
Cell State and Cell Type: Deconvoluting Circulating Tumor Cell Populations in Liquid Biopsies by Multi-Omics
 , , , , , , , , and
, , , , , , , , and  add
Show full author list
add
Show full author list
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Samples
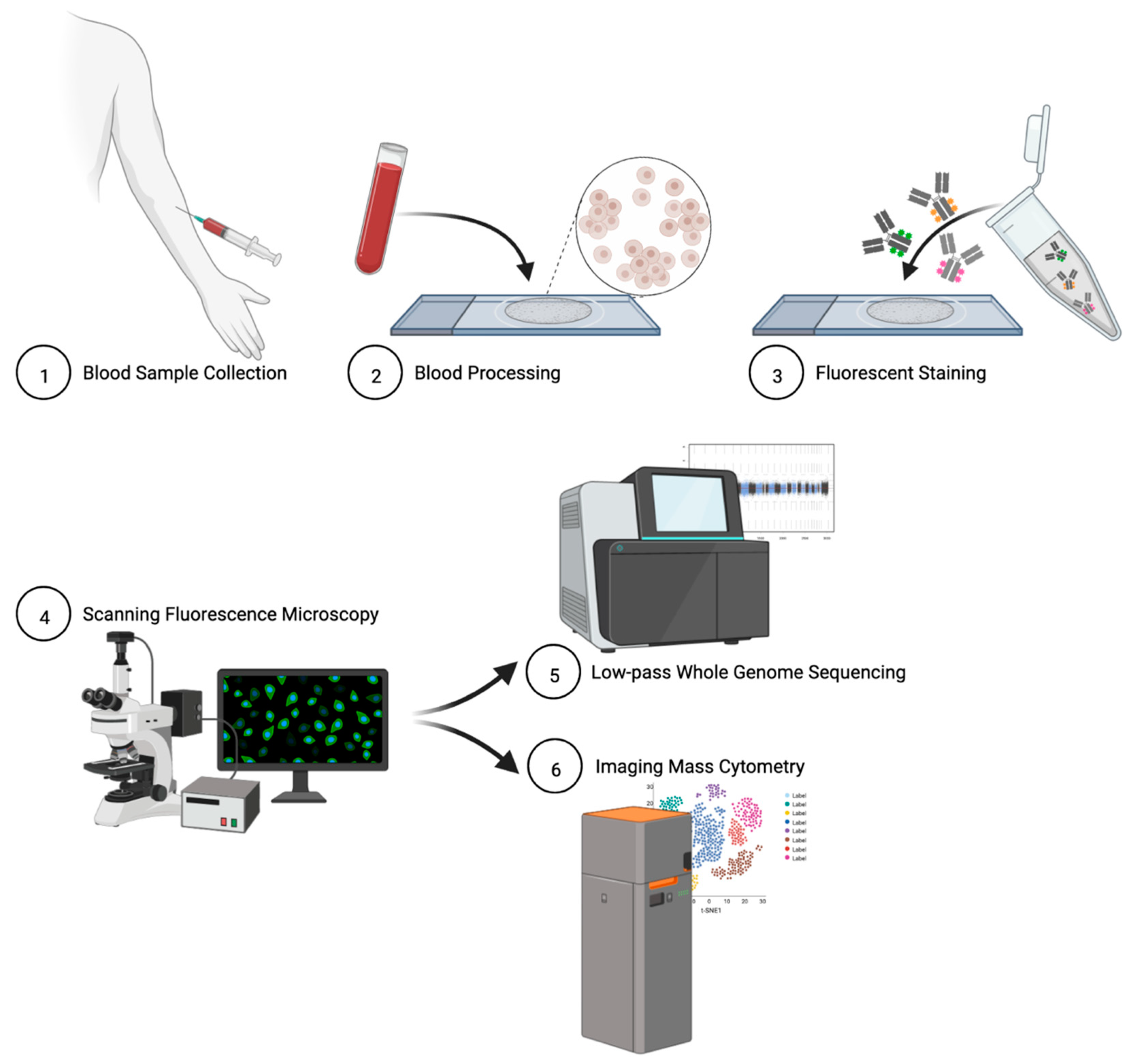
2.2. Blood Sample Collection and Processing
2.3. Cell Culture and Contrived Sample Generation
2.4. Immunofluorescent Staining of Patient Slides
2.5. Rare Cell Identification and Characterization
2.6. Single-Cell Next-Generation Sequencing and Bioinformatic Analysis
2.7. Single-Cell Targeted Proteomics and Data Analysis
2.8. Data Analysis and Visualization
3. Results
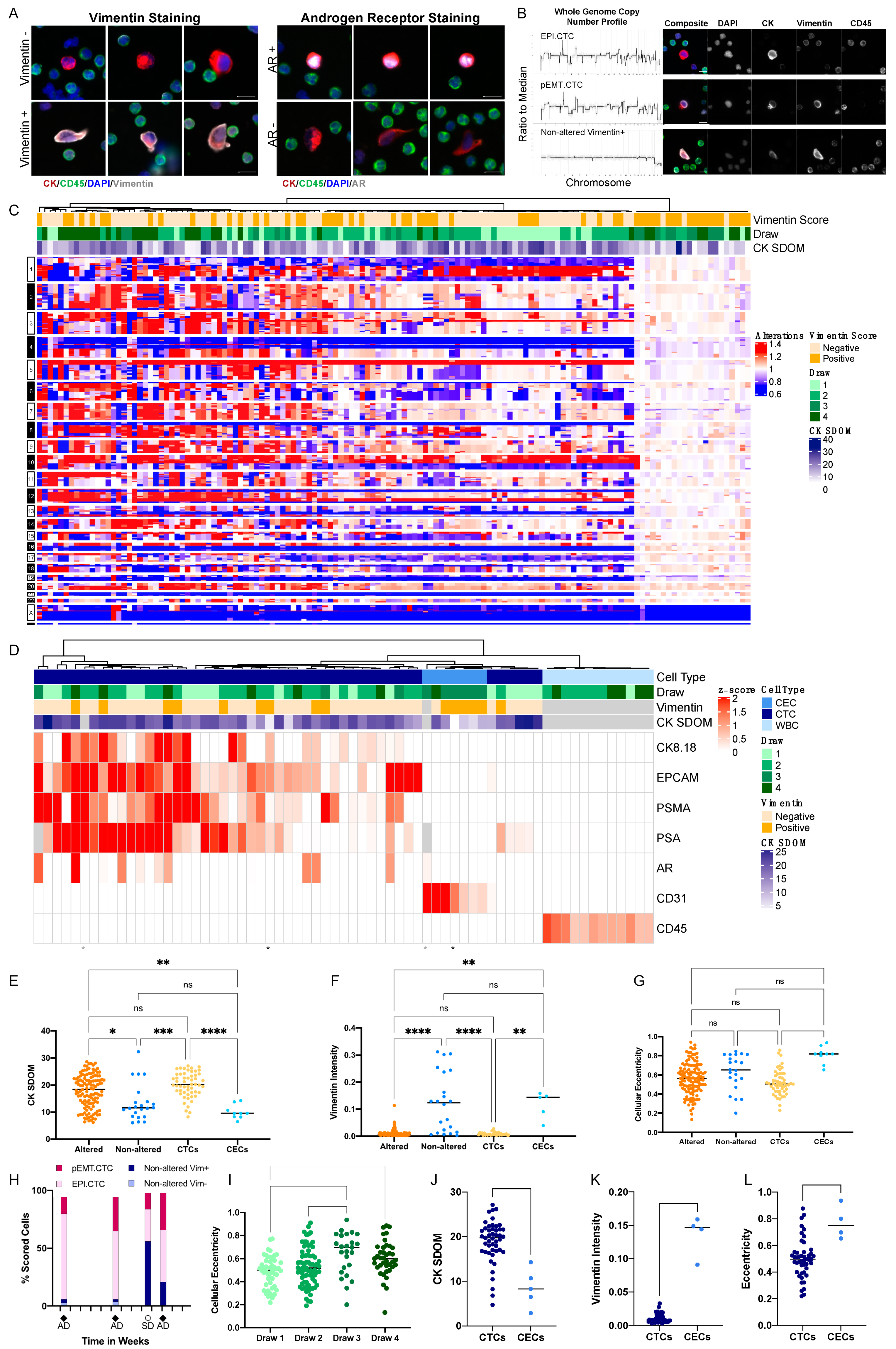
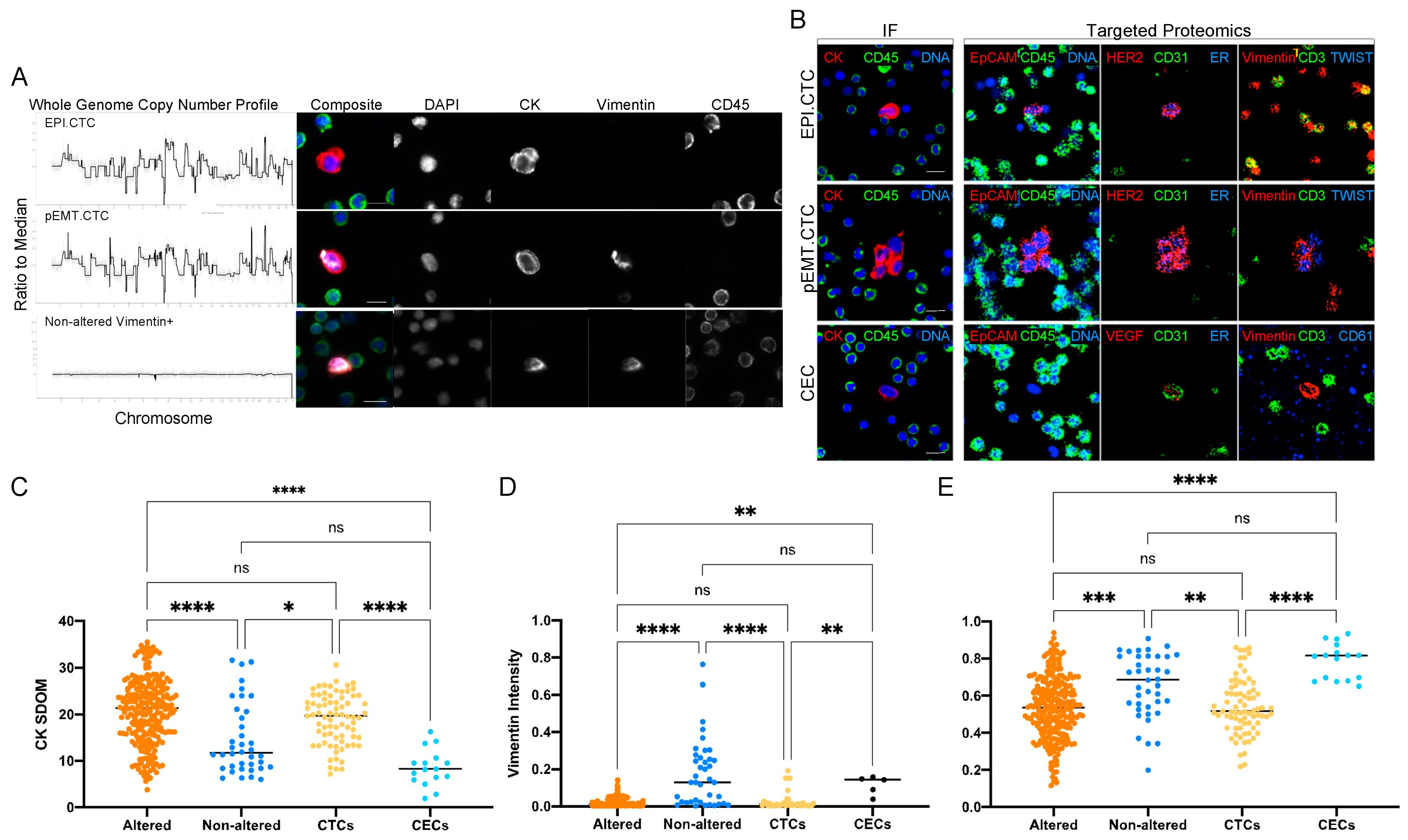
3.1. Characterization of Circulating Rare Cells Appearing at Time Points of Progression and Stable Disease of a Metastatic Prostate Cancer Patient
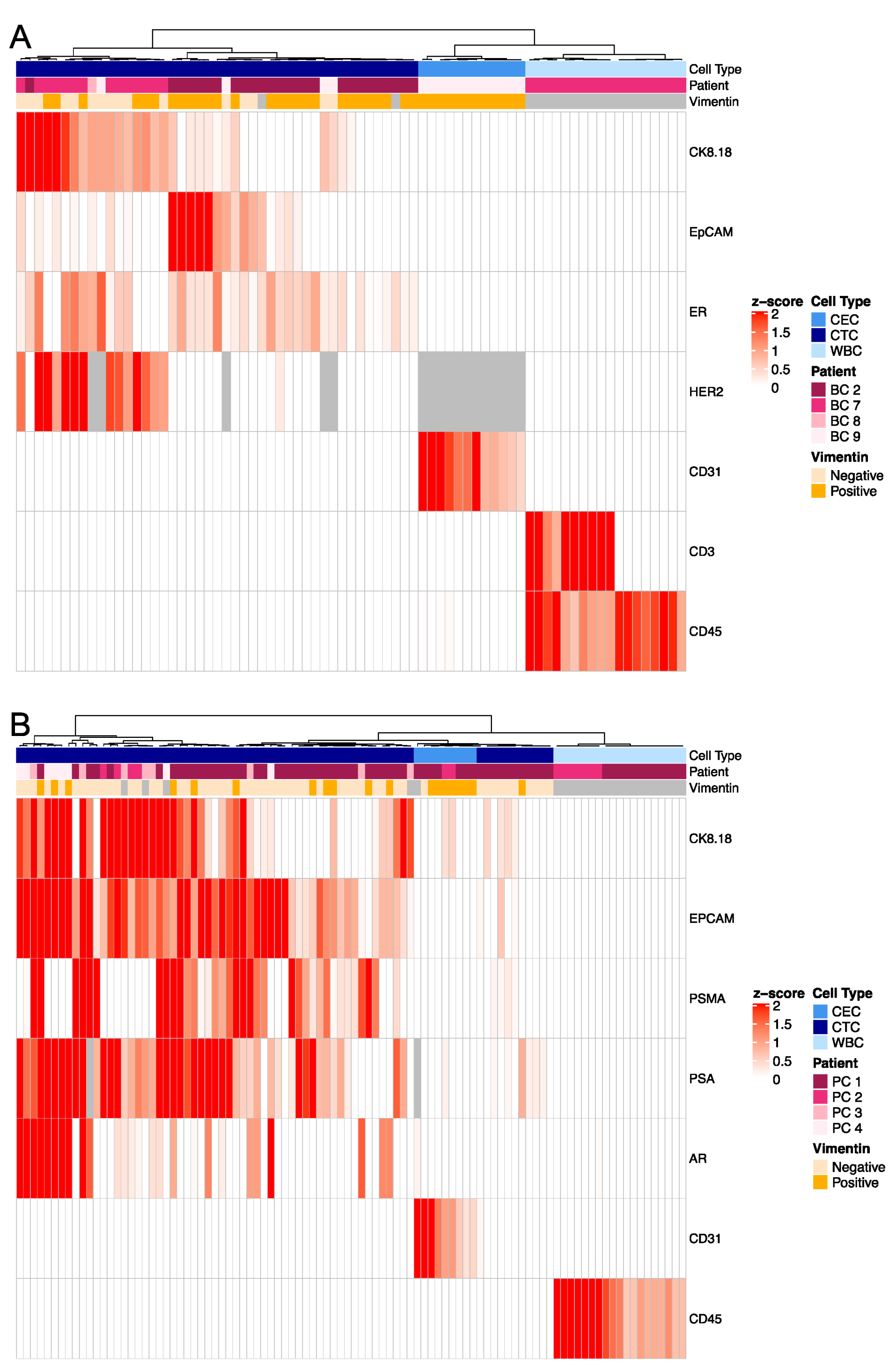
3.2. Inter-Patient Assessment of Circulating Cell Types and Cell States in the Liquid Biopsy
3.3. Targeted Proteomics Identifies Distinct Phenotypes
3.4. Morphometrics and Multi-Omics to Separate Cell Types
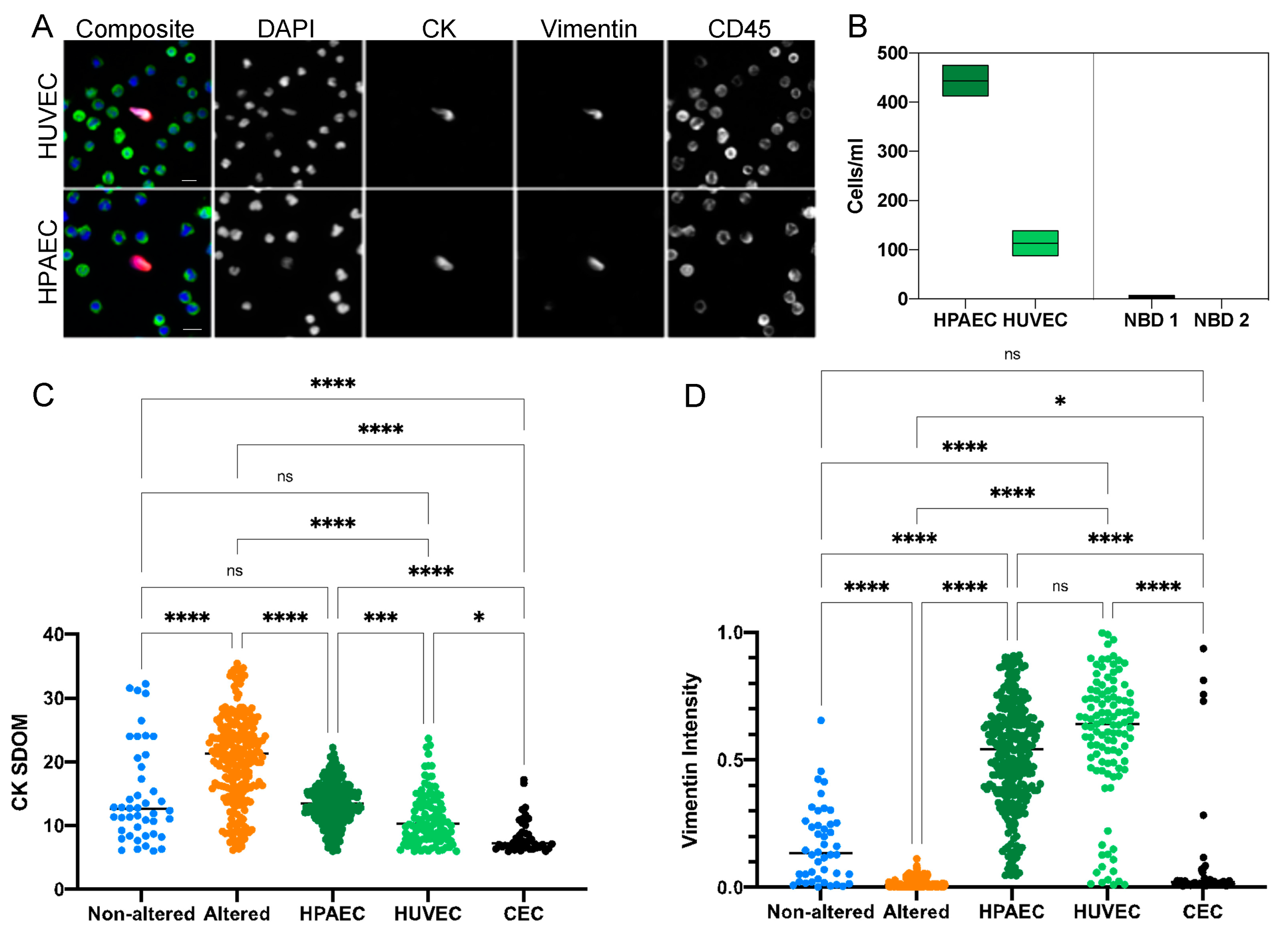
3.5. Characterization of CECs
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bremnes, R.M.; Dønnem, T.; Al-Saad, S.; Al-Shibli, K.; Andersen, S.; Sirera, R.; Camps, C.; Marinez, I.; Busund, L.T. The role of tumor stroma in cancer progression and prognosis: Emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 209–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, S.; Matsumoto, N.; Storgard, R.; Peng, C.C.; Aparicio, A.; Ormseth, B.; Rappard, K.; Cunningham, K.; Kolatkar, A.; Nevarez, R.; et al. Platelet-Coated Circulating Tumor Cells Are a Predictive Biomarker in Patients with Metastatic Castrate-Resistant Prostate Cancer. Mol. Cancer Res. 2021, 19, 2036–2045. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, B.; Wu, A.; Wetterskog, D.; Attard, G. Blood-based liquid biopsies for prostate cancer: Clinical opportunities and challenges. Br. J. Cancer 2022, 127, 1394–1402. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.; Mu, Z.; Rademaker, A.W.; Austin, L.K.; Strickland, K.S.; Costa, R.L.B.; Nagy, R.J.; Zagonel, V.; Taxter, T.J.; Behdad, A.; et al. Cell-Free DNA and Circulating Tumor Cells: Comprehensive Liquid Biopsy Analysis in Advanced Breast Cancer. Clin. Cancer Res. 2018, 24, 560–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alimirzaie, S.; Bagherzadeh, M.; Akbari, M.R. Liquid biopsy in breast cancer: A comprehensive review. Clin. Genet. 2019, 95, 643–660. [Google Scholar] [CrossRef]
- FDA. 1510(k) Substantial Equivalence Determination Decision Summary. Available online: https://www.accessdata.fda.gov/cdrh_docs/reviews/K050245.pdf (accessed on 15 May 2021).
- Sciences, E. Available online: https://www.epicsciences.com/ar-v7-test/ (accessed on 27 May 2021).
- Bhatia, S.; Wang, P.; Toh, A.; Thompson, E.W. New Insights Into the Role of Phenotypic Plasticity and EMT in Driving Cancer Progression. Front. Mol. Biosci. 2020, 7, 71. [Google Scholar] [CrossRef] [Green Version]
- Williamson, S.C.; Metcalf, R.L.; Trapani, F.; Mohan, S.; Antonello, J.; Abbott, B.; Leong, H.S.; Chester, C.P.; Simms, N.; Polanski, R.; et al. Vasculogenic mimicry in small cell lung cancer. Nat. Commun. 2016, 7, 13322. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Ginther, C.; Kim, J.; Mosher, N.; Chung, S.; Slamon, D.; Vadgama, J.V. Expression of Wnt3 activates Wnt/beta-catenin pathway and promotes EMT-like phenotype in trastuzumab-resistant HER2-overexpressing breast cancer cells. Mol. Cancer Res. 2012, 10, 1597–1606. [Google Scholar] [CrossRef] [Green Version]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Liu, S.; Liu, Z.; Huang, J.; Pu, X.; Li, J.; Yang, D.; Deng, H.; Yang, N.; Xu, J. Classification of circulating tumor cells by epithelial-mesenchymal transition markers. PLoS ONE 2015, 10, e0123976. [Google Scholar] [CrossRef]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681–695.e4. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Kim, Y.M. Tumor endothelial cells as a potential target of metronomic chemotherapy. Arch. Pharm. Res. 2019, 42, 1–13. [Google Scholar] [CrossRef]
- Bethel, K.; Luttgen, M.S.; Damani, S.; Kolatkar, A.; Lamy, R.; Sabouri-Ghomi, M.; Topol, S.; Topol, E.J.; Kuhn, P. Fluid phase biopsy for detection and characterization of circulating endothelial cells in myocardial infarction. Phys. Biol. 2014, 11, 016002. [Google Scholar] [CrossRef] [Green Version]
- Damani, S.; Bacconi, A.; Libiger, O.; Chourasia, A.H.; Serry, R.; Gollapudi, R.; Goldberg, R.; Rapeport, K.; Haaser, S.; Topol, S.; et al. Characterization of circulating endothelial cells in acute myocardial infarction. Sci. Transl. Med. 2012, 4, 126ra33. [Google Scholar] [CrossRef] [Green Version]
- Beerepoot, L.V.; Radema, S.A.; Witteveen, E.O.; Thomas, T.; Wheeler, C.; Kempin, S.; Voest, E.E. Phase I clinical evaluation of weekly administration of the novel vascular-targeting agent, ZD6126, in patients with solid tumors. J. Clin. Oncol. 2006, 24, 1491–1498. [Google Scholar] [CrossRef]
- Rahbari, N.N.; Scholch, S.; Bork, U.; Kahlert, C.; Schneider, M.; Rahbari, M.; Buchler, M.W.; Weitz, J.; Reissfelder, C. Prognostic value of circulating endothelial cells in metastatic colorectal cancer. Oncotarget 2017, 8, 37491–37501. [Google Scholar] [CrossRef] [Green Version]
- Rowand, J.L.; Martin, G.; Doyle, G.V.; Miller, M.C.; Pierce, M.S.; Connelly, M.C.; Rao, C.; Terstappen, L.W. Endothelial cells in peripheral blood of healthy subjects and patients with metastatic carcinomas. Cytometry A 2007, 71, 105–113. [Google Scholar] [CrossRef]
- Setayesh, S.M.; Hart, O.; Naghdloo, A.; Higa, N.; Nieva, J.; Lu, J.; Hwang, S.; Wilkinson, K.; Kidd, M.; Anderson, A.; et al. Multianalyte liquid biopsy to aid the diagnostic workup of breast cancer. NPJ Breast Cancer 2022, 8, 112. [Google Scholar] [CrossRef] [PubMed]
- Calleri, A.; Bono, A.; Bagnardi, V.; Quarna, J.; Mancuso, P.; Rabascio, C.; Dellapasqua, S.; Campagnoli, E.; Shaked, Y.; Goldhirsch, A.; et al. Predictive Potential of Angiogenic Growth Factors and Circulating Endothelial Cells in Breast Cancer Patients Receiving Metronomic Chemotherapy Plus Bevacizumab. Clin. Cancer Res. 2009, 15, 7652–7657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simkens, L.H.J.; Tol, J.; Terstappen, L.; Teerenstra, S.; Punt, C.J.A.; Nagtegaal, I.D. The predictive and prognostic value of circulating endothelial cells in advanced colorectal cancer patients receiving first-line chemotherapy and bevacizumab. Ann. Oncol. 2010, 21, 2447–2448. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.; Kato, T.; Ogura, T.; Sekine, A.; Oda, T.; Masuda, N.; Igawa, S.; Katono, K.; Otani, S.; Yamada, K.; et al. Phase II study of bevacizumab, cisplatin, and docetaxel plus maintenance bevacizumab as first-line treatment for patients with advanced non-squamous non-small-cell lung cancer combined with exploratory analysis of circulating endothelial cells: Thoracic Oncology Research Group (TORG)1016. BMC Cancer 2018, 18, 241. [Google Scholar]
- Bidard, F.C.; Mathiot, C.; Degeorges, A.; Etienne-Grimaldi, M.C.; Delva, R.; Pivot, X.; Veyret, C.; Bergougnoux, L.; de Cremoux, P.; Milano, G.; et al. Clinical value of circulating endothelial cells and circulating tumor cells in metastatic breast cancer patients treated first line with bevacizumab and chemotherapy. Ann. Oncol. 2010, 21, 1765–1771. [Google Scholar] [CrossRef]
- Marrinucci, D.; Bethel, K.; Kolatkar, A.; Luttgen, M.S.; Malchiodi, M.; Baehring, F.; Voigt, K.; Lazar, D.; Nieva, J.; Bazhenova, L.; et al. Fluid biopsy in patients with metastatic prostate, pancreatic and breast cancers. Phys. Biol. 2012, 9, 016003. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Lee, M.; Kolatkar, A.; McCormick, M.; Dago, A.D.; Kendall, J.; Carlsson, N.A.; Bethel, K.; Greenspan, E.J.; Hwang, S.E.; Waitman, K.R.; et al. Effect of Blood Collection Tube Type and Time to Processing on the Enumeration and High-Content Characterization of Circulating Tumor Cells Using the High-Definition Single-Cell Assay. Arch. Pathol. Lab. Med. 2018, 142, 198–207. [Google Scholar] [CrossRef] [Green Version]
- Dago, A.E.; Stepansky, A.; Carlsson, A.; Luttgen, M.; Kendall, J.; Baslan, T.; Kolatkar, A.; Wigler, M.; Bethel, K.; Gross, M.E.; et al. Rapid phenotypic and genomic change in response to therapeutic pressure in prostate cancer inferred by high content analysis of single circulating tumor cells. PLoS ONE 2014, 9, e101777. [Google Scholar] [CrossRef] [Green Version]
- Thiele, J.A.; Pitule, P.; Hicks, J.; Kuhn, P. Single-Cell Analysis of Circulating Tumor Cells. Methods Mol. Biol. 2019, 1908, 243–264. [Google Scholar]
- Baslan, T.; Kendall, J.; Ward, B.; Cox, H.; Leotta, A.; Rodgers, L.; Riggs, M.; D’Italia, S.; Sun, G.; Yong, M.; et al. Optimizing sparse sequencing of single cells for highly multiplex copy number profiling. Genome Res. 2015, 25, 714–724. [Google Scholar] [CrossRef] [Green Version]
- Baslan, T.; Kendall, J.; Rodgers, L.; Cox, H.; Riggs, M.; Stepansky, A.; Troge, J.; Ravi, K.; Esposito, D.; Lakshmi, B.; et al. Genome-wide copy number analysis of single cells. Nat. Protoc. 2012, 7, 1024–1041. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Kaur, P.; Porras, T.B.; Ring, A.; Carpten, J.D.; Lang, J.E. Comparison of TCGA and GENIE genomic datasets for the detection of clinically actionable alterations in breast cancer. Sci. Rep. 2019, 9, 1482. [Google Scholar] [CrossRef] [Green Version]
- Gerdtsson, E.; Pore, M.; Thiele, J.A.; Gerdtsson, A.S.; Malihi, P.D.; Nevarez, R.; Kolatkar, A.; Velasco, C.R.; Wix, S.; Singh, M.; et al. Multiplex protein detection on circulating tumor cells from liquid biopsies using imaging mass cytometry. Converg. Sci. Phys. Oncol. 2018, 4, 015002. [Google Scholar] [CrossRef]
- Berg, S.; Kutra, D.; Kroeger, T.; Straehle, C.N.; Kausler, B.X.; Haubold, C.; Schiegg, M.; Ales, J.; Beier, T.; Rudy, M.; et al. Ilastik: Interactive machine learning for (bio)image analysis. Nat. Methods 2019, 16, 1226–1232. [Google Scholar] [CrossRef]
- Carpenter, A.E.; Jones, T.R.; Lamprecht, M.R.; Clarke, C.; Kang, I.H.; Friman, O.; Guertin, D.A.; Chang, J.H.; Lindquist, R.A.; Moffat, J.; et al. CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006, 7, R100. [Google Scholar] [CrossRef] [Green Version]
- Catena, R.; Montuenga, L.M.; Bodenmiller, B. Ruthenium counterstaining for imaging mass cytometry. J. Pathol. 2018, 244, 479–484. [Google Scholar] [CrossRef]
- Chi, J.T.; Chang, H.Y.; Haraldsen, G.; Jahnsen, F.L.; Troyanskaya, O.G.; Chang, D.S.; Wang, Z.; Rockson, S.G.; van de Rijn, M.; Botstein, D.; et al. Endothelial cell diversity revealed by global expression profiling. Proc. Natl. Acad. Sci. USA 2003, 100, 10623–10628. [Google Scholar] [CrossRef]
- Remotti, F.; Fetsch, J.F.; Miettinen, M. Keratin 1 expression in endothelia and mesenchymal tumors: An immunohistochemical analysis of normal and neoplastic tissues. Hum. Pathol. 2001, 32, 873–879. [Google Scholar] [CrossRef]
- Xia, B.; Zhang, H.; Yang, M.; Du, S.; Wei, J.; Ding, L. Leukamenin E Induces K8/18 Phosphorylation and Blocks the Assembly of Keratin Filament Networks through ERK Activation. Int. J. Mol. Sci. 2020, 21, 3164. [Google Scholar] [CrossRef]
- Mattey, D.L.; Nixon, N.; Wynn-Jones, C.; Dawes, P.T. Demonstration of cytokeratin in endothelial cells of the synovial microvasculature in situ and in vitro. Br. J. Rheumatol. 1993, 32, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.; Fetsch, J.F. Distribution of keratins in normal endothelial cells and a spectrum of vascular tumors: Implications in tumor diagnosis. Hum. Pathol. 2000, 31, 1062–1067. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Jacot, W.; Yamashita, T.; Sohn, J.; Vidal, M.; Tokunaga, E.; Tsurutani, J.; Ueno, N.T.; Prat, A.; Chae, Y.S.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. N. Engl. J. Med. 2022, 387, 9–20. [Google Scholar] [CrossRef] [PubMed]
- de Wit, S.; van Dalum, G.; Lenferink, A.T.; Tibbe, A.G.; Hiltermann, T.J.; Groen, H.J.; van Rijn, C.J.; Terstappen, L.W. The detection of EpCAM(+) and EpCAM(−) circulating tumor cells. Sci. Rep. 2015, 5, 12270. [Google Scholar] [CrossRef] [Green Version]
- Nicolazzo, C.; Gradilone, A.; Loreni, F.; Raimondi, C.; Gazzaniga, P. EpCAM (low) Circulating Tumor Cells: Gold in the Waste. Dis. Markers 2019, 2019, 1718920. [Google Scholar] [CrossRef] [Green Version]
- Gires, O.; Stoecklein, N.H. Dynamic EpCAM expression on circulating and disseminating tumor cells: Causes and consequences. Cell Mol. Life Sci. 2014, 71, 4393–4402. [Google Scholar] [CrossRef]
- Grover, P.K.; Cummins, A.G.; Price, T.J.; Roberts-Thomson, I.C.; Hardingham, J.E. Circulating tumour cells: The evolving concept and the inadequacy of their enrichment by EpCAM-based methodology for basic and clinical cancer research. Ann. Oncol. 2014, 25, 1506–1516. [Google Scholar] [CrossRef]
- Clarke, L.A.; Shah, V.; Arrigoni, F.; Eleftheriou, D.; Hong, Y.; Halcox, J.; Klein, N.; Brogan, P.A. Quantitative detection of circulating endothelial cells in vasculitis: Comparison of flow cytometry and immunomagnetic bead extraction. J. Thromb. Haemost. 2008, 6, 1025–1032. [Google Scholar] [CrossRef]
- Shaffer, R.G.; Greene, S.; Arshi, A.; Supple, G.; Bantly, A.; Moore, J.S.; Mohler, E.R., 3rd. Flow cytometric measurement of circulating endothelial cells: The effect of age and peripheral arterial disease on baseline levels of mature and progenitor populations. Cytom. B Clin. Cytom. 2006, 70, 56–62. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Cancer Type | Stage | IRB # |
|---|---|---|---|
| PC 1 | Prostate | Metastatic | UP-16-00691 |
| PC 2 | Prostate | Metastatic | UP-16-00643 |
| PC 3 | Prostate | Metastatic | UP-16-00691 |
| PC 4 | Prostate | Metastatic | UP-16-00691 |
| Patient ID | Cancer Type | Stage | Cancer Subtype | IRB # |
|---|---|---|---|---|
| BC 1 | Breast | Metastatic | ER+/HER2− | UP-14-00592 |
| BC 2 | Breast | Metastatic | Triple Negative | UP-16-0070 |
| BC 3 | Breast | Metastatic | Triple Negative | UP-16-0070 |
| BC 4 | Breast | Metastatic | ER+/HER2− | UP-17-00882 |
| BC 5 | Breast | Metastatic | Triple Negative | UP-16-0070 |
| BC 6 | Breast | Metastatic | Triple Negative | UP-16-0070 |
| BC 7 | Breast | Metastatic | ER+/HER2− | UP-14-00182 |
| BC 8 | Breast | Metastatic | ER+/HER2− | UP-14-00523 |
| BC 9 | Breast | Metastatic | ER+/HER2− | UP-14-00523 |
| Patient ID | Disease Type | IRB # |
|---|---|---|
| MI 1 | MI | IRB-09-5287 |
| MI 2 | MI | IRB-09-5287 |
| MI 3 | MI | IRB-09-5287 |
| Metal Tag | Target | Antibody Clone | Final Dilution |
|---|---|---|---|
| Yb174 | CK8/18 | C51 | 1:50 |
| Pr141 | EpCAM | 9C4 | 1:100 |
| Dy163 | ER | D8H8 | 1:100 |
| Tb174 | HER2 | 42c/erbB2-2 | 1:100 |
| Nd148 | HER2 | 29D8 | 1:100 |
| Y89 | CD45 | HI30 | 1:200 |
| Tm169 | PSMA | 460420 | 1:100 |
| Gd160 | PSA | TD11B3-4 | 1:100 |
| Sm154 | AR | G122-434 | 1:100 |
| Ir193 | DNA | ||
| In115 | Membrane |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Welter, L.; Zheng, S.; Setayesh, S.M.; Morikado, M.; Agrawal, A.; Nevarez, R.; Naghdloo, A.; Pore, M.; Higa, N.; Kolatkar, A.; et al. Cell State and Cell Type: Deconvoluting Circulating Tumor Cell Populations in Liquid Biopsies by Multi-Omics. Cancers 2023, 15, 3949. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15153949
Welter L, Zheng S, Setayesh SM, Morikado M, Agrawal A, Nevarez R, Naghdloo A, Pore M, Higa N, Kolatkar A, et al. Cell State and Cell Type: Deconvoluting Circulating Tumor Cell Populations in Liquid Biopsies by Multi-Omics. Cancers. 2023; 15(15):3949. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15153949
Chicago/Turabian StyleWelter, Lisa, Serena Zheng, Sonia Maryam Setayesh, Michael Morikado, Arushi Agrawal, Rafael Nevarez, Amin Naghdloo, Milind Pore, Nikki Higa, Anand Kolatkar, and et al. 2023. "Cell State and Cell Type: Deconvoluting Circulating Tumor Cell Populations in Liquid Biopsies by Multi-Omics" Cancers 15, no. 15: 3949. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15153949