

Improved Characterization of Circulating Tumor Cells and Cancer-Associated Fibroblasts in One-Tube Assay in Breast Cancer Patients Using Imaging Flow Cytometry
 , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Breast Cancer Patients and Non-Cancer Volunteer Cohort
2.2. Blood Collection and Processing for CTC Enrichment
2.3. Cell Culture
2.4. Immunofluorescent Staining for Simultaneous CTC and cCAF Detection
2.5. Imaging Flow Cytometry Analysis
2.6. Statistics
3. Results
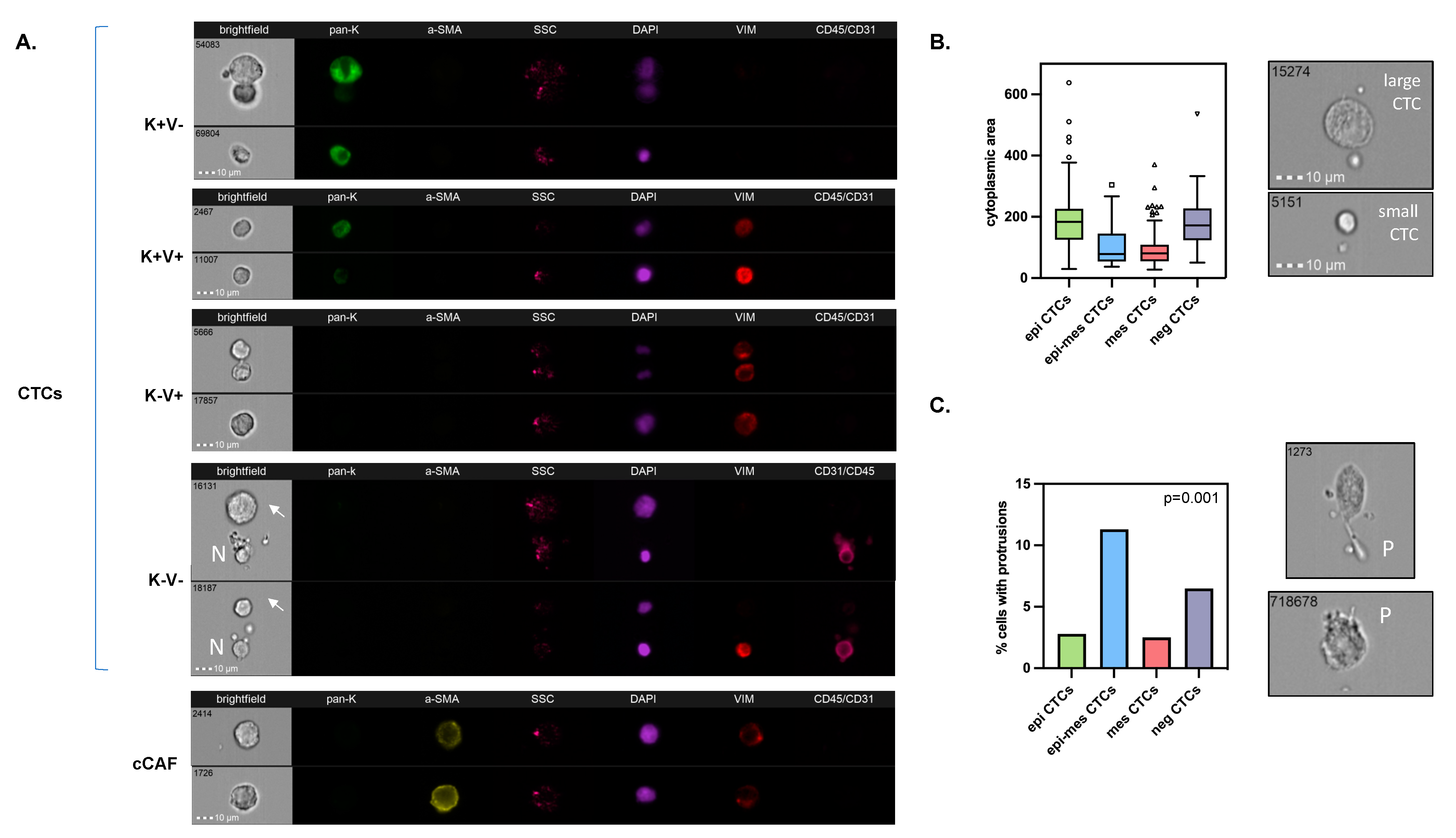
3.1. Detection and Characterization of Different Phenotypes of Circulating Tumor Cells and Circulating Cancer-Associated Fibroblasts
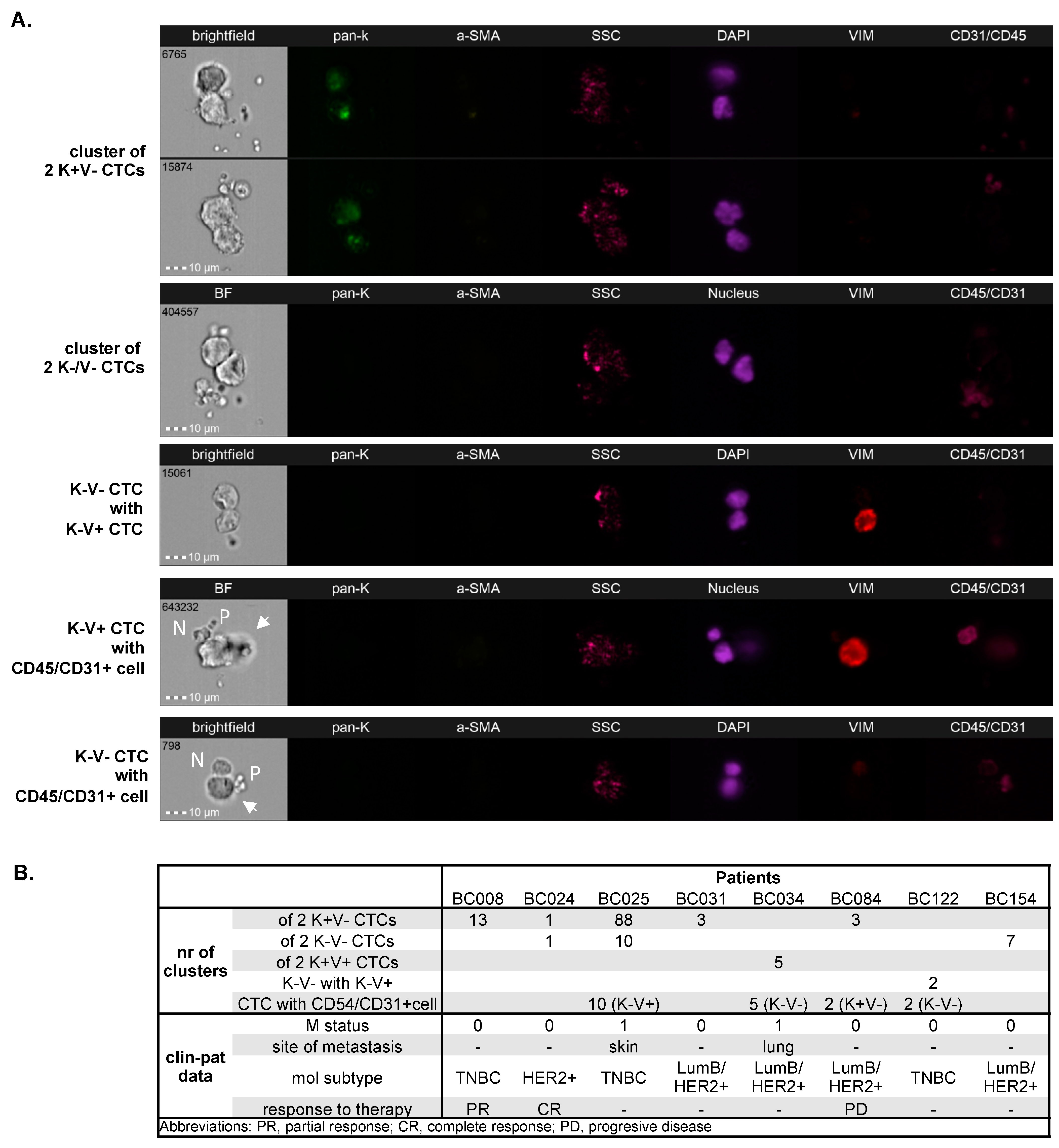
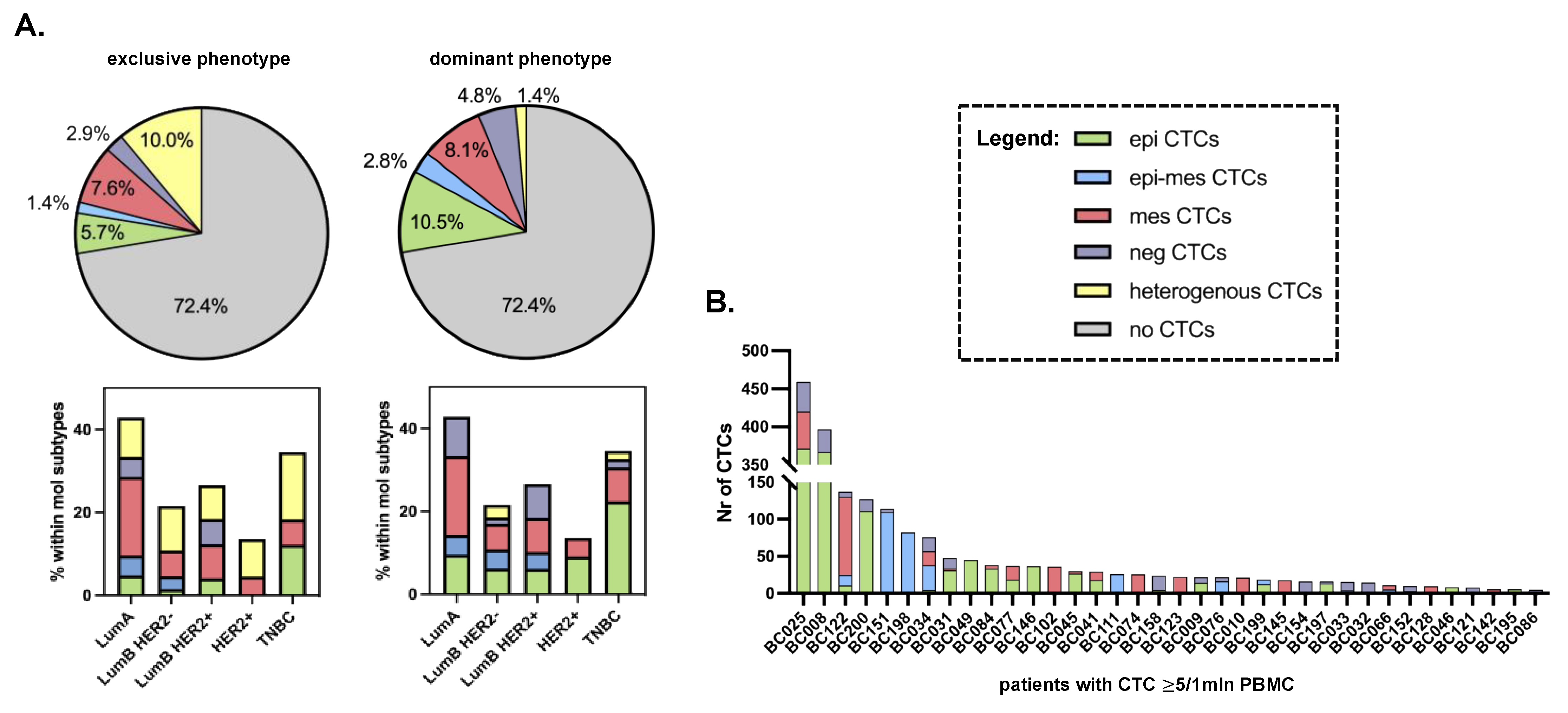
3.2. Distribution of Different Phenotypes of Circulating Tumor Cells and Circulating Cancer-Associated Fibroblasts
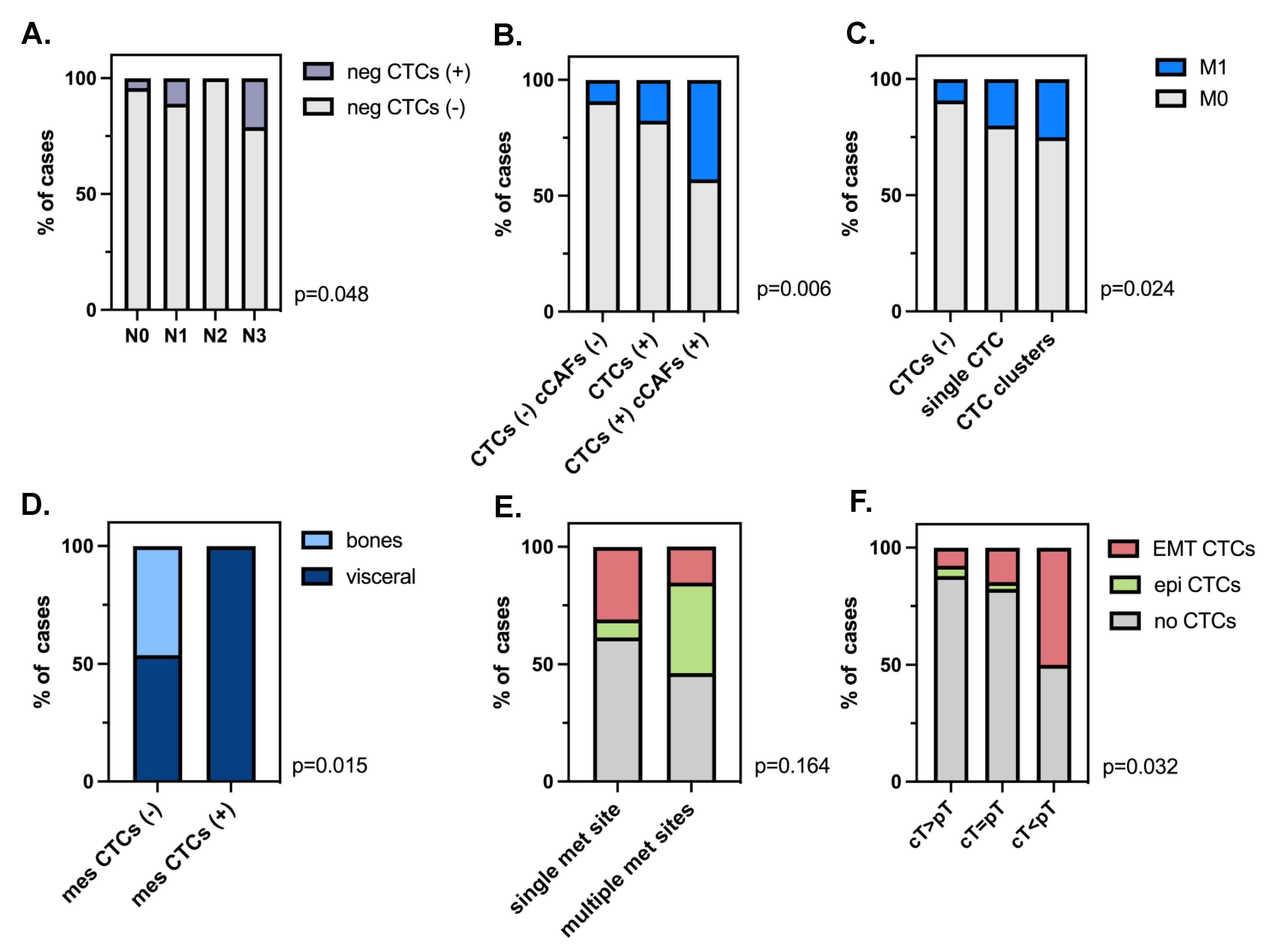
3.3. Clinical Relevance of Different Phenotypes of Circulating Tumor Cells and Circulating Cancer-Associated Fibroblasts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pantel, K.; Alix-Panabières, C. Liquid Biopsy and Minimal Residual Disease—Latest Advances and Implications for Cure. Nat. Rev. Clin. Oncol. 2019, 16, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Bednarz-Knoll, N.; Alix-Panabières, C.; Pantel, K. Plasticity of Disseminating Cancer Cells in Patients with Epithelial Malignancies. Cancer Metastasis Rev. 2012, 31, 673–687. [Google Scholar] [CrossRef]
- Topa, J.; Grešner, P.; Żaczek, A.J.; Markiewicz, A. Breast Cancer Circulating Tumor Cells with Mesenchymal Features—An Unreachable Target? Cell. Mol. Life Sci. 2022, 79, 81. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.L.; Adams, D.K.; Alpaugh, R.K.; Cristofanilli, M.; Martin, S.S.; Chumsri, S.; Tang, C.-M.; Marks, J.R. Circulating Cancer-Associated Macrophage-Like Cells Differentiate Malignant Breast Cancer and Benign Breast Conditions. Cancer Epidemiol. Biomark. Prev. 2016, 25, 1037–1042. [Google Scholar] [CrossRef]
- Lin, P.P. Aneuploid Circulating Tumor-Derived Endothelial Cell (CTEC): A Novel Versatile Player in Tumor Neovascularization and Cancer Metastasis. Cells 2020, 9, 1539. [Google Scholar] [CrossRef]
- Ao, Z.; Shah, S.H.; Machlin, L.M.; Parajuli, R.; Miller, P.C.; Rawal, S.; Williams, A.J.; Cote, R.J.; Lippman, M.E.; Datar, R.H.; et al. Identification of Cancer-Associated Fibroblasts in Circulating Blood from Patients with Metastatic Breast Cancer. Cancer Res. 2015, 75, 4681–4687. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Shen, L.; Luo, M.; Zhang, K.; Li, J.; Yang, Q.; Zhu, F.; Zhou, D.; Zheng, S.; Chen, Y.; et al. Circulating Tumor Cells: Biology and Clinical Significance. Signal Transduct. Target. Ther. 2021, 6, 404. [Google Scholar] [CrossRef] [PubMed]
- Matikas, A.; Kotsakis, A.; Apostolaki, S.; Politaki, H.; Perraki, M.; Kalbakis, K.; Nikolaou, M.; Economopoulou, P.; Hatzidaki, D.; Georgoulias, V. Detection of Circulating Tumour Cells before and Following Adjuvant Chemotherapy and Long-Term Prognosis of Early Breast Cancer. Br. J. Cancer 2022, 126, 1563–1569. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Stopeck, A.; Reuben, J.M. Circulating Tumor Cells, Disease Progression, and Survival in Metastatic Breast Cancer. N. Engl. J. Med. 2004, 351, 781–791. [Google Scholar] [CrossRef]
- Cohen, S.J.; Alpaugh, R.K.; Gross, S.; O’Hara, S.M.; Smirnov, D.A.; Terstappen, L.W.M.M.; Allard, W.J.; Bilbee, M.; Cheng, J.D.; Hoffman, J.P.; et al. Isolation and Characterization of Circulating Tumor Cells in Patients with Metastatic Colorectal Cancer. Clin. Color. Cancer 2006, 6, 125–132. [Google Scholar] [CrossRef]
- Lorente, D.; Olmos, D.; Mateo, J.; Bianchini, D.; Seed, G.; Fleisher, M.; Danila, D.C.; Flohr, P.; Crespo, M.; Figueiredo, I.; et al. Decline in Circulating Tumor Cell Count and Treatment Outcome in Advanced Prostate Cancer. Eur. Urol. 2016, 70, 985–992. [Google Scholar] [CrossRef] [PubMed]
- Grisanti, S.; Almici, C.; Consoli, F.; Buglione, M.; Verardi, R.; Bolzoni-Villaret, A.; Bianchetti, A.; Ciccarese, C.; Mangoni, M.; Ferrari, L.; et al. Circulating Tumor Cells in Patients with Recurrent or Metastatic Head and Neck Carcinoma: Prognostic and Predictive Significance. PLoS ONE 2014, 9, e103918. [Google Scholar] [CrossRef] [PubMed]
- Markiewicz, A.; Nagel, A.; Szade, J.; Majewska, H.; Skokowski, J.; Seroczynska, B.; Stokowy, T.; Welnicka-Jaskiewicz, M.; Zaczek, A.J. Aggressive Phenotype of Cells Disseminated via Hematogenous and Lymphatic Route in Breast Cancer Patients. Transl. Oncol. 2018, 11, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Duda, D.G.; Duyverman, A.M.M.J.; Kohno, M.; Snuderl, M.; Steller, E.J.A.; Fukumura, D.; Jain, R.K. Malignant Cells Facilitate Lung Metastasis by Bringing Their Own Soil. Proc. Natl. Acad. Sci. USA 2010, 107, 21677–21682. [Google Scholar] [CrossRef]
- Jones, M.L.; Siddiqui, J.; Pienta, K.J.; Getzenberg, R.H. Circulating Fibroblast-like Cells in Men with Metastatic Prostate Cancer. Prostate 2013, 73, 176–181. [Google Scholar] [CrossRef]
- Sharma, U.; Medina-Saenz, K.; Miller, P.C.; Troness, B.; Spartz, A.; Sandoval-Leon, A.; Parke, D.N.; Seagroves, T.N.; Lippman, M.E.; El-Ashry, D. Heterotypic Clustering of Circulating Tumor Cells and Circulating Cancer-Associated Fibroblasts Facilitates Breast Cancer Metastasis. Breast Cancer Res. Treat. 2021, 189, 63–80. [Google Scholar] [CrossRef]
- Agarwal, A.; Balic, M.; El-Ashry, D.; Cote, R.J. Circulating Tumor Cells: Strategies for Capture, Analyses, and Propagation. Cancer J. 2018, 24, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Muchlińska, A.; Smentoch, J.; Żaczek, A.J.; Bednarz-Knoll, N. Detection and Characterization of Circulating Tumor Cells Using Imaging Flow Cytometry—A Perspective Study. Cancers 2022, 14, 4178. [Google Scholar] [CrossRef]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open Source Software for Digital Pathology Image Analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef]
- Pierga, J.-Y.; Hajage, D.; Bachelot, T.; Delaloge, S.; Brain, E.; Campone, M.; Diéras, V.; Rolland, E.; Mignot, L.; Mathiot, C.; et al. High Independent Prognostic and Predictive Value of Circulating Tumor Cells Compared with Serum Tumor Markers in a Large Prospective Trial in First-Line Chemotherapy for Metastatic Breast Cancer Patients. Ann. Oncol. 2012, 23, 618–624. [Google Scholar] [CrossRef]
- Janni, W.J.; Rack, B.; Terstappen, L.W.M.M.; Pierga, J.-Y.; Taran, F.-A.; Fehm, T.; Hall, C.; de Groot, M.R.; Bidard, F.-C.; Friedl, T.W.P.; et al. Pooled Analysis of the Prognostic Relevance of Circulating Tumor Cells in Primary Breast Cancer. Clin. Cancer Res. 2016, 22, 2583–2593. [Google Scholar] [CrossRef]
- Rack, B.; Schindlbeck, C.; Jückstock, J.; Andergassen, U.; Hepp, P.; Zwingers, T.; Friedl, T.W.P.; Lorenz, R.; Tesch, H.; Fasching, P.A.; et al. Circulating Tumor Cells Predict Survival in Early Average-to-High Risk Breast Cancer Patients. JNCI J. Natl. Cancer Inst. 2014, 106, dju066. [Google Scholar] [CrossRef]
- Strati, A.; Zavridou, M.; Kallergi, G.; Politaki, E.; Kuske, A.; Gorges, T.M.; Riethdorf, S.; Joosse, S.A.; Koch, C.; Bohnen, A.-L.; et al. A Comprehensive Molecular Analysis of in Vivo Isolated EpCAM-Positive Circulating Tumor Cells in Breast Cancer. Clin. Chem. 2021, 67, 1395–1405. [Google Scholar] [CrossRef]
- Markiewicz, A.; Żaczek, A.J. The Landscape of Circulating Tumor Cell Research in the Context of Epithelial-Mesenchymal Transition. Pathobiology 2017, 84, 264–283. [Google Scholar] [CrossRef]
- Alix-Panabières, C.; Mader, S.; Pantel, K. Epithelial-Mesenchymal Plasticity in Circulating Tumor Cells. J. Mol. Med. 2017, 95, 133–142. [Google Scholar] [CrossRef]
- Papadaki, M.A.; Kallergi, G.; Zafeiriou, Z.; Manouras, L.; Theodoropoulos, P.A.; Mavroudis, D.; Georgoulias, V.; Agelaki, S. Co-Expression of Putative Stemness and Epithelial-to-Mesenchymal Transition Markers on Single Circulating Tumour Cells from Patients with Early and Metastatic Breast Cancer. BMC Cancer 2014, 14, 651. [Google Scholar] [CrossRef]
- Szczerba, B.M.; Castro-Giner, F.; Vetter, M.; Krol, I.; Gkountela, S.; Landin, J.; Scheidmann, M.C.; Donato, C.; Scherrer, R.; Singer, J.; et al. Neutrophils Escort Circulating Tumour Cells to Enable Cell Cycle Progression. Nature 2019, 566, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating Tumor Cell Clusters Are Oligoclonal Precursors of Breast Cancer Metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Otero, N.; Marshall, J.R.; Lash, B.; King, M.R. Chemotherapy-Induced Release of Circulating-Tumor Cells into the Bloodstream in Collective Migration Units with Cancer-Associated Fibroblasts in Metastatic Cancer Patients. BMC Cancer 2020, 20, 873. [Google Scholar] [CrossRef] [PubMed]
- Patteson, A.E.; Pogoda, K.; Byfield, F.J.; Mandal, K.; Ostrowska-Podhorodecka, Z.; Charrier, E.E.; Galie, P.A.; Deptuła, P.; Bucki, R.; McCulloch, C.A.; et al. Loss of Vimentin Enhances Cell Motility through Small Confining Spaces. Small 2019, 15, 1903180. [Google Scholar] [CrossRef]
- Liu, C.-Y.; Lin, H.-H.; Tang, M.-J.; Wang, Y.-K. Vimentin Contributes to Epithelial-Mesenchymal Transition Cancer Cell Mechanics by Mediating Cytoskeletal Organization and Focal Adhesion Maturation. Oncotarget 2015, 6, 15966–15983. [Google Scholar] [CrossRef]
- Mendez, M.G.; Restle, D.; Janmey, P.A. Vimentin Enhances Cell Elastic Behavior and Protects against Compressive Stress. Biophys. J. 2014, 107, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef]
- López-Riquelme, N.; Minguela, A.; Villar-Permuy, F.; Ciprian, D.; Castillejo, A.; Álvarez-López, M.-R.; Soto, J.-L. Imaging Cytometry for Counting Circulating Tumor Cells: Comparative Analysis of the CellSearch vs ImageStream Systems. APMIS 2013, 121, 1139–1143. [Google Scholar] [CrossRef]
- Mourtzikou, A.; Stamouli, M.; Karakitsos, P.; Athanasas, A. Colorectal Cancer CTCs Detection Using Two FCM Protocol Approaches. Clin. Lab. 2019, 65. [Google Scholar] [CrossRef] [PubMed]
- Merugu, S.; Chen, L.; Gavens, E.; Gabra, H.; Brougham, M.; Makin, G.; Ng, A.; Murphy, D.; Gabriel, A.S.; Robinson, M.L.; et al. Detection of Circulating and Disseminated Neuroblastoma Cells Using the ImageStream Flow Cytometer for Use as Predictive and Pharmacodynamic Biomarkers. Clin. Cancer Res. 2020, 26, 122–134. [Google Scholar] [CrossRef]
- Carter, T.J.; Jeyaneethi, J.; Kumar, J.; Karteris, E.; Glynne-Jones, R.; Hall, M. Identification of Cancer-Associated Circulating Cells in Anal Cancer Patients. Cancers 2020, 12, 2229. [Google Scholar] [CrossRef] [PubMed]
- Semaan, A.; Bernard, V.; Kim, D.U.; Lee, J.J.; Huang, J.; Kamyabi, N.; Stephens, B.M.; Qiao, W.; Varadhachary, G.R.; Katz, M.H.; et al. Characterisation of Circulating Tumour Cell Phenotypes Identifies a Partial-EMT Sub-Population for Clinical Stratification of Pancreatic Cancer. Br. J. Cancer 2021, 124, 1970–1977. [Google Scholar] [CrossRef]
- Staudte, S.; Klinghammer, K.; Jurmeister, P.S.; Jank, P.; Blohmer, J.-U.; Liebs, S.; Rhein, P.; Hauser, A.E.; Tinhofer, I. Multiparametric Phenotyping of Circulating Tumor Cells for Analysis of Therapeutic Targets, Oncogenic Signaling Pathways and DNA Repair Markers. Cancers 2022, 14, 2810. [Google Scholar] [CrossRef]
- Ligthart, S.T.; Coumans, F.A.W.; Bidard, F.-C.; Simkens, L.H.J.; Punt, C.J.A.; de Groot, M.R.; Attard, G.; de Bono, J.S.; Pierga, J.-Y.; Terstappen, L.W.M.M. Circulating Tumor Cells Count and Morphological Features in Breast, Colorectal and Prostate Cancer. PLoS ONE 2013, 8, e67148. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CTC | cCAF | ||||||
|---|---|---|---|---|---|---|---|
| Variable | Total n | CTC neg | CTC pos | p-Value | cCAF neg | cCAF pos | p-Value |
| Age | |||||||
| <50 | 84 (40%) | 68 (44.7%) | 16 (27.6%) | 0.02 | 83 (40.9%) | 1 (14.3%) | 0.25 F |
| ≥50 | 126 (60%) | 84 (55.3%) | 42 (72.4%) | 120 (59.1%) | 6 (85.75) | ||
| cT stage | |||||||
| cT1-2 | 126 (71.6%) | 99 (72.8%) | 27 (67.5%) | 0.51 | 125 (71.8%) | 1 (50%) | 0.49 F |
| cT3-4 | 50 (28.4%) | 37 (27.2%) | 13 (32.5%) | 49 (28.2%) | 1 (50%) | ||
| cN stage | |||||||
| cN0 | 70 (40.2%) | 55 (41%) | 15 (37.5%) | 0.69 | 70 (40.7%) | 0 (0%) | 0.52 F |
| cN1 | 104 (59.8%) | 79 (59%) | 25 (62.5%) | 102 (59.3%) | 2 (100%) | ||
| M stage | |||||||
| M0 | 184 (87.6%) | 138 (90.8%) | 46 (79.3%) | 0.02 | 180 (88.7%) | 4 (57.1%) | 0.04 F |
| M1 | 26 (12.4%) | 14 (9.2%) | 12 (20.7%) | 23 (11.3%) | 3 (42.9%) | ||
| Grading | |||||||
| 1 | 11 (5.7%) | 8 (5.5%) | 3 (6.3%) | 0.58 | 11 (5.8%) | 0 (0%) | 0.54 |
| 2 | 93 (48.2%) | 67 (46.2%) | 26 (54.2%) | 90 (47.6%) | 3 (75%) | ||
| 3 | 89 (46.1%) | 70 (48.3%) | 19 (39.6%) | 88 (46.6%) | 1 (25%) | ||
| Molecular type | |||||||
| Luminal A | 21 (10.2%) | 12 (8%) | 9 (16.1%) | 0.13 | 20 (10.1%) | 1 (14.3%) | 0.25 |
| Luminal B HER2− | 65 (31.6%) | 51 (34%) | 14 (25%) | 65 (32.7%) | 0 (0%) | ||
| Luminal B HER2+ | 49 (23.8%) | 36 (24%) | 13 (23.2%) | 46 (23.1%) | 3 (42.9%) | ||
| Non-luminal HER2+ | 22 (10.7%) | 19 (12.7%) | 3 (5.4%) | 22 (11.1%) | 0 (0%) | ||
| Triple-negative | 49 (23.8%) | 32 (21.3%) | 17 (30.4%) | 46 (23.1%) | 3 (42.9%) | ||
| Response to treatment | |||||||
| Stable disease | 21 (51.1%) | 15 (13.6%) | 6 (20.7%) | 0.62 | 21 (15.2%) | 0 (0%) | 0.8 |
| Complete response | 39 28.1%) | 30 (27.3%) | 9 (31%) | 39 (28.3%) | 0 (0%) | ||
| Partial response | 69 (49.6%) | 56 (50.9%) | 13 (44.8%) | 68 (49.3%) | 1 (100%) | ||
| Progressive disease | 10 (7.2%) | 9 (8.2%) | 1 (3.4%) | 10 (7.2%) | 0 (0%) | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muchlińska, A.; Wenta, R.; Ścińska, W.; Markiewicz, A.; Suchodolska, G.; Senkus, E.; Żaczek, A.J.; Bednarz-Knoll, N. Improved Characterization of Circulating Tumor Cells and Cancer-Associated Fibroblasts in One-Tube Assay in Breast Cancer Patients Using Imaging Flow Cytometry. Cancers 2023, 15, 4169. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15164169
Muchlińska A, Wenta R, Ścińska W, Markiewicz A, Suchodolska G, Senkus E, Żaczek AJ, Bednarz-Knoll N. Improved Characterization of Circulating Tumor Cells and Cancer-Associated Fibroblasts in One-Tube Assay in Breast Cancer Patients Using Imaging Flow Cytometry. Cancers. 2023; 15(16):4169. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15164169
Chicago/Turabian StyleMuchlińska, Anna, Robert Wenta, Wiktoria Ścińska, Aleksandra Markiewicz, Grażyna Suchodolska, Elżbieta Senkus, Anna J. Żaczek, and Natalia Bednarz-Knoll. 2023. "Improved Characterization of Circulating Tumor Cells and Cancer-Associated Fibroblasts in One-Tube Assay in Breast Cancer Patients Using Imaging Flow Cytometry" Cancers 15, no. 16: 4169. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15164169