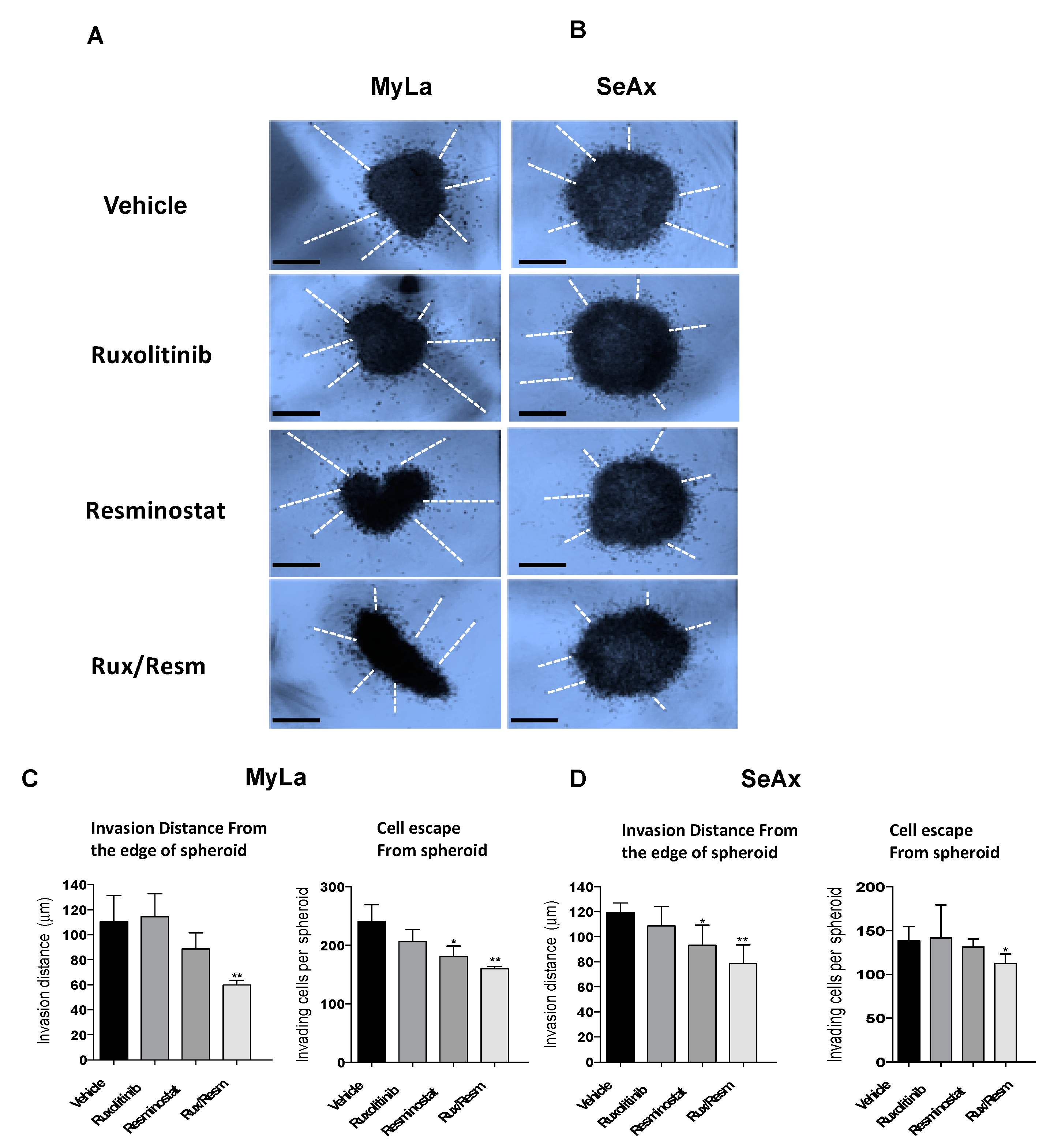
3.3. Combined Treatment of Resminostat and Ruxolitinib Impairs CTCL Migration and Invasion
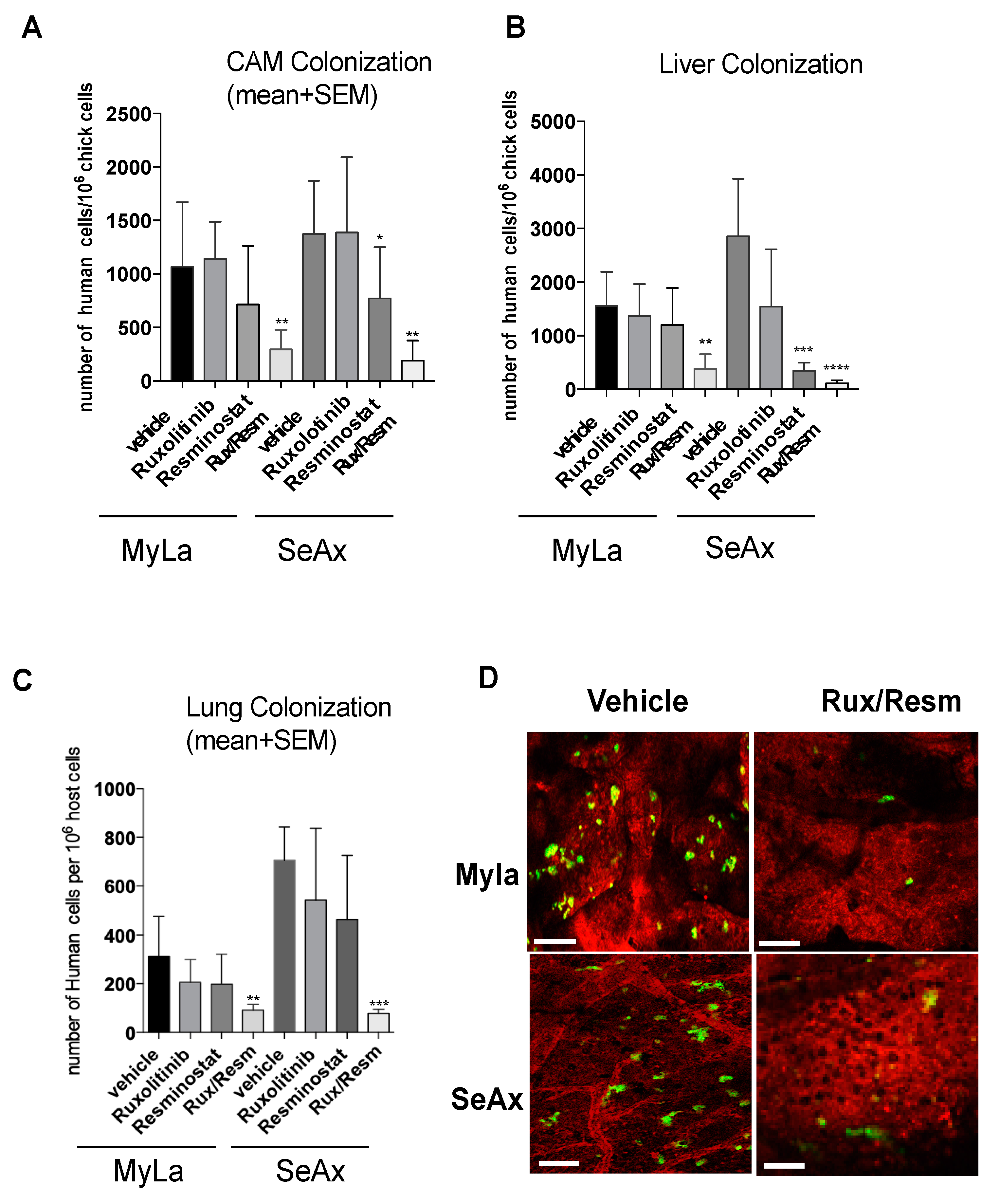
3.4. Combined Treatment of Resminostat and Ruxolitinib Reduce CTCL Extravasation CAM Assay
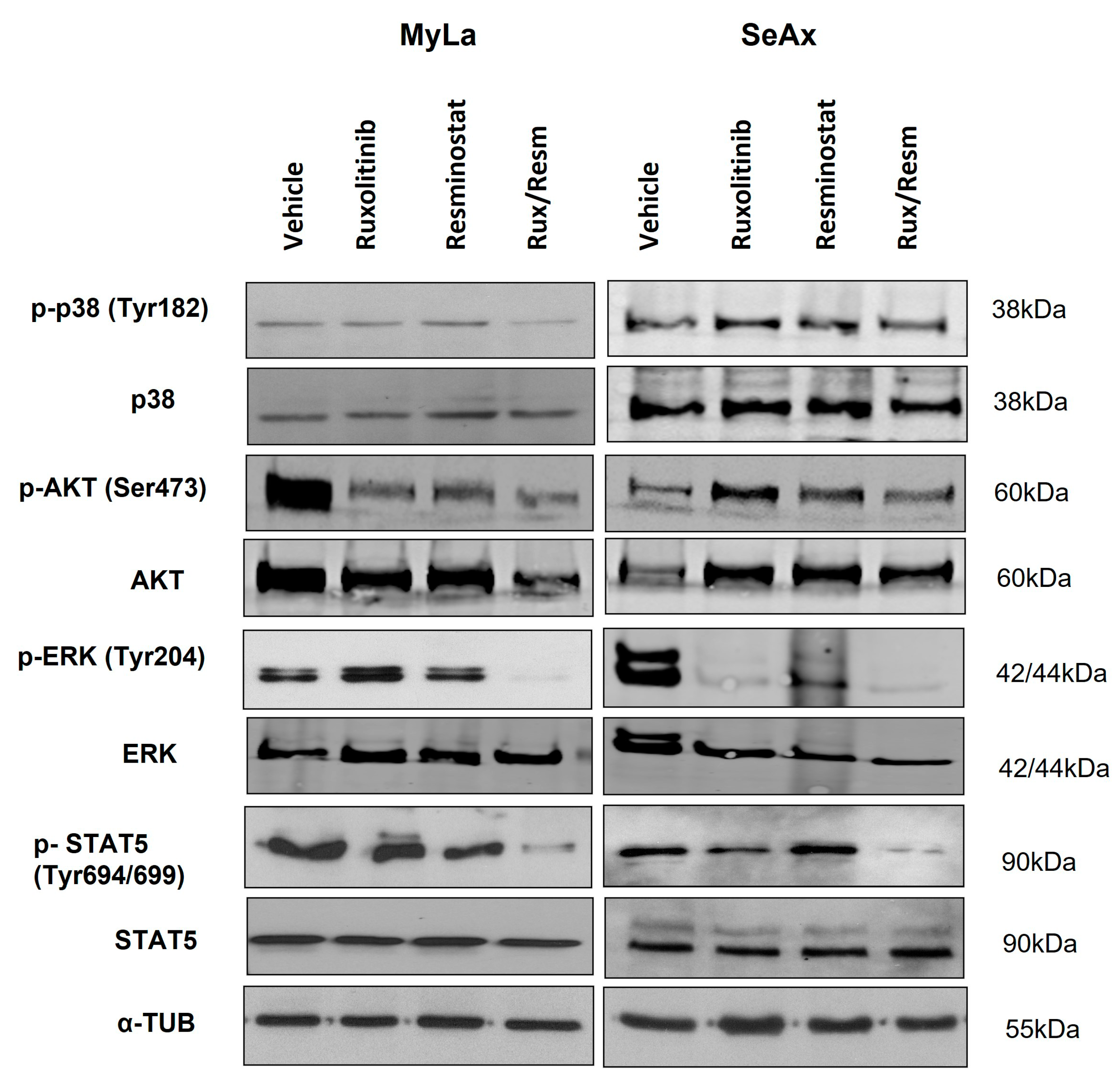
3.5. Resminostat, Ruxolitinib, and Their Combination Inhibit Key Signaling Pathways in CTCL Xenografted Tumors
4. Discussion
5. Conclusions—Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Willemze, R.; Jaffe, E.S.; Burg, G.; Cerroni, L.; Berti, E.; Swerdlow, S.H.; Ralfkiaer, E.; Chimenti, S.; Diaz-Perez, J.L.; Duncan, L.M.; et al. WHO-EORTC Classification for Cutaneous Lymphomas. Blood 2005, 105, 3768–3785. [Google Scholar] [CrossRef] [Green Version]
- Girardi, M.; Heald, P.W.; Wilson, L.D. The Pathogenesis of Mycosis Fungoides. N. Engl. J. Med. 2004, 350, 1978–1988. [Google Scholar] [CrossRef] [PubMed]
- Querfeld, C.; Guitart, J.; Kuzel, T.M.; Rosen, S.T. Primary Cutaneous Lymphomas: A Review with Current Treatment Options. Blood Rev. 2003, 17, 131–142. [Google Scholar] [CrossRef]
- Willemze, R.; Cerroni, L.; Kempf, W.; Berti, E.; Facchetti, F.; Swerdlow, S.H.; Jaffe, E.S. The 2018 Update of the WHO-EORTC Classification for Primary Cutaneous Lymphomas. Blood 2019, 133, 1703–1714. [Google Scholar] [CrossRef]
- Arulogun, S.O.; Prince, H.M.; Ng, J.; Lade, S.; Ryan, G.F.; Blewitt, O.; McCormack, C. Long-Term Outcomes of Patients with Advanced-Stage Cutaneous T-Cell Lymphoma and Large Cell Transformation. Blood 2008, 112, 3082–3087. [Google Scholar] [CrossRef] [Green Version]
- Pavlidis, A.; Piperi, C.; Papadavid, E. Novel Therapeutic Approaches for Cutaneous T Cell Lymphomas. Expert Rev. Clin. Immunol. 2021, 17, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Sawas, A. Antibody-Directed Therapies: Toward a Durable and Tolerable Treatment Platform for CTCL. Front. Oncol. 2019, 9, 645. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Miyagaki, T. Novel and Future Therapeutic Drugs for Advanced Mycosis Fungoides and Sézary Syndrome. Front. Med. 2019, 6, 116. [Google Scholar] [CrossRef]
- Samimi, S.; Morrissey, K.; Anshelevich, S.; Evans, K.; Gardner, J.; Musiek, A.; Vittorio, C.; Rook, A.; Kim, E. Romidepsin and Interferon Gamma: A Novel Combination for Refractory Cutaneous T-Cell Lymphoma. J. Am. Acad. Dermatol. 2013, 68, e5. [Google Scholar] [CrossRef]
- Kim, S.R.; Lewis, J.M.; Cyrenne, B.M.; Monico, P.F.; Mirza, F.N.; Carlson, K.R.; Foss, F.M.; Girardi, M. BET Inhibition in Advanced Cutaneous T Cell Lymphoma Is Synergistically Potentiated by BCL2 Inhibition or HDAC Inhibition. Oncotarget 2018, 9, 29193–29207. [Google Scholar] [CrossRef] [Green Version]
- Froehlich, T.C.; Müller-Decker, K.; Braun, J.D.; Albrecht, T.; Schroeder, A.; Gülow, K.; Goerdt, S.; Krammer, P.H.; Nicolay, J.P. Combined Inhibition of Bcl-2 and NFκB Synergistically Induces Cell Death in Cutaneous T-Cell Lymphoma. Blood 2019, 134, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Okhovat, J.P.; Hong, E.K.; Kim, Y.H.; Wood, G.S. Preclinical Studies Support Combined Inhibition of BET Family Proteins and Histone Deacetylases as Epigenetic Therapy for Cutaneous T-Cell Lymphoma. Neoplasia 2019, 21, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Vu, K.; Wu, C.H.; Yang, C.Y.; Zhan, A.; Cavallone, E.; Berry, W.; Heeter, P.; Pincus, L.; Wieduwilt, M.J.; William, B.M.; et al. Romidepsin Plus Liposomal Doxorubicin Is Safe and Effective in Patients with Relapsed or Refractory T-Cell Lymphoma: Results of a Phase I Dose-Escalation Study. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 1000–1008. [Google Scholar] [CrossRef] [PubMed]
- Phyo, Z.H.; Shanbhag, S.; Rozati, S. Update on Biology of Cutaneous T-Cell Lymphoma. Front. Oncol. 2020, 10, 765. [Google Scholar] [CrossRef] [PubMed]
- Dulmage, B.O.; Geskin, L.J. Lessons Learned from Gene Expression Profiling of Cutaneous T-Cell Lymphoma. Br. J. Dermatol. 2013, 169, 1188–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koboldt, D.C.; Steinberg, K.M.; Larson, D.E.; Wilson, R.K.; Mardis, E.R. The Next-Generation Sequencing Revolution and Its Impact on Genomics. Cell 2013, 155, 27. [Google Scholar] [CrossRef] [Green Version]
- van Doorn, R.; van Kester, M.S.; Dijkman, R.; Vermeer, M.H.; Mulder, A.A.; Szuhai, K.; Knijnenburg, J.; Boer, J.M.; Willemze, R.; Tensen, C.P. Oncogenomic Analysis of Mycosis Fungoides Reveals Major Differences with Sezary Syndrome. Blood 2009, 113, 127–136. [Google Scholar] [CrossRef] [Green Version]
- Meyer, S.C.; Keller, M.D.; Chiu, S.; Koppikar, P.; Guryanova, O.A.; Rapaport, F.; Xu, K.; Manova, K.; Pankov, D.; O’Reilly, R.J.; et al. CHZ868, a Type II JAK2 Inhibitor, Reverses Type I JAK Inhibitor Persistence and Demonstrates Efficacy in Myeloproliferative Neoplasms. Cancer Cell 2015, 28, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Mullally, A.; Ebert, B.L. Janus Reveals Another Face: The Biologic Rationale for Targeting Janus Kinase 2 in Lymphoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 4168–4170. [Google Scholar] [CrossRef]
- Karagianni, F.; Piperi, C.; Mpakou, V.; Spathis, A.; Foukas, P.G.; Dalamaga, M.; Pappa, V.; Papadavid, E. Ruxolitinib with Resminostat Exert Synergistic Antitumor Effects in Cutaneous T-Cell Lymphoma. PLoS ONE 2021, 16, e0248298. [Google Scholar] [CrossRef]
- Merlos Rodrigo, M.A.; Casar, B.; Michalkova, H.; Jimenez Jimenez, A.M.; Heger, Z.; Adam, V. Extending the Applicability of In Ovo and Ex Ovo Chicken Chorioallantoic Membrane Assays to Study Cytostatic Activity in Neuroblastoma Cells. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Crespo, P.; Casar, B. The Chick Embryo Chorioallantoic Membrane as an in Vivo Model to Study Metastasis. BIO-PROTOCOL 2016, 6, e1962. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, B.B.; da Silva, M.V.; de Morais Ribeiro, L.N. The Chicken Embryo as an in Vivo Experimental Model for Drug Testing: Advantages and Limitations. Lab Anim. 2021, 50, 138–139. [Google Scholar] [CrossRef] [PubMed]
- Weber, W.T.; Mausner, R. Migration Patterns of Avian Embryonic Bone Marrow Cells and Their Differentiation to Functional T and B Cells. Adv. Exp. Med. Biol. 1977, 88, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Janković, B.D.; Isaković, K.; Lukić, M.L.; Vujanović, N.L.; Petrović, S.; Marković, B.M. Immunological capacity of the chicken embryo. I. Relationship between the maturation of lymphoid tissues and the occurrence of cell-mediated immunity in the developing chicken embryo. Immunology 1975, 29, 497. [Google Scholar] [PubMed]
- Marga Janse, E.; Jeurissen, S.H.M. Ontogeny and Function of Two Non-Lymphoid Cell Populations in the Chicken Embryo. Immunobiology 1991, 182, 472–481. [Google Scholar] [CrossRef]
- Koop, S.; Khokha, R.; Schmidt, E.E.; MacDonald, I.C.; Morris, V.L.; Chambers, A.F.; Groom, A.C. Overexpression of metalloproteinase inhibitor in B16F10 cells does not affect extravasation but reduces tumor growth. Cancer Res. 1994, 54, 4791–4797. [Google Scholar] [PubMed]
- Schneider-Stock, R.; Ribatti, D. The CAM Assay as an Alternative In Vivo Model for Drug Testing. Handb. Exp. Pharmacol. 2021, 265, 303–323. [Google Scholar] [CrossRef]
- Chou, T.C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Zackheim, H.S.; Kashani-Sabet, M. Treatment of Cutaneous T Cell Lymphoma: Current Status and Future Directions. Am. J. Clin. Dermatol. 2002, 3, 103–119. [Google Scholar] [CrossRef]
- Ramelyte, E.; Dummer, R.; Guenova, E. Investigative Drugs for the Treatment of Cutaneous T-Cell Lymphomas (CTCL): An Update. Expert Opin. Investig. Drugs 2019, 28, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Quoc Trung, L.; Espinoza, J.L.; Takami, A.; Nakao, S. Resveratrol Induces Cell Cycle Arrest and Apoptosis in Malignant NK Cells via JAK2/STAT3 Pathway Inhibition. PLoS ONE 2013, 8, e55183. [Google Scholar] [CrossRef] [Green Version]
- Duvic, M.; Lemak, N.A.; Redman, J.R.; Eifel, P.J.; Tucker, S.L.; Cabanillas, F.F.; Kurzrock, R. Combined Modality Therapy for Cutaneous T-Cell Lymphoma. J. Am. Acad. Dermatol. 1996, 34, 1022–1029. [Google Scholar] [CrossRef]
- Braverman, I.M.; Bruce Yager, N.; Chen, M.; Cadman, E.C.; Hait, W.N.; Maynard, T. Combined Total Body Electron Beam Irradiation and Chemotherapy for Mycosis Fungoides. J. Am. Acad. Dermatol. 1987, 16, 45–60. [Google Scholar] [CrossRef]
- Winkler, C.F.; Sausville, E.A.; Ihde, D.C.; Fischmann, A.B.; Schechter, G.P.; Kumar, P.P.; Nibhanupdi, J.R.; Minna, J.D.; Makuch, R.W.; Eddy, J.L. Combined Modality Treatment of Cutaneous T Cell Lymphoma: Results of a 6-Year Follow-Up. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1986, 4, 1094–1100. [Google Scholar] [CrossRef]
- Hallahan, D.E.; Griem, M.L.; Griem, S.F.; Medenica, M.; Soltani, K.; Lorincz, A.L.; Baron, J.M. Combined Modality Therapy for Tumor Stage Mycosis Fungoides: Results of a 10-Year Follow-Up. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1988, 6, 1177–1183. [Google Scholar] [CrossRef]
- Kaye, F.J.; Bunn, P.A.; Steinberg, S.M.; Stocker, J.L.; Ihde, D.C.; Fischmann, A.B.; Glatstein, E.J.; Schechter, G.P.; Phelps, R.M.; Foss, F.M.; et al. A Randomized Trial Comparing Combination Electron-Beam Radiation and Chemotherapy with Topical Therapy in the Initial Treatment of Mycosis Fungoides. N. Engl. J. Med. 1989, 321, 1784–1790. [Google Scholar] [CrossRef]
- Zinzani, P.L.; Baliva, G.; Magagnoli, M.; Bendandi, M.; Modugno, G.; Gherlinzoni, F.; Orcioni, G.F.; Ascani, S.; Simoni, R.; Pileri, S.A.; et al. Gemcitabine Treatment in Pretreated Cutaneous T-Cell Lymphoma: Experience in 44 Patients. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2000, 18, 2603–2606. [Google Scholar] [CrossRef]
- Wilson, L.D.; Jones, G.W.; Kim, D.; Rosenthal, D.; Christensen, I.R.; Edelson, R.L.; Heald, P.W.; Kacinski, B.M. Experience with Total Skin Electron Beam Therapy in Combination with Extracorporeal Photopheresis in the Management of Patients with Erythrodermic (T4) Mycosis Fungoides. J. Am. Acad. Dermatol. 2000, 43, 54–60. [Google Scholar] [CrossRef]
- Wilson, L.D.; Licata, A.L.; Braverman, I.M.; Edelson, R.L.; Heald, P.W.; Feldman, A.M.; Kacinski, B.M. Systemic Chemotherapy and Extracorporeal Photochemotherapy for T3 and T4 Cutaneous T-Cell Lymphoma Patients Who Have Achieved a Complete Response to Total Skin Electron Beam Therapy. Int. J. Radiat. Oncol. Biol. Phys. 1995, 32, 987–995. [Google Scholar] [CrossRef]
- Quirós, P.A.; Jones, G.W.; Kacinski, B.M.; Braverman, I.M.; Heald, P.W.; Edelson, R.L.; Wilson, L.D. Total Skin Electron Beam Therapy Followed by Adjuvant Psoralen/Ultraviolet-A Light in the Management of Patients with T1 and T2 Cutaneous T-Cell Lymphoma (Mycosis Fungoides). Int. J. Radiat. Oncol. Biol. Phys. 1997, 38, 1027–1035. [Google Scholar] [CrossRef]
- Chinn, D.M.; Chow, S.; Kim, Y.H.; Hoppe, R.T. Total Skin Electron Beam Therapy with or without Adjuvant Topical Nitrogen Mustard or Nitrogen Mustard Alone as Initial Treatment of T2 and T3 Mycosis Fungoides. Int. J. Radiat. Oncol. Biol. Phys. 1999, 43, 951–958. [Google Scholar] [CrossRef]
- Kaltoft, K.; Bisballe, S.; Rasmussen, H.F.; Thestrup-Pedersen, K.; Thomsen, K.; Sterry, W. A Continuous T-Cell Line from a Patient with Sézary Syndrome. Arch. Dermatol. Res. 1987, 279, 293–298. [Google Scholar] [CrossRef]
- Kaltoft, K.; Bisballe, S.; Dyrberg, T.; Boel, E.; Rasmussen, P.B.; Thestrup-Pedersen, K. Establishment of Two Continuous T-Cell Strains from a Single Plaque of a Patient with Mycosis Fungoides. In Vitr. Cell. Dev. Biol. J. Tissue Cult. Assoc. 1992, 28A, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Charley, M.R.; Tharp, M.; Locker, J.; Deng, J.S.; Goslen, J.B.; Mauro, T.; McCoy, P.; Abell, E.; Jegasothy, B. Establishment of a Human Cutaneous T-Cell Lymphoma in C.B-17 SCID Mice. J. Investig. Dermatol. 1990, 94, 381–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaler, S.; Burger, A.M.; Schulz, T.; Brill, B.; Bittner, A.; Oberholzer, P.A.; Dummer, R.; Schnierle, B.S. Establishment of a Mouse Xenograft Model for Mycosis Fungoides. Exp. Dermatol. 2004, 13, 406–412. [Google Scholar] [CrossRef]
- Beylot-Barry, M.; Sibaud, V.; Thiebaut, R.; Vergier, B.; Beylot, C.; Delaunay, M.; Chene, G.; Dubus, P.; Merlio, J.P. Evidence That an Identical T Cell Clone in Skin and Peripheral Blood Lymphocytes Is an Independent Prognostic Factor in Primary Cutaneous T Cell Lymphomas. J. Investig. Dermatol. 2001, 117, 920–926. [Google Scholar] [CrossRef] [Green Version]
- Andrique, L.; Poglio, S.; Prochazkova-Carlotti, M.; Kadin, M.E.; Giese, A.; Idrissi, Y.; Beylot-Barry, M.; Merlio, J.P.; Chevret, E. Intrahepatic Xenograft of Cutaneous T-Cell Lymphoma Cell Lines: A Useful Model for Rapid Biological and Therapeutic Evaluation. Am. J. Pathol. 2016, 186, 1775–1785. [Google Scholar] [CrossRef] [Green Version]
- Begley, C.G.; Ellis, L.M. Drug Development: Raise Standards for Preclinical Cancer Research. Nature 2012, 483, 531–533. [Google Scholar] [CrossRef]
- Perrin, S. Preclinical Research: Make Mouse Studies Work. Nature 2014, 507, 423–425. [Google Scholar] [CrossRef]
- Ribatti, D.; Annese, T.; Tamma, R. The Use of the Chick Embryo CAM Assay in the Study of Angiogenic Activiy of Biomaterials. Microvasc. Res. 2020, 131, 104026. [Google Scholar] [CrossRef] [PubMed]
- Naik, M.; Brahma, P.; Dixit, M. A Cost-Effective and Efficient Chick Ex-Ovo CAM Assay Protocol to Assess Angiogenesis. Methods Protoc. 2018, 1, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nebbioso, A.; Dell’Aversana, C.; Bugge, A.; Sarno, R.; Valente, S.; Rotili, D.; Manzo, F.; Teti, D.; Mandrup, S.; Ciana, P.; et al. HDACs Class II-Selective Inhibition Alters Nuclear Receptor-Dependent Differentiation. J. Mol. Endocrinol. 2010, 45, 219–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benedetti, R.; Conte, M.; Altucci, L. Targeting Histone Deacetylases in Diseases: Where Are We? Antioxid. Redox Signal. 2015, 23, 99–126. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.J.; Snowden, J.A.; Zeidler, M.P.; Danson, S.J. The Role of JAK/STAT Signalling in the Pathogenesis, Prognosis and Treatment of Solid Tumours. Br. J. Cancer 2015, 113, 365–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Fiskus, W.; Chong, D.G.; Buckley, K.M.; Natarajan, K.; Rao, R.; Joshi, A.; Balusu, R.; Koul, S.; Chen, J.; et al. Cotreatment with Panobinostat and JAK2 Inhibitor TG101209 Attenuates JAK2V617F Levels and Signaling and Exerts Synergistic Cytotoxic Effects against Human Myeloproliferative Neoplastic Cells. Blood 2009, 114, 5024–5033. [Google Scholar] [CrossRef]
- Lafave, L.M.; Levine, R.L. JAK2 the Future: Therapeutic Strategies for JAK-Dependent Malignancies. Trends Pharmacol. Sci. 2012, 33, 574–582. [Google Scholar] [CrossRef]
- Fiskus, W.; Verstovsek, S.; Manshouri, T.; Rao, R.; Balusu, R.; Venkannagari, S.; Nalabothula, N.R.; Ha, K.; Smith, J.E.; Hembruff, S.L.; et al. Heat Shock Protein 90 Inhibitor Is Synergistic with JAK2 Inhibitor and Overcomes Resistance to JAK2-TKI in Human Myeloproliferative Neoplasm Cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 7347–7358. [Google Scholar] [CrossRef] [Green Version]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer Activities of Histone Deacetylase Inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Nolan, L.; Johnson, P.W.M.; Ganesan, A.; Packham, G.; Crabb, S.J. Will Histone Deacetylase Inhibitors Require Combination with Other Agents to Fulfil Their Therapeutic Potential? Br. J. Cancer 2008, 99, 689–694. [Google Scholar] [CrossRef] [Green Version]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination Therapy with Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindemann, R.K.; Gabrielli, B.; Johnstone, R.W. Histone-deacetylase inhibitors for the treatment of cancer. Cell Cycle 2004, 3, 777–786. [Google Scholar] [CrossRef]
- Verstovsek, S.; Gotlib, J.; Mesa, R.A.; Vannucchi, A.M.; Kiladjian, J.J.; Cervantes, F.; Harrison, C.N.; Paquette, R.; Sun, W.; Naim, A.; et al. Long-Term Survival in Patients Treated with Ruxolitinib for Myelofibrosis: COMFORT-I and -II Pooled Analyses. J. Hematol. Oncol. 2017, 10, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzacurati, L.; Lambert, Q.T.; Pradhan, A.; Griner, L.N.; Huszar, D.; Reuther, G.W. The PIM Inhibitor AZD1208 Synergizes with Ruxolitinib to Induce Apoptosis of Ruxolitinib Sensitive and Resistant JAK2-V617F-Driven Cells and Inhibit Colony Formation of Primary MPN Cells. Oncotarget 2015, 6, 40141–40157. [Google Scholar] [CrossRef] [Green Version]
- Pérez, C.; González-Rincón, J.; Onaindia, A.; Almaráz, C.; García-Díaz, N.; Pisonero, H.; Curiel-Olmo, S.; Gómez, S.; Cereceda, L.; Madureira, R.; et al. Mutated JAK Kinases and Deregulated STAT Activity Are Potential Therapeutic Targets in Cutaneous T-Cell Lymphoma. Haematologica 2015, 100, e450–e453. [Google Scholar] [CrossRef] [Green Version]
- Civallero, M.; Cosenza, M.; Pozzi, S.; Sacchi, S. Ruxolitinib Combined with Vorinostat Suppresses Tumor Growth and Alters Metabolic Phenotype in Hematological Diseases. Oncotarget 2017, 8, 103797. [Google Scholar] [CrossRef] [Green Version]
- Brunetto, A.T.; Ang, J.E.; Lal, R.; Olmos, D.; Molife, L.R.; Kristeleit, R.; Parker, A.; Casamayor, I.; Olaleye, M.; Mais, A.; et al. First-in-Human, Pharmacokinetic and Pharmacodynamic Phase I Study of Resminostat, an Oral Histone Deacetylase Inhibitor, in Patients with Advanced Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 5494–5504. [Google Scholar] [CrossRef] [Green Version]
- Yumeen, S.; Mirza, F.N.; Lewis, J.M.; King, A.L.O.; Kim, S.R.; Carlson, K.R.; Umlauf, S.R.; Surovtseva, Y.V.; Foss, F.M.; Girardi, M. JAK Inhibition Synergistically Potentiates BCL2, BET, HDAC, and Proteasome Inhibition in Advanced CTCL. Blood Adv. 2020, 4, 2213–2226. [Google Scholar] [CrossRef]
- Levidou, G.; Siakantaris, M.; Papadaki, T.; Papadavid, E.; Vassilakopoulos, T.P.; Angelopoulou, M.K.; Marinos, L.; Nikolaou, V.; Economidi, A.; Antoniou, C.; et al. A Comprehensive Immunohistochemical Approach of AKT/MTOR Pathway and p-STAT3 in Mycosis Fungoides. J. Am. Acad. Dermatol. 2013, 69, 375–384. [Google Scholar] [CrossRef]
- Papadavid, E.; Korkolopoulou, P.; Levidou, G.; Saetta, A.A.; Papadaki, T.; Siakantaris, M.; Nikolaou, V.; Oikonomidi, A.; Chatziandreou, I.; Marinos, L.; et al. In Situ Assessment of PI3K and PTEN Alterations in Mycosis Fungoides: Correlation with Clinicopathological Features. Exp. Dermatol. 2014, 23, 931–933. [Google Scholar] [CrossRef]
- de Masson, A.; O’Malley, J.T.; Elco, C.P.; Garcia, S.S.; Divito, S.J.; Lowry, E.L.; Tawa, M.; Fisher, D.C.; Devlin, P.M.; Teague, J.E.; et al. High-Throughput Sequencing of the T Cell Receptor β Gene Identifies Aggressive Early-Stage Mycosis Fungoides. Sci. Transl. Med. 2018, 10, eaar5894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, J.J.; Clark, R.A.; Watanabe, R.; Kupper, T.S. Sezary Syndrome and Mycosis Fungoides Arise from Distinct T-Cell Subsets: A Biologic Rationale for Their Distinct Clinical Behaviors. Blood 2010, 116, 767–771. [Google Scholar] [CrossRef] [PubMed]
- Barrio, S.; Gallardo, M.; Arenas, A.; Ayala, R.; Rapado, I.; Rueda, D.; Jimenez, A.; Albizua, E.; Burgaleta, C.; Gilsanz, F.; et al. Inhibition of Related JAK/STAT Pathways with Molecular Targeted Drugs Shows Strong Synergy with Ruxolitinib in Chronic Myeloproliferative Neoplasm. Br. J. Haematol. 2013, 161, 667–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karagianni, F.; Piperi, C.; Casar, B.; de la Fuente-Vivas, D.; García-Gómez, R.; Lampadaki, K.; Pappa, V.; Papadavid, E. Combination of Resminostat with Ruxolitinib Exerts Antitumor Effects in the Chick Embryo Chorioallantoic Membrane Model for Cutaneous T Cell Lymphoma. Cancers 2022, 14, 1070. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14041070
Karagianni F, Piperi C, Casar B, de la Fuente-Vivas D, García-Gómez R, Lampadaki K, Pappa V, Papadavid E. Combination of Resminostat with Ruxolitinib Exerts Antitumor Effects in the Chick Embryo Chorioallantoic Membrane Model for Cutaneous T Cell Lymphoma. Cancers. 2022; 14(4):1070. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14041070
Chicago/Turabian StyleKaragianni, Fani, Christina Piperi, Berta Casar, Dalia de la Fuente-Vivas, Rocío García-Gómez, Kyriaki Lampadaki, Vasiliki Pappa, and Evangelia Papadavid. 2022. "Combination of Resminostat with Ruxolitinib Exerts Antitumor Effects in the Chick Embryo Chorioallantoic Membrane Model for Cutaneous T Cell Lymphoma" Cancers 14, no. 4: 1070. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14041070