
The Role of SMAD4 Inactivation in Epithelial–Mesenchymal Plasticity of Pancreatic Ductal Adenocarcinoma: The Missing Link?

 , , , , ,
, , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Literature Review
3. Discovery and General Structure of SMAD4
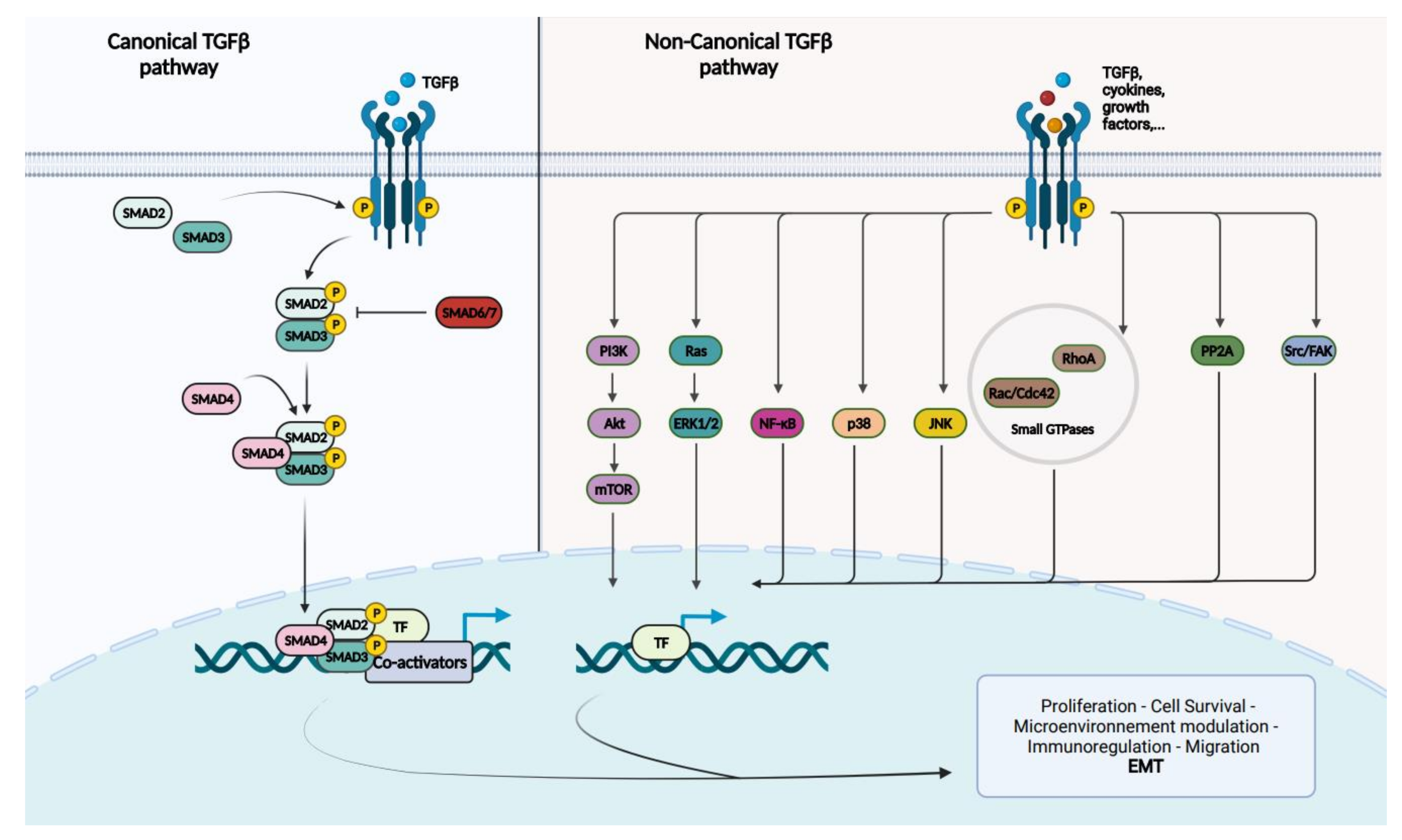
4. SMAD4 and TGF-Beta Signaling Pathway
5. The Role of the TGF-Beta Signaling Pathway in PDAC: In Brief
6. SMAD4 and Pancreatic Ductal Adenocarcinoma
7. An Introduction to Epithelial–Mesenchymal Transition
8. Epithelial–Mesenchymal Transition in PDAC: What Is Known
9. SMAD4 Alterations and EMT in PDAC
10. Controversies Regarding SMAD4 Alterations and its Potential Role in PDAC EMP
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 5’UTR | 5’ untranslated region |
| Beta-CAT | beta-catenin |
| BMP | bone morphogenic protein |
| CDK | cyclin-dependent kinase |
| ChIP | chromatin immunoprecipitation |
| Co-Smad | common-Smad |
| CTC | circulaing tumor cell |
| DFS | disease-free survival |
| DPC4 | deleted in pancreatic cancer, locus 4 |
| DAP-kinase | death-associated protein kinase |
| E-CAD | E-cadherin |
| EMP | epithelial–mesenchymal plasticity |
| EMT | epithelial–mesenchymal transition |
| EMT-TF | epithelial–mesenchymal transition transcription factor |
| GDF | growth and differentiation factor |
| GEMM | genetically engineered mouse model |
| IF | immunofluorescence |
| IHC | immunohistochemistry |
| I-Smads | inhibitory-Smads |
| CK19 | cytokeratin 19 |
| CK7 | cytokeratin 7 |
| CKAE1AE3 | cytokeratin AE1AE3 |
| LOH | loss of heterozygosity |
| Mad | Mothers against decapentaplegic |
| MH1 | Mad homology 1 |
| MH2 | Mad homology 2 |
| NB | Northern blot |
| N-CAD | N-Cadherin |
| NGS | next-generation sequencing |
| NK | natural killer |
| OS | overall survival |
| PDAC | pancreatic ductal adenocarcinoma |
| RNA-Seq | RNA Sequencing |
| R-Smad | receptor-regulated Smad |
| RT-PCR | reverse transcription polymerase chain reaction |
| RT-qPCR | reverse transcription quantitative polymerase chain reaction |
| scRNA-Seq | single-cell RNA-Sequencing |
| shRNAi | short hairpin RNA interference |
| siRNA | small interfering RNA |
| sma | small worm phenotype |
| SMA | smooth muscle actin |
| shRNA | short hairpin RNA |
| TEMTIA | The Epithelial–Mesenchymal Transition International Association |
| TF | transcription factor |
| TGF-beta | transforming growth factor-beta |
| TGFBR | transforming growth factor-beta receptor |
| TMA | tissue microarray |
| VIM | vimentin |
| WB | Western blot |
| WGS | whole-genome sequencing |
| WTS | whole-transcriptiome sequencing |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [Green Version]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primer 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Balaban, E.P.; Mangu, P.B.; Khorana, A.A.; Shah, M.A.; Mukherjee, S.; Crane, C.H.; Javle, M.M.; Eads, J.R.; Allen, P.; Ko, A.H.; et al. Locally Advanced, Unresectable Pancreatic Cancer: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2016, 34, 2654–2668. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.; Schwarz, L.; Borbath, I.; Henry, A.; Van Laethem, J.-L.; Malka, D.; Ducreux, M.; Conroy, T. An update on treatment options for pancreatic adenocarcinoma. Ther. Adv. Med. Oncol. 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worni, M.; Guller, U.; White, R.R.; Castleberry, A.W.; Pietrobon, R.; Cerny, T.; Gloor, B.; Koeberle, D. Modest Improvement in Overall Survival for Patients With Metastatic Pancreatic Cancer: A Trend Analysis Using the Surveillance, Epidemiology, and End Results Registry From 1988 to 2008. Pancreas 2013, 42, 1157–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- StatBite, U.S. Pancreatic Cancer Rates. JNCI J. Natl. Cancer Inst. 2010, 102, 1822. [Google Scholar]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.-L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.-C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.-M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.-C.; Mansour, J.; Mollaee, M.; Wagner, K.-U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef] [PubMed]
- Notta, F.; Chan-Seng-Yue, M.; Lemire, M.; Li, Y.; Wilson, G.W.; Connor, A.A.; Denroche, R.E.; Liang, S.-B.; Brown, A.M.K.; Kim, J.C.; et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 2016, 538, 378–382. [Google Scholar] [CrossRef]
- Australian Pancreatic Cancer Genome Initiative; Waddell, N.; Pajic, M.; Patch, A.-M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connor, A.A.; Denroche, R.E.; Jang, G.H.; Lemire, M.; Zhang, A.; Chan-Seng-Yue, M.; Wilson, G.; Grant, R.C.; Merico, D.; Lungu, I.; et al. Integration of Genomic and Transcriptional Features in Pancreatic Cancer Reveals Increased Cell Cycle Progression in Metastases. Cancer Cell 2019, 35, 267.e7–282.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Getz, G. Cancer Genome Atlas Research Network. Electronic address: [email protected], Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185.e13–203.e13. [Google Scholar] [CrossRef] [Green Version]
- Dreyer, S.; Chang, D.K.; Bailey, P.; Biankin, A.V. Pancreatic Cancer Genomes: Implications for Clinical Management and Therapeutic Development. Clin. Cancer Res. 2017, 23, 1638–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, A.; Hong, J.; Iacobuzio-Donahue, C.A. The pancreatic cancer genome revisited. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Hosoda, W.; Chianchiano, P.; Griffin, J.F.; Pittman, M.E.; Brosens, L.A.; Noë, M.; Yu, J.; Shindo, K.; Suenaga, M.; Rezaee, N.; et al. Genetic analyses of isolated high-grade pancreatic intraepithelial neoplasia (HG-PanIN) reveal paucity of alterations in TP53 and SMAD4. J. Pathol. 2017, 242, 16–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilentz, R.E.; Iacobuzio-Donahue, C.A.; Argani, P.; McCarthy, D.M.; Parsons, J.L.; Yeo, C.J.; Kern, S.E.; Hruban, R.H. Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: Evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res. 2000, 60, 2002–2006. [Google Scholar] [PubMed]
- Ahmed, S.; Bradshaw, A.-D.; Gera, S.; Dewan, M.Z.; Xu, R. The TGF-β/Smad4 Signaling Pathway in Pancreatic Carcinogenesis and Its Clinical Significance. J. Clin. Med. 2017, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S.A.; Schutte, M.; Hoque, A.T.M.S.; Moskaluk, C.A.; da Costa, L.T.; Rozenblum, E.; Weinstein, C.L.; Fischer, A.; Yeo, C.J.; Hruban, R.H.; et al. DPC4, A Candidate Tumor Suppressor Gene at Human Chromosome 18q21.1. Science 1996, 271, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Cohen, C.; Little, J.V.; Sequeira, J.H.; Mosunjac, M.B.; Siddiqui, M.T. The utility of SMAD4 as a diagnostic immunohistochemical marker for pancreatic adenocarcinoma, and its expression in other solid tumors. Diagn. Cytopathol. 2007, 35, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Mishra, L.; Deng, C.X. The role of TGF-β/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef] [Green Version]
- Miyaki, M.; Kuroki, T. Role of Smad4 (DPC4) inactivation in human cancer. Biochem. Biophys. Res. Commun. 2003, 306, 799–804. [Google Scholar] [CrossRef]
- Wendt, M.K.; Tian, M.; Schiemann, W.P. Deconstructing the mechanisms and consequences of TGF-β-induced EMT during cancer progression. Cell Tissue Res. 2011, 347, 85–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352, Correction in Nat. Rev. Mol. Cell Biol. 2021, 22, 834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilentz, R.E.; Su, G.; Le Dai, J.; Sparks, A.B.; Argani, P.; Sohn, T.A.; Yeo, C.J.; Kern, S.E.; Hruban, R.H. Immunohistochemical Labeling for Dpc4 Mirrors Genetic Status in Pancreatic Adenocarcinomas: A New Marker of DPC4 Inactivation. Am. J. Pathol. 2000, 156, 37–43. [Google Scholar] [CrossRef]
- Jj, S.; Sj, N.; La, R.; Eh, C.; Wm, G. Genetic Characterization and Cloning of Mothers against Dpp, a Gene Required for Decapentaplegic Function in Drosophila melanogaster. Genetics 1995, 139, 1347–1358. Available online: https://pubmed.ncbi.nlm.nih.gov/7768443/ (accessed on 4 January 2022).
- Xia, X.; Wu, W.; Huang, C.; Cen, G.; Jiang, T.; Cao, J.; Huang, K.; Qiu, Z. SMAD4 and its role in pancreatic cancer. Tumor Biol. 2014, 36, 111–119. [Google Scholar] [CrossRef]
- Schmierer, B.; Hill, C.S. TGFbeta-SMAD signal transduction: Molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 2007, 8, 970–982. [Google Scholar] [CrossRef] [PubMed]
- Gough, N.R.; Xiang, X.; Mishra, L. TGF-β Signaling in Liver, Pancreas, and Gastrointestinal Diseases and Cancer. Gastroenterology 2021, 161, 434.e15–452.e15. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Heldin, C.-H. The regulation of TGFβ signal transduction. Development 2009, 136, 3699–3714. [Google Scholar] [CrossRef] [Green Version]
- Massagué, J. TGFβ in Cancer. Cell 2008, 134, 215–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Glazer, E.; Welsh, E.; Pimiento, J.M.; Teer, J.K.; Malafa, M.P. TGFβ1 overexpression is associated with improved survival and low tumor cell proliferation in patients with early-stage pancreatic ductal adenocarcinoma. Oncotarget 2016, 8, 999–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Chen, X.-Q.; Li, P. The Role of TGF-β and Its Receptors in Gastrointestinal Cancers. Transl. Oncol. 2019, 12, 475–484. [Google Scholar] [CrossRef]
- Javle, M.; Li, Y.; Tan, D.; Dong, X.; Chang, P.; Kar, S.; Li, D. Biomarkers of TGF-β Signaling Pathway and Prognosis of Pancreatic Cancer. PLoS ONE 2014, 9, e85942. [Google Scholar] [CrossRef]
- Park, H.; Bang, J.; Nam, A.; Park, J.E.; Jin, M.H.; Bang, Y.; Oh, D. The prognostic role of soluble TGF-beta and its dynamics in unresectable pancreatic cancer treated with chemotherapy. Cancer Med. 2019, 9, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-beta signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.-W.; Chen, C.-H.; Chen, C.-C.; Chen, J.; Su, Y.-H.; Chen, R.-H. TGF-beta induces apoptosis through Smad-mediated expression of DAP-kinase. Nat. Cell Biol. 2002, 4, 51–58. [Google Scholar] [CrossRef]
- Dai, J.L.; Bansal, R.K.; Kern, S.E. G1 cell cycle arrest and apoptosis induction by nuclear Smad4/Dpc4: Phenotypes reversed by a tumorigenic mutation. Proc. Natl. Acad. Sci. USA 1999, 96, 1427–1432. [Google Scholar] [CrossRef] [Green Version]
- Atfi, A.; Buisine, M.; Mazars, A.; Gespach, C. Induction of apoptosis by DPC4, a transcriptional factor regulated by transforming growth factor-beta through stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK) signaling pathway. J. Biol. Chem. 1997, 272, 24731–24734. [Google Scholar] [CrossRef] [Green Version]
- Bulle, A.; Lim, K.-H. Beyond just a tight fortress: Contribution of stroma to epithelial-mesenchymal transition in pancreatic cancer. Signal Transduct. Target. Ther. 2020, 5, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Schutte, M.; Hruban, R.H.; Hedrick, L.; Cho, K.; Nadasdy, G.M.; Weinstein, C.L.; Bova, G.S.; Isaacs, W.B.; Cairns, P.; Nawroz, H.; et al. DPC4 gene in various tumor types. Cancer Res. 1996, 56, 2527–2530. [Google Scholar] [PubMed]
- Shi, Y.; Hata, A.; Lo, R.S.; Massagué, J.; Pavletich, N.P. A structural basis for mutational inactivation of the tumour suppressor Smad4. Nature 1997, 388, 87–93. [Google Scholar] [CrossRef]
- Xu, J.; Attisano, L. Mutations in the tumor suppressors Smad2 and Smad4 inactivate transforming growth factor beta signaling by targeting Smads to the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. USA 2000, 97, 4820–4825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demagny, H.; De Robertis, E.M. Point mutations in the tumor suppressor Smad4/DPC4 enhance its phosphorylation by GSK3 and reversibly inactivate TGF-β signaling. Mol. Cell Oncol. 2016, 3, e1025181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, M.; Huang, J.; Jhala, N.C.; Tytler, E.M.; Yang, L.; Vickers, S.M.; Tang, Y.; Lu, C.; Wang, N.; Cao, X. SCF(beta-TrCP1) controls Smad4 protein stability in pancreatic cancer cells. Am. J. Pathol. 2005, 166, 1379–1392. [Google Scholar] [CrossRef]
- Wang, J.-D.; Jin, K.; Chen, X.-Y.; Lv, J.-Q.; Ji, K.-W. Clinicopathological significance of SMAD4 loss in pancreatic ductal adenocarcinomas: A systematic review and meta-analysis. Oncotarget 2016, 8, 16704–16711. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, Y.; Esaka, S.; Suzuki, A.; Hamashima, Y.; Imaizumi, M.; Matsukawa, M.; Fujii, Y.; Aida, J.; Takubo, K.; Ishiwata, T.; et al. Abnormal immunolabelling of SMAD 4 in cell block specimens to distinguish malignant and benign pancreatic cells. Cytopathology 2018, 30, 201–208. [Google Scholar] [CrossRef]
- Park, J.Y.; King, J.; Reber, H.; Hines, O.J.; Mederos, M.A.; Wang, H.L.; Dawson, D.; Wainberg, Z.; Donahue, T.; Girgis, M. Poorly differentiated histologic grade correlates with worse survival in SMAD4 negative pancreatic adenocarcinoma patients. J. Surg. Oncol. 2020, 123, 389–398. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Y.; Zhan, S.; Zhang, L.; Zhang, S.; Tang, Q.; Li, M.; Tan, Z.; Liu, S.; Xing, X. SMAD4 Y353C promotes the progression of PDAC. BMC Cancer 2019, 19, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, Z. Loss of DPC4 expression and its correlation with clinicopathological parameters in pancreatic carcinoma. World J. Gastroenterol. 2003, 9, 2764–2767. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.; Serrano, O.K.; Wolfgang, C.L.; Parmigiani, G.; Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Eshleman, J.R.; et al. SMAD4 Gene Mutations Are Associated with Poor Prognosis in Pancreatic Cancer. Clin. Cancer Res. 2009, 15, 4674–4679. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; He, C.; Geng, S.; Sheng, H.; Shen, X.; Zhang, X.; Li, H.; Zhu, S.; Chen, X.; Yang, C.; et al. RhoT1 and Smad4 Are Correlated with Lymph Node Metastasis and Overall Survival in Pancreatic Cancer. PLoS ONE 2012, 7, e42234. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Fujii, T.; Shimoyama, Y.; Kanda, M.; Nakayama, G.; Sugimoto, H.; Koike, M.; Nomoto, S.; Fujiwara, M.; Nakao, A.; et al. SMAD4 Expression Predicts Local Spread and Treatment Failure in Resected Pancreatic Cancer. Pancreas 2015, 44, 660–664. [Google Scholar] [CrossRef]
- Held, T.; Verbeke, C.S.; Strobel, O.; Rutkowski, W.; Villard, C.; Moro, C.F.; Del Chiaro, M.; Büchler, M.; Heuchel, R.; Löhr, M. Immunohistochemical profiling of liver metastases and matched-pair analysis in patients with metastatic pancreatic ductal adenocarcinoma. Pancreatology 2019, 19, 963–970. [Google Scholar] [CrossRef]
- Tascilar, M.; Skinner, H.G.; Rosty, C.; Sohn, T.; Wilentz, R.E.; Offerhaus, G.J.; Adsay, V.; Abrams, R.A.; Cameron, J.L.; Kern, S.E.; et al. The SMAD4 protein and prognosis of pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2001, 7, 4115–4121. [Google Scholar] [PubMed]
- Shin, S.H.; Kim, H.J.; Hwang, D.W.; Lee, J.H.; Song, K.B.; Jun, E.; Shim, I.K.; Hong, S.; Kim, H.J.; Park, K.W.; et al. The DPC4/SMAD4 genetic status determines recurrence patterns and treatment outcomes in resected pancreatic ductal adenocarcinoma: A prospective cohort study. Oncotarget 2017, 8, 17945–17959. [Google Scholar] [CrossRef] [Green Version]
- Kadera, B.E.; Sunjaya, D.B.; Isacoff, W.H.; Li, L.; Hines, O.J.; Tomlinson, J.S.; Dawson, D.W.; Rochefort, M.M.; Donald, G.W.; Clerkin, B.M.; et al. Locally Advanced Pancreatic Cancer: Association Between Prolonged Preoperative Treatment and Lymph-Node Negativity and Overall Survival. JAMA Surg. 2014, 149, 145. [Google Scholar] [CrossRef] [Green Version]
- Oshima, M.; Okano, K.; Muraki, S.; Haba, R.; Maeba, T.; Suzuki, Y.; Yachida, S. Immunohistochemically Detected Expression of 3 Major Genes (CDKN2A/p16, TP53, and SMAD4/DPC4) Strongly Predicts Survival in Patients with Resectable Pancreatic Cancer. Ann. Surg. 2013, 258, 336–346. [Google Scholar] [CrossRef]
- Toga, T.; Nio, Y.; Hashimoto, K.; Higami, T.; Maruyama, R. The dissociated expression of protein and messenger RNA of DPC4 in human invasive ductal carcinoma of the pancreas and their implication for patient outcome. Anticancer Res. 2004, 24, 1173–1178. [Google Scholar] [PubMed]
- Ottenhof, N.A.; Morsink, F.H.M.; Kate, F.T.; Van Noorden, C.J.F.; Offerhaus, G.J.A. Multivariate analysis of immunohistochemical evaluation of protein expression in pancreatic ductal adenocarcinoma reveals prognostic significance for persistent Smad4 expression only. Cell. Oncol. 2012, 35, 119–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Liu, L.; Xu, H.; Wu, C.; Xiang, J.; Xu, J.; Liu, C.; Long, J.; Ni, Q.; Yu, X. Infiltrating immune cells and gene mutations in pancreatic ductal adenocarcinoma. Br. J. Surg. 2016, 103, 1189–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khorana, A.A.; Hu, Y.C.; Ryan, C.K.; Komorowski, R.A.; Hostetter, G.; Ahrendt, S.A. Vascular Endothelial Growth Factor and DPC4 Predict Adjuvant Therapy Outcomes in Resected Pancreatic Cancer. J. Gastrointest. Surg. 2005, 9, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Bachet, J.B.; Maréchal, R.; Demetter, P.; Bonnetain, F.; Couvelard, A.; Svrcek, M.; Bardier-Dupas, A.; Hammel, P.; Sauvanet, A.; Louvet, C.; et al. Contribution of CXCR4 and SMAD4 in predicting disease progression pattern and benefit from adjuvant chemotherapy in resected pancreatic adenocarcinoma. Ann. Oncol. 2012, 23, 2327–2335. [Google Scholar] [CrossRef] [PubMed]
- Ormanns, S.; Haas, M.; Remold, A.; Kruger, S.; Holdenrieder, S.; Kirchner, T.; Heinemann, V.; Boeck, S. The Impact of SMAD4 Loss on Outcome in Patients with Advanced Pancreatic Cancer Treated with Systemic Chemotherapy. Int. J. Mol. Sci. 2017, 18, 1094. [Google Scholar] [CrossRef]
- Winter, J.M.; Tang, L.H.; Klimstra, D.S.; Liu, W.; Linkov, I.; Brennan, M.F.; D’Angelica, M.I.; DeMatteo, R.P.; Fong, Y.; Jarnagin, W.R.; et al. Failure Patterns in Resected Pancreas Adenocarcinoma: Lack of Predicted Benefit to SMAD4 Expression. Ann. Surg. 2013, 258, 331–335. [Google Scholar] [CrossRef]
- Qian, Z.R.; Rubinson, D.A.; Nowak, J.A.; Morales-Oyarvide, V.; Dunne, R.F.; Kozak, M.M.; Welch, M.W.; Brais, L.K.; Da Silva, A.; Li, T.; et al. Association of Alterations in Main Driver Genes With Outcomes of Patients With Resected Pancreatic Ductal Adenocarcinoma. JAMA Oncol. 2018, 4, e173420. [Google Scholar] [CrossRef]
- Pokataev, I.; Kudaibergenova, A.; Artemyeva, A.; Popova, A.; Rumyantsev, A.; Podluzhny, D.; Kudashkin, N.; Fedyanin, M.; Tryakin, A.; Tjulandin, S. Intratumoral Heterogeneity of SMAD4 Immunohistochemical Expression and Its Role in Prediction of Recurrence Pattern in Patients with Resectable Pancreatic Cancer. J. Gastrointest. Cancer 2019, 50, 478–484. [Google Scholar] [CrossRef]
- Lu, J.; Yu, R.; Liu, R.; Liang, X.; Sun, J.; Zhang, H.; Wu, H.; Zhang, Z.; Shao, Y.W.; Guo, J.; et al. Genetic aberrations in Chinese pancreatic cancer patients and their association with anatomic location and disease outcomes. Cancer Med. 2020, 10, 933–943. [Google Scholar] [CrossRef]
- Hsieh, Y.-Y.; Liu, T.-P.; Chou, C.-J.; Chen, H.-Y.; Lee, K.-H.; Yang, P.-M. Integration of Bioinformatics Resources Reveals the Therapeutic Benefits of Gemcitabine and Cell Cycle Intervention in SMAD4-Deleted Pancreatic Ductal Adenocarcinoma. Genes 2019, 10, 766. [Google Scholar] [CrossRef] [Green Version]
- Pen, S.-L.; Shan, Y.-S.; Hsiao, C.-F.; Liu, T.-W.; Chen, J.-S.; Ho, C.-L.; Chou, W.-C.; Hsieh, R.-K.; Chen, L.-T.; Ch’Ang, H.-J. High expression of krüppel-like factor 10 or Smad4 predicts clinical benefit of adjuvant chemoradiotherapy in curatively resected pancreatic adenocarcinoma: From a randomized phase III trial. Radiother. Oncol. 2021, 158, 146–154. [Google Scholar] [CrossRef]
- Cuneo, K.C.; Morgan, M.A.; Griffith, K.A.; Hawkins, P.G.; Greenson, J.K.; Ben-Josef, E.; Lawrence, T.S.; Zalupski, M.M. Prognostic Value of c-MET Expression in Patients With Pancreatic Cancer Receiving Adjuvant and Neoadjuvant Chemoradiation Therapy. Int. J. Radiat. Oncol. 2017, 100, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Yokose, T.; Kitago, M.; Matsuda, S.; Sasaki, Y.; Masugi, Y.; Nakamura, Y.; Shinoda, M.; Yagi, H.; Abe, Y.; Oshima, G.; et al. Combination of KRAS and SMAD4 mutations in formalin-fixed paraffin-embedded tissues as a biomarker for pancreatic cancer. Cancer Sci. 2020, 111, 2174–2182. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.M.; Jabbour, S.K.; Lin, S.H.; Deek, M.P.; Hsu, C.C.; Fishman, E.K.; Kim, S.; Cameron, J.L.; Chekmareva, M.; Laheru, D.A.; et al. Smad4 Loss Correlates With Higher Rates of Local and Distant Failure in Pancreatic Adenocarcinoma Patients Receiving Adjuvant Chemoradiation. Pancreas 2018, 47, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Lee, S.H.; Qiu, F.; Zhou, L.; Wang, X.; Ye, T.; Hu, X. Association of SMAD4 loss with drug resistance in clinical cancer patients: A systematic meta-analysis. PLoS ONE 2021, 16, e0250634. [Google Scholar] [CrossRef]
- Kassardjian, A.; Wang, H.L. SMAD4-Expressing Pancreatic Ductal Adenocarcinomas Have Better Response to Neoadjuvant Therapy and Significantly Lower Lymph Node Metastasis Rates. Pancreas 2020, 49, 1153–1160. [Google Scholar] [CrossRef]
- Paiella, S.; Malleo, G.; Cataldo, I.; Gasparini, C.; De Pastena, M.; De Marchi, G.; Marchegiani, G.; Rusev, B.; Scarpa, A.; Girelli, R.; et al. Radiofrequency ablation for locally advanced pancreatic cancer: SMAD4 analysis segregates a responsive subgroup of patients. Langenbeck’s Arch. Surg. 2017, 403, 213–220. [Google Scholar] [CrossRef]
- Wang, F.; Xia, X.; Yang, C.; Shen, J.; Mai, J.; Kim, H.-C.; Kirui, D.; Kang, Y.; Fleming, J.B.; Koay, E.J.; et al. SMAD4 Gene Mutation Renders Pancreatic Cancer Resistance to Radiotherapy through Promotion of Autophagy. Clin. Cancer Res. 2018, 24, 3176–3185. [Google Scholar] [CrossRef] [Green Version]
- Iacobuzio-Donahue, C.A.; Fu, B.; Yachida, S.; Luo, M.; Abe, H.; Henderson, C.M.; Vilardell, F.; Wang, Z.; Keller, J.W.; Banerjee, P.; et al. DPC4 Gene Status of the Primary Carcinoma Correlates With Patterns of Failure in Patients With Pancreatic Cancer. J. Clin. Oncol. 2009, 27, 1806–1813. [Google Scholar] [CrossRef] [Green Version]
- Boone, B.A.; Sabbaghian, S.; Zenati, M.; Marsh, J.W.; Moser, A.J.; Zureikat, A.H.; Singhi, A.D.; Zeh, H.J., III; Krasinskas, A.M. Loss of SMAD4 staining in pre-operative cell blocks is associated with distant metastases following pancreaticoduodenectomy with venous resection for pancreatic cancer: Pre-Op SMAD4 in PDA With Venous Invasion. J. Surg. Oncol. 2014, 110, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Crane, C.H.; Varadhachary, G.R.; Yordy, J.S.; Staerkel, G.A.; Javle, M.M.; Safran, H.; Haque, W.; Hobbs, B.D.; Krishnan, S.; Fleming, J.B.; et al. Phase II Trial of Cetuximab, Gemcitabine, and Oxaliplatin Followed by Chemoradiation With Cetuximab for Locally Advanced (T4) Pancreatic Adenocarcinoma: Correlation of Smad4(Dpc4) Immunostaining With Pattern of Disease Progression. J. Clin. Oncol. 2011, 29, 3037–3043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gits, H.C.; Tang, A.H.; Harmsen, W.S.; Bamlet, W.R.; Graham, R.P.; Petersen, G.M.; Smyrk, T.C.; Mahipal, A.; Kowalchuk, R.O.; Ashman, J.B.; et al. Intact SMAD-4 is a predictor of increased locoregional recurrence in upfront resected pancreas cancer receiving adjuvant therapy. J. Gastrointest. Oncol. 2021, 12, 2275–2286. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; Iacobuzio-Donahue, C. Evolution and dynamics of pancreatic cancer progression. Oncogene 2013, 32, 5253–5260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardeesy, N.; Cheng, K.-h.; Berger, J.H.; Chu, G.C.; Pahler, J.; Olson, P.; Hezel, A.F.; Horner, J.; Lauwers, G.Y.; Hanahan, D.; et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 2006, 20, 3130–3146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, C.; Rennhack, J.P.; Arnoff, T.E.; Thaker, M.; Younger, S.T.; Doench, J.G.; Huang, A.Y.; Yang, A.; Aguirre, A.J.; Wang, B.; et al. SMAD4 represses FOSL1 expression and pancreatic cancer metastatic colonization. Cell Rep. 2021, 36, 109443. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Potts, J.D.; Runyan, R. Epithelial-mesenchymal cell transformation in the embryonic heart can be mediated, in part, by transforming growth factor β. Dev. Biol. 1989, 134, 392–401. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Somarelli, J.A.; Sheth, M.; Biddle, A.; Tripathi, S.C.; Armstrong, A.J.; Hanash, S.M.; Bapat, S.A.; Rangarajan, A.; Levine, H. Hybrid epithelial/mesenchymal phenotypes promote metastasis and therapy resistance across carcinomas. Pharmacol. Ther. 2018, 194, 161–184. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Pastushenko, I.; Skibinski, A.; Blanpain, C.; Kuperwasser, C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 2018, 24, 65–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bronsert, P.; Enderle-Ammour, K.; Bader, M.; Timme, S.; Kuehs, M.; Csanadi, A.; Kayser, G.; Kohler, I.; Bausch, D.; Hoeppner, J.; et al. Cancer cell invasion and EMT marker expression: A three-dimensional study of the human cancer-host interface: 3D cancer-host interface. J. Pathol. 2014, 234, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Kurahara, H.; Takao, S.; Maemura, K.; Mataki, Y.; Kuwahata, T.; Maeda, K.; Ding, Q.; Sakoda, M.; Iino, S.; Ishigami, S.; et al. Epithelial-mesenchymal transition and mesenchymal-epithelial transition via regulation of ZEB-1 and ZEB-2 expression in pancreatic cancer: EMT and MET in Pancreatic Cancer. J. Surg. Oncol. 2012, 105, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Galván, J.A.; Zlobec, I.; Wartenberg, M.; Lugli, A.; Gloor, B.; Perren, A.; Karamitopoulou, E. Expression of E-cadherin repressors SNAIL, ZEB1 and ZEB2 by tumour and stromal cells influences tumour-budding phenotype and suggests heterogeneity of stromal cells in pancreatic cancer. Br. J. Cancer 2015, 112, 1944–1950. [Google Scholar] [CrossRef] [Green Version]
- Bronsert, P.; Kohler, I.; Timme, S.; Kiefer, S.; Werner, M.; Schilling, O.; Vashist, Y.; Makowiec, F.; Brabletz, T.; Hopt, U.T.; et al. Prognostic significance of Zinc finger E-box binding homeobox 1 (ZEB1) expression in cancer cells and cancer-associated fibroblasts in pancreatic head cancer. Surgery 2014, 156, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Kohler, I.; Bronsert, P.; Timme, S.; Werner, M.; Brabletz, T.; Hopt, U.T.; Schilling, O.; Bausch, D.; Keck, T.; Wellner, U.F. Detailed analysis of epithelial-mesenchymal transition and tumor budding identifies predictors of long-term survival in pancreatic ductal adenocarcinoma. J. Gastroenterol. Hepatol. 2015, 30, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Masugi, Y.; Yamazaki, K.; Hibi, T.; Aiura, K.; Kitagawa, Y.; Sakamoto, M. Solitary cell infiltration is a novel indicator of poor prognosis and epithelial-mesenchymal transition in pancreatic cancer. Hum. Pathol. 2010, 41, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Chan-Seng-Yue, M.; Kim, J.C.; Wilson, G.W.; Ng, K.; Figueroa, E.F.; O’Kane, G.M.; Connor, A.A.; Denroche, R.E.; Grant, R.C.; McLeod, J.; et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat. Genet. 2020, 52, 231–240. [Google Scholar] [CrossRef]
- Lin, W.; Noel, P.; Borazanci, E.H.; Lee, J.; Amini, A.; Han, I.W.; Heo, J.S.; Jameson, G.S.; Fraser, C.; Steinbach, M.; et al. Single-cell transcriptome analysis of tumor and stromal compartments of pancreatic ductal adenocarcinoma primary tumors and metastatic lesions. Genome Med. 2020, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-M.; Li, A.; Olino, K.; Wolfgang, C.L.; Herman, J.M.; Schulick, R.D.; Iacobuzio-Donahue, C.; Hruban, R.H.; Goggins, M. Loss of E-cadherin expression and outcome among patients with resectable pancreatic adenocarcinomas. Mod. Pathol. 2011, 24, 1237–1247. [Google Scholar] [CrossRef] [Green Version]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and Dissemination Precede Pancreatic Tumor Formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Handler, J.; Cullis, J.; Avanzi, A.; Vucic, E.A.; Bar-Sagi, D. Pre-neoplastic pancreas cells enter a partially mesenchymal state following transient TGF-β exposure. Oncogene 2018, 37, 4334–4342. [Google Scholar] [CrossRef]
- Zheng, X.; Carstens, J.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittle, M.C.; Izeradjene, K.; Rani, P.G.; Feng, L.; Carlson, M.A.; DelGiorno, K.E.; Wood, L.D.; Goggins, M.; Hruban, R.H.; Chang, A.E.; et al. RUNX3 Controls a Metastatic Switch in Pancreatic Ductal Adenocarcinoma. Cell 2015, 161, 1345–1360. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Navarro-Serer, B.; Jeong, Y.J.; Chianchiano, P.; Xia, L.; Luchini, C.; Veronese, N.; Dowiak, C.; Ng, T.; Trujillo, M.A.; et al. Pattern of Invasion in Human Pancreatic Cancer Organoids Is Associated with Loss of SMAD4 and Clinical Outcome. Cancer Res. 2020, 80, 2804–2817. [Google Scholar] [CrossRef]
- Krebs, A.M.; Mitschke, J.; Lasierra Losada, M.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotz, B.; Arndt, M.; Dullat, S.; Bhargava, S.; Buhr, H.-J.; Hotz, H.G. Epithelial to Mesenchymal Transition: Expression of the Regulators Snail, Slug, and Twist in Pancreatic Cancer. Clin. Cancer Res. 2007, 13, 4769–4776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, A.N.; Summy, J.M.; Zhang, J.; Park, S.I.; Parikh, N.U.; Gallick, G.E. Development and Characterization of Gemcitabine-Resistant Pancreatic Tumor Cells. Ann. Surg. Oncol. 2007, 14, 3629–3637. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, T.; Ramachandran, V.; Fournier, K.F.; Wang, H.; Marquis, L.; Abbruzzese, J.L.; Gallick, G.E.; Logsdon, C.D.; McConkey, D.J.; Choi, W. Epithelial to Mesenchymal Transition Contributes to Drug Resistance in Pancreatic Cancer. Cancer Res. 2009, 69, 5820–5828. [Google Scholar] [CrossRef] [Green Version]
- David, C.J.; Huang, Y.-H.; Chen, M.; Su, J.; Zou, Y.; Bardeesy, N.; Iacobuzio-Donahue, C.A.; Massagué, J. TGF-β Tumor Suppression through a Lethal EMT. Cell 2016, 164, 1015–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Venkatasubbarao, K.; Lazor, J.W.; Sperry, J.; Jin, C.; Cao, L.; Freeman, J.W. Inhibition of STAT3 Tyr705 phosphorylation by Smad4 suppresses transforming growth factor beta-mediated invasion and metastasis in pancreatic cancer cells. Cancer Res. 2008, 68, 4221–4228. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-W.; Hsiao, P.-J.; Weng, C.-C.; Kuo, K.-K.; Kuo, T.-L.; Wu, D.-C.; Hung, W.-C.; Cheng, K.-H. SMAD4 Loss triggers the phenotypic changes of pancreatic ductal adenocarcinoma cells. BMC Cancer 2014, 14, 181. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.; Ling, J.; Suzuki, R.; Roife, D.; Chopin-Laly, X.; Truty, M.J.; Chatterjee, D.; Wang, H.; Thomas, R.M.; Katz, M.H.; et al. SMAD4 Regulates Cell Motility through Transcription of N-Cadherin in Human Pancreatic Ductal Epithelium. PLoS ONE 2014, 9, e107948. [Google Scholar] [CrossRef]
- Shichi, Y.; Sasaki, N.; Michishita, M.; Hasegawa, F.; Matsuda, Y.; Arai, T.; Gomi, F.; Aida, J.; Takubo, K.; Toyoda, M.; et al. Enhanced morphological and functional differences of pancreatic cancer with epithelial or mesenchymal characteristics in 3D culture. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faheem, M.M.; Rasool, R.; Ahmad, S.M.; Jamwal, V.L.; Chakraborty, S.; Katoch, A.; Gandhi, S.G.; Bhagat, M.; Goswami, A. Par-4 mediated Smad4 induction in PDAC cells restores canonical TGF-β/Smad4 axis driving the cells towards lethal EMT. Eur. J. Cell Biol. 2020, 99, 151076. [Google Scholar]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [Green Version]
- Aguirre, A.J.; Bardeesy, N.; Sinha, M.; Lopez, L.; Tuveson, D.A.; Horner, J.; Redston, M.S.; DePinho, R.A. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003, 17, 3112–3126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wartenberg, M.; Cibin, S.; Zlobec, I.; Vassella, E.; Eppenberger-Castori, S.; Terracciano, L.; Eichmann, M.; Worni, M.; Gloor, B.; Perren, A.; et al. Integrated Genomic and Immunophenotypic Classification of Pancreatic Cancer Reveals Three Distinct Subtypes with Prognostic/Predictive Significance. Clin. Cancer Res. 2018, 24, 4444–4454. [Google Scholar] [CrossRef] [Green Version]
- Levy, L.; Hill, C.S. Smad4 Dependency Defines Two Classes of Transforming Growth Factor β (TGF-β) Target Genes and Distinguishes TGF-β-Induced Epithelial-Mesenchymal Transition from Its Antiproliferative and Migratory Responses. Mol. Cell. Biol. 2005, 25, 8108–8125. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial–mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, J.J.; Rajasekaran, A.K. Reassessing Epithelial to Mesenchymal Transition as a Prerequisite for Carcinoma Invasion and Metastasis. Cancer Res. 2006, 66, 8319–8326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, P.; Srinivasan, R.; Wig, J.D. SMAD4 Genetic Alterations Predict a Worse Prognosis in Patients With Pancreatic Ductal Adenocarcinoma. Pancreas 2012, 41, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Masugi, Y.; Yamazaki, K.; Emoto, K.; Effendi, K.; Tsujikawa, H.; Kitago, M.; Itano, O.; Kitagawa, Y.; Sakamoto, M. Upregulation of integrin β4 promotes epithelial–mesenchymal transition and is a novel prognostic marker in pancreatic ductal adenocarcinoma. Lab. Investig. 2015, 95, 308–319. [Google Scholar] [CrossRef] [Green Version]
- White, B.D.; Chien, A.J.; Dawson, D.W. Dysregulation of Wnt/β-Catenin Signaling in Gastrointestinal Cancers. Gastroenterology 2012, 142, 219–232. [Google Scholar] [CrossRef] [Green Version]
- Kang, E.; Seo, J.; Yoon, H.; Cho, S. The Post-Translational Regulation of Epithelial–Mesenchymal Transition-Inducing Transcription Factors in Cancer Metastasis. Int. J. Mol. Sci. 2021, 22, 3591. [Google Scholar] [CrossRef]
- Moreno-Bueno, G.; Portillo, F.; Cano, A. Transcriptional regulation of cell polarity in EMT and cancer. Oncogene 2008, 27, 6958–6969. [Google Scholar] [CrossRef] [Green Version]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681.e4–695.e4. [Google Scholar] [CrossRef] [Green Version]
- Rice, A.J.; Cortes, E.; Lachowski, D.; Cheung, B.C.H.; Karim, S.A.; Morton, J.; Hernández, A.D.R. Matrix stiffness induces epithelial–mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis 2017, 6, e352. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Chen, X.; Li, W.; Shan, T.; Lin, W.R.; Ma, J.; Cui, X.; Yang, W.; Cao, G.; Li, Y.; et al. Conversion of epithelial-to-mesenchymal transition to mesenchymal-to-epithelial transition is mediated by oxygen concentration in pancreatic cancer cells. Oncol. Lett. 2018, 15, 7144–7152. [Google Scholar] [CrossRef] [PubMed]
- Shan, T.; Chen, S.; Chen, X.; Lin, W.R.; Li, W.; Ma, J.; Wu, T.; Ji, H.; Li, Y.; Cui, X.; et al. Prometastatic mechanisms of CAF-mediated EMT regulation in pancreatic cancer cells. Int. J. Oncol. 2016, 50, 121–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zadran, S.; Arumugam, R.; Herschman, H.; Phelps, M.E.; Levine, R.D. Surprisal analysis characterizes the free energy time course of cancer cells undergoing epithelial-to-mesenchymal transition. Proc. Natl. Acad. Sci. USA 2014, 111, 13235–13240. [Google Scholar] [CrossRef] [Green Version]
- Whittle, M.C.; Hingorani, S.R. Disconnect between EMT and metastasis in pancreas cancer. Oncotarget 2015, 6, 30445–30446. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [Green Version]
- Kleeff, J.; Maruyama, H.; Ishiwata, T.; Sawhney, H.; Friess, H.; Büchler, M.W.; Korc, M. Bone morphogenetic protein 2 exerts diverse effects on cell growth in vitro and is expressed in human pancreatic cancer in vivo. Gastroenterology 1999, 116, 1202–1216. [Google Scholar] [CrossRef]
- Gordon, K.J.; Kirkbride, K.C.; How, T.; Blobe, G.C. Bone morphogenetic proteins induce pancreatic cancer cell invasiveness through a Smad1-dependent mechanism that involves matrix metalloproteinase-2. Carcinogenesis 2008, 30, 238–248. [Google Scholar] [CrossRef] [Green Version]
- Hamada, S.; Satoh, K.; Hirota, M.; Kimura, K.; Kanno, A.; Masamune, A.; Shimosegawa, T. Bone morphogenetic protein 4 induces epithelial-mesenchymal transition through MSX2 induction on pancreatic cancer cell line. J. Cell. Physiol. 2007, 213, 768–774. [Google Scholar] [CrossRef]
- Voorneveld, P.W.; Stache, V.; Jacobs, R.J.; Smolders, E.; Sitters, A.I.; Liesker, A.; Korkmaz, K.S.; Lam, S.M.; De Miranda, N.F.C.C.; Morreau, H.; et al. Reduced expression of bone morphogenetic protein receptor IA in pancreatic cancer is associated with a poor prognosis. Br. J. Cancer 2013, 109, 1805–1812. [Google Scholar] [CrossRef] [Green Version]
- Dardare, J.; Witz, A.; Merlin, J.-L.; Bochnakian, A.; Toussaint, P.; Gilson, P.; Harlé, A. Epithelial to Mesenchymal Transition in Patients with Pancreatic Ductal Adenocarcinoma: State-of-the-Art and Therapeutic Opportunities. Pharmaceuticals 2021, 14, 740. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Authors | Year | Material | Methods | Results |
|---|---|---|---|---|
| EMT requires intact SMAD4 | ||||
| Bardeesy N et al. [86] | 2006 | PDAC GEMM | WB (E-CAD, SLUG and SMAD4) IHC (E-CAD, CK19 and SMAD4) IF (E-CAD, SLUG) Cell proliferation assay reagent, wound-healing assay | SMAD4-null tumors displayed significantly more prominent epithelial identity including higher E-CAD and lower SLUG expression, upholding a role of SMAD4 in promoting EMT. |
| Zhao S et al. [114] | 2008 | Isogenically matched PDAC cell lines (BxPC3, Capan-2, MIAPaCa-2, CFPAC-1, PANC-1 and UK PAN-1) Orthotopic mouse model | WB (Beta-CAT, E-CAD, ERK1/2, phosphorylated ERK1/2Thr202/Tyr204, SMAD2/3, SMAD4, phospho-Smad2, STAT3, STAT3Ser727, phospho-STAT3Tyr705 and VIM) IHC (phospho-STAT3Tyr705) shRNA (STAT3) Clonogenic assay, Matrigel invasion assay | Cells expressing SMAD4 showed an enhanced TGF-beta-mediated EMT as determined by the increased expression of VIM and decreased expression of Beta-CAT and E-CAD. |
| Chen YW et al. [115] | 2014 | PDAC cell lines (AsPC-1, CFPAC-1 and PANC-1) Mouse model | WB (Akt/p-AKT, CD133/44, CD133/1 c-Jun/p-c-Jun, Fast-1, Fos, E-CAD, EGFR/p-EGFR Hes1, nestin, NF-kB, p-p44/42, PTEN, SMAD2/3, SMAD4, Sp1, TGF-beta1, VIM) IHC (CD133, E-CAD, EGFR, nestin, SMAD4) IF (SMAD4) RT-qPCR (CD133, CD44, E-CAD, EGFR, N-CAD, SMAD4, VEGF and VIM) shRNA (SMAD4) Transient transfections, luciferase reporter assays, cell proliferation assay, wound-healing assay and Transwell migration assay | SMAD4 deficiency in vitro promotes an epithelial phenotype and induced chemoresistance. SMAD4 restoration in vivo increased migration and EMT markers (VIM and SMA). |
| Kang Y et al. [116] | 2014 | PDAC cell line (PANC-1) Human cell line (HPNE) | WB (phosphor-Akt, CK19, phosphor-MEK1/2, MEK1/2, N-CAD, p21, phospho-SMAD2, phosphor-SMAD3, SMAD2/3, SMAD4, Tak1, VIM and Wafl/Cip1) IHC (N-CAD, SMAD4) IF (CK19, SMAD4) RT-PCR, RT-qPCR (Beta-CAT, E-CAD, FN1, N-CAD, TWIST1, TWIST2, VIM and ZEB1) ChIP (N-CAD, SMAD4) shRNAi (SMAD4) Modified Boyden chamber invasion and migration assay, electrophoretic mobility shift assay and luciferase reporter assay | SMAD4 is necessary for the upregulation of N-CAD. Knocked down SMAD4 reduces N-CAD protein levels and inhibits invasion and migration. |
| Whittle MC et al. [107] | 2015 | PDAC cell lines (CFPAC-1, PANC-1 and MiaPaCa-2) PDAC GEMM NOD SCID/NCr mice Human TMA | WB (p16, p19, p21, Parp, RUNX3 and SMAD4) IHC (amylase, cleaved caspase 3, insulin, CK19, RUNX3 and SMAD4) IF (E-CAD) RT-qPCR (Col6a1, RUNX3 and Spp1) ELISA (Spp1) shRNAi (Col6a1, RUNX3) Cell proliferation assay, migration assay, Matrigel invasion assay, soft agar assay and luciferase reporter assay | Pancreas-specific homozygous deletion of SMAD4 in the mouse model of PDAC abrogates the TGF-beta-induced EMT of cancer epithelia but does not impair metastasis. |
| David JC et al. [113] | 2016 | Cell lines from GEMM PDAC GEMM | WB (Cdx2, cleaved caspase 3, E-CAD, Foxa2, KLF5, Pdx1, SMAD2/3, SMAD4, Snail, SOX4 and ZEB1) IHC (cleaved caspase 3, E-CAD, CK19, KLF5 and SOX4) IF (cleaved caspase 3, E-CAD, CK19, KLF5 and SOX4) ChIP (E-CAD, Klf5, Serpine, SMAD2/3 and SMAD7) shRNA screening (Foxa2, Klf5, Renill, Snail, SOX4 and ZEB1) Cleaved caspase activity measurements | The TGF-beta-/SMAD4-dependent pathway induces EMT and then apoptosis, implicating SOX4 and KLF5. |
| Shichi Y et al. [117] | 2019 | PDAC cell lines (PANC-1, MiaPaCa-2 and PK-1) | IHC (CA19.9, CEA, E-CAD, CKAE1AE3, CK7, Ki-67, phospho-SMAD2L/3L, SMAD4, TGF-beta receptor II, trypsin, and VIM) RT-qPCR (E-CAD, N-CAD, Snail and VIM) Sphere-forming assays, scanning electron microscopic analysis and transmission electron microscopic analysis | Cells with loss of SMAD4 maintain an epithelial phenotype. |
| Mohd Faheem M et al. [118] | 2020 | PDAC cell lines (PANC-1, MiaPaCa-2 and BxPC3) | WB (Akt/pAkt, Bax, Bcl2, E-CAD, CDK2, cleaved caspase 3, caspase 3, cyclin A, cyclin E, NM23H1, p21, p27, Par-4, SMAD4, Snail, STRAP and VIM) IF (E-CAD, NM23H1, Par-4, SMAD4 and Snail) RT-qPCR (Akt/pAkt, Bax, Bcl2, E-CAD, CDK2, cleaved caspase 3, caspase 3, cyclin A, cyclin E, NM23H1, p21, p27, Par-4, SMAD4, Snail, STRAP and VIM) siRNA (NM23H1, Par-4 and SMAD4) Co-immunoprecipitation assay (NM23H1, STRAP) Fluorescent gelatin degradation assay, transient transfections with N3-secAnnexinV-mVenus construct and D spheroid migration assay | Par-4 induces SMAD4 lethal EMT. |
| Chan-Seng-Yue M et al. [101] | 2020 | Tissue bulk analysis of human PDAC specimens | WGS, WTS, RNA-Seq and scRNA-Seq | Complete loss of SMAD4 is more frequent in “Classical A/B forms” and, therefore, is less associated with the upregulation of EMT markers, which are associated with “Basal A/B forms”. |
| EMT requires SMAD4 alteration | ||||
| Yamada S et al. [56] | 2015 | Retrospective clinical trial | IHC (E-CAD, SMAD4 and VIM) | SMAD4 inactivation was associated with tumor progression, pattern of failure and EMT. SMAD4-negative tumors correlated with a mesenchymal phenotype. |
| Wartenberg M et al. [121] | 2018 | Retrospective clinical trial | IHC (CD3, CD4, CD8, CD20, FOXP3, MLH1, MSH2, MSH6, p63, PD-L1 and PMS2) NGS (APC, ATM, CDKN2A, EGFR, FGFR3, GNAS, JAK3, KRAS, MET, PIK3CA, SMAD4, SMARCB1, STK11 and TP53) | EMT-like tumor budding and SMAD4 inactivation are associated with the “immune-escape” phenotype. |
| Wang Z et al. [52] | 2019 | Retrospective clinical trial PDAC cell lines (PANC-1, SW1990) | WB (E-CAD, SMAD4 and VIM) IHC (SMAD4) RT-qPCR (E-CAD, SMAD4 and VIM) Sanger sequencing (SMAD4) Cell proliferation assay, transwell migration assay and wound-healing assay | The SMAD4 Y353C mutation leads to increased cell migration as well as invasion and EMT promotion in vitro. |
| EMT is SMAD4 independent | ||||
| Levy L et al. [122] | 2005 | PDAC cell line (Colo-357) Human keratinocyte cell line (HaCaT) | WB (HA, PAI-1, p21, SMAD2/3, phosphor-SMAD2, phosphor-SMAD3, SMAD4 and Smurf1) IF (E-CAD, VIM) RT-PCR siRNA (SMAD4) Luciferase assay, cell cycle analysis and scratch assays | SMAD4 is necessary for TGF-beta-induced cell cycle arrest and migration but is not involved in the TGF-beta-induced EMT. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Racu, M.-L.; Lebrun, L.; Schiavo, A.A.; Van Campenhout, C.; De Clercq, S.; Absil, L.; Minguijon Perez, E.; Maris, C.; Decaestecker, C.; Salmon, I.; et al. The Role of SMAD4 Inactivation in Epithelial–Mesenchymal Plasticity of Pancreatic Ductal Adenocarcinoma: The Missing Link? Cancers 2022, 14, 973. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14040973
Racu M-L, Lebrun L, Schiavo AA, Van Campenhout C, De Clercq S, Absil L, Minguijon Perez E, Maris C, Decaestecker C, Salmon I, et al. The Role of SMAD4 Inactivation in Epithelial–Mesenchymal Plasticity of Pancreatic Ductal Adenocarcinoma: The Missing Link? Cancers. 2022; 14(4):973. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14040973
Chicago/Turabian StyleRacu, Marie-Lucie, Laetitia Lebrun, Andrea Alex Schiavo, Claude Van Campenhout, Sarah De Clercq, Lara Absil, Esmeralda Minguijon Perez, Calliope Maris, Christine Decaestecker, Isabelle Salmon, and et al. 2022. "The Role of SMAD4 Inactivation in Epithelial–Mesenchymal Plasticity of Pancreatic Ductal Adenocarcinoma: The Missing Link?" Cancers 14, no. 4: 973. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14040973