Flow-Through Macroporous Polymer Monoliths Containing Artificial Catalytic Centers Mimicking Chymotrypsin Active Site

 , and
, and
Abstract
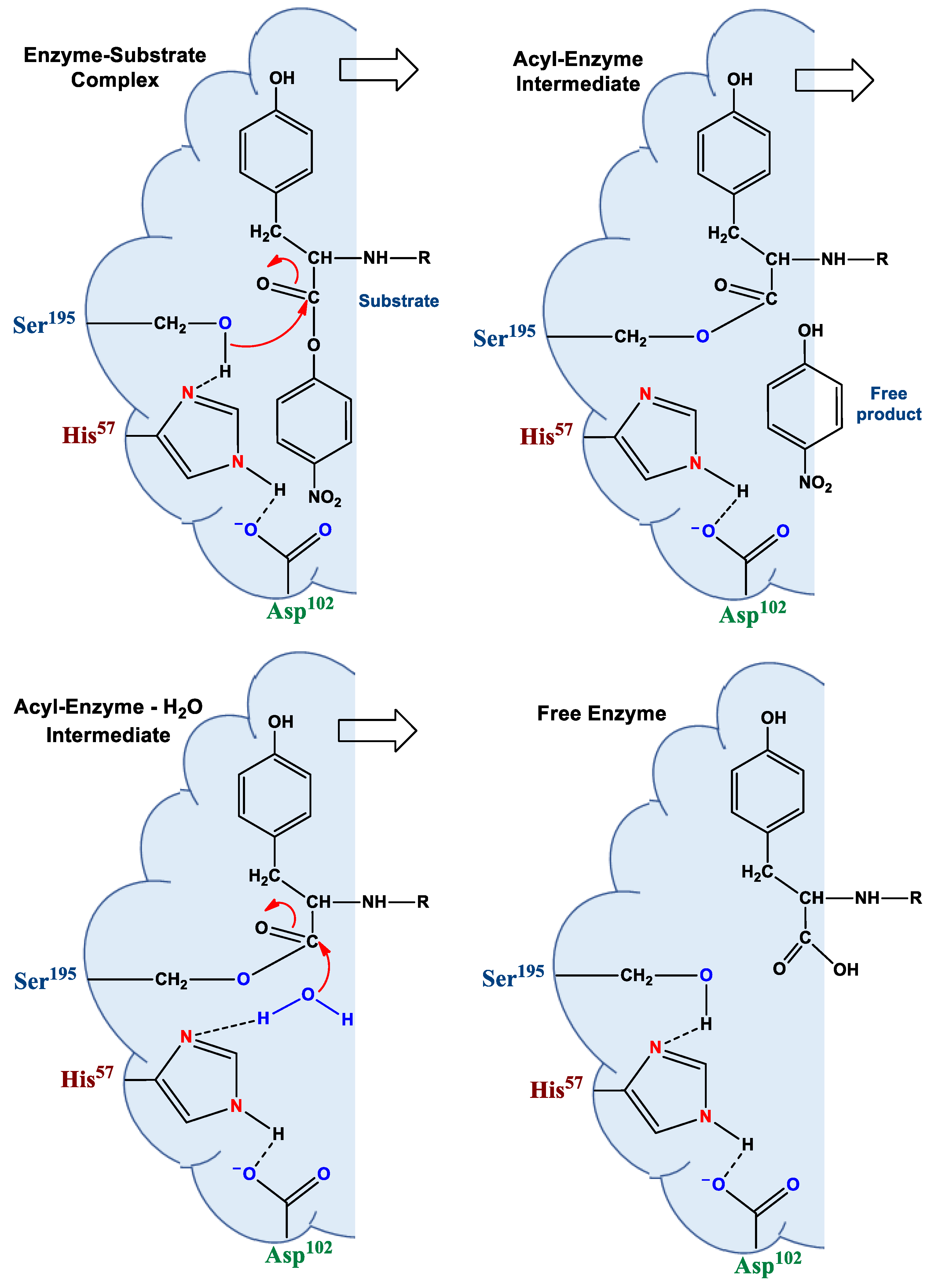
:1. Introduction
2. Results and Discussion
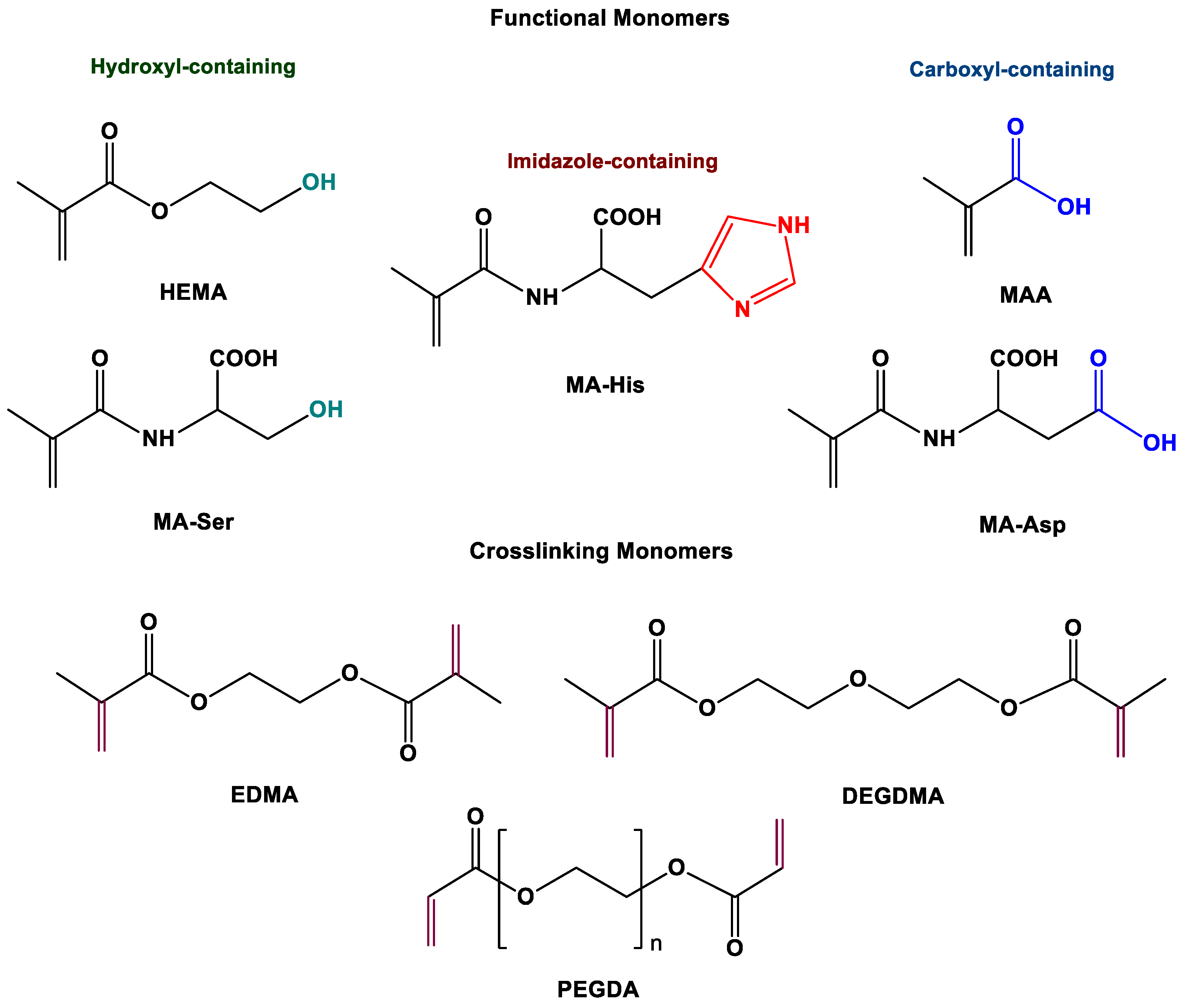
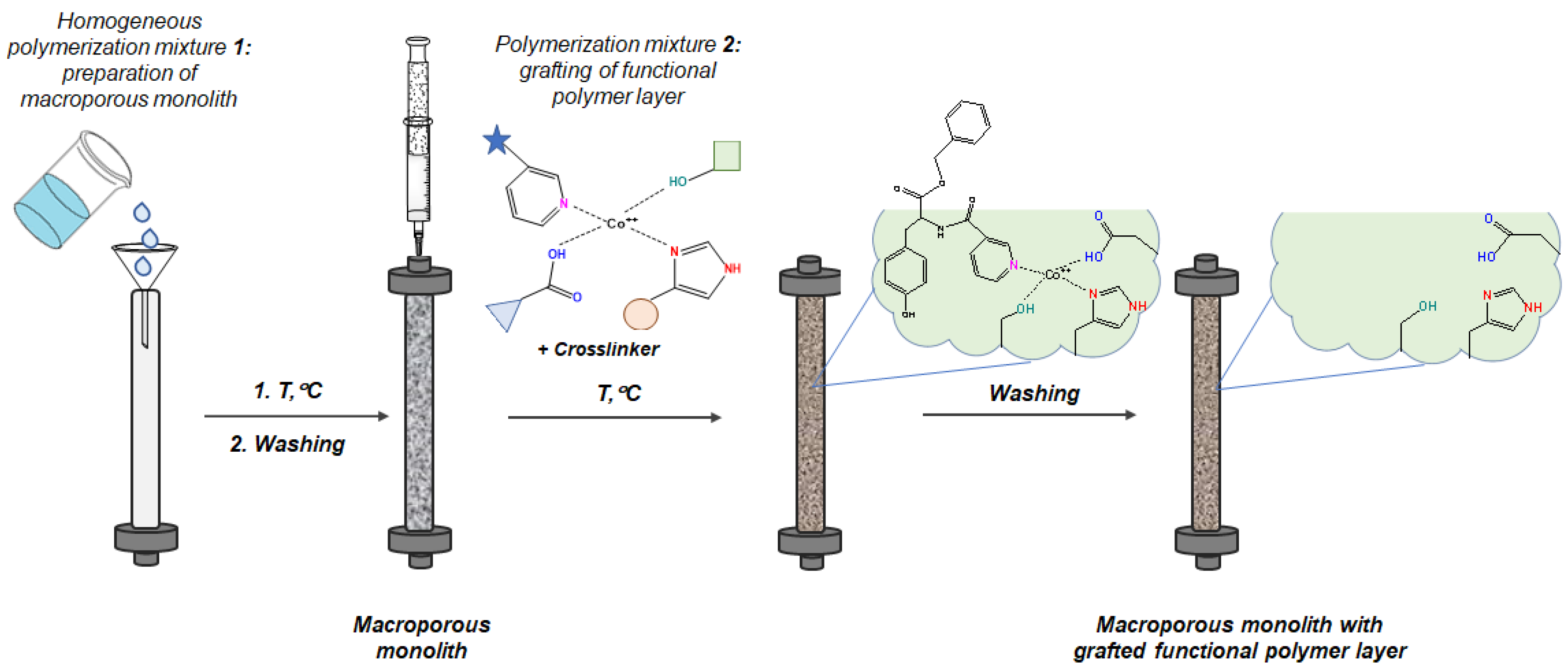
2.1. Preparation and Characterization of Macroporous Monolithic MICs
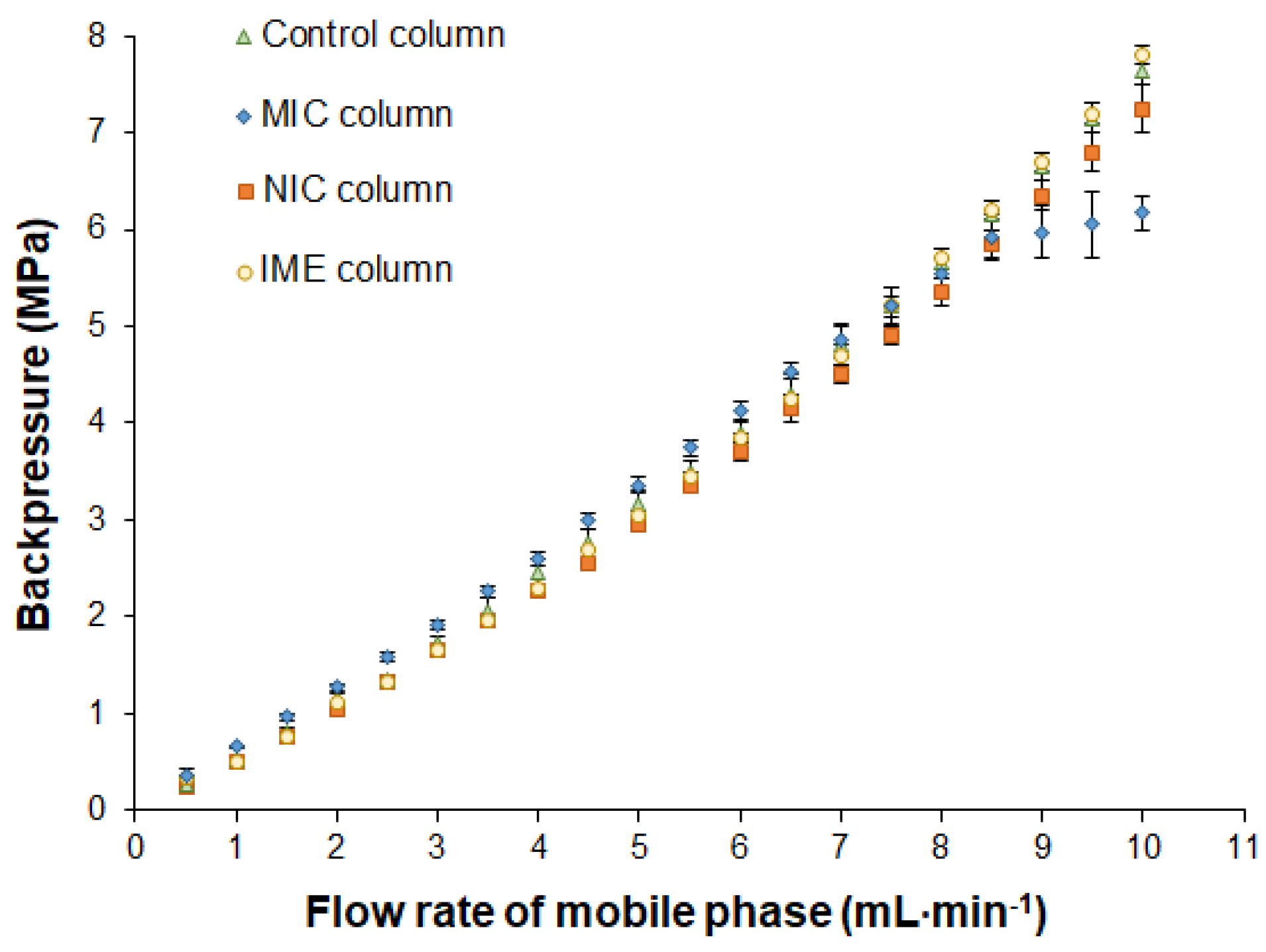
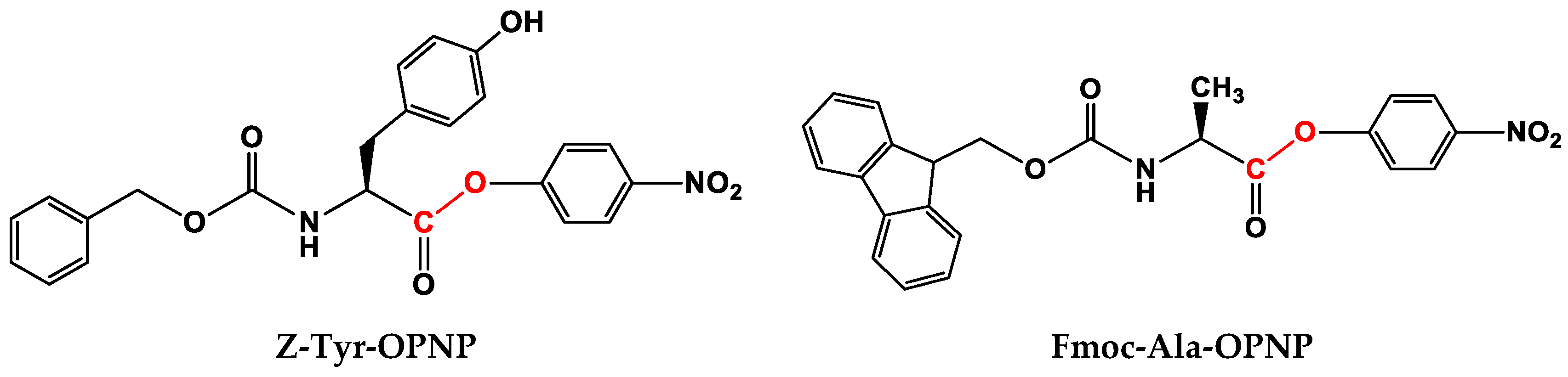
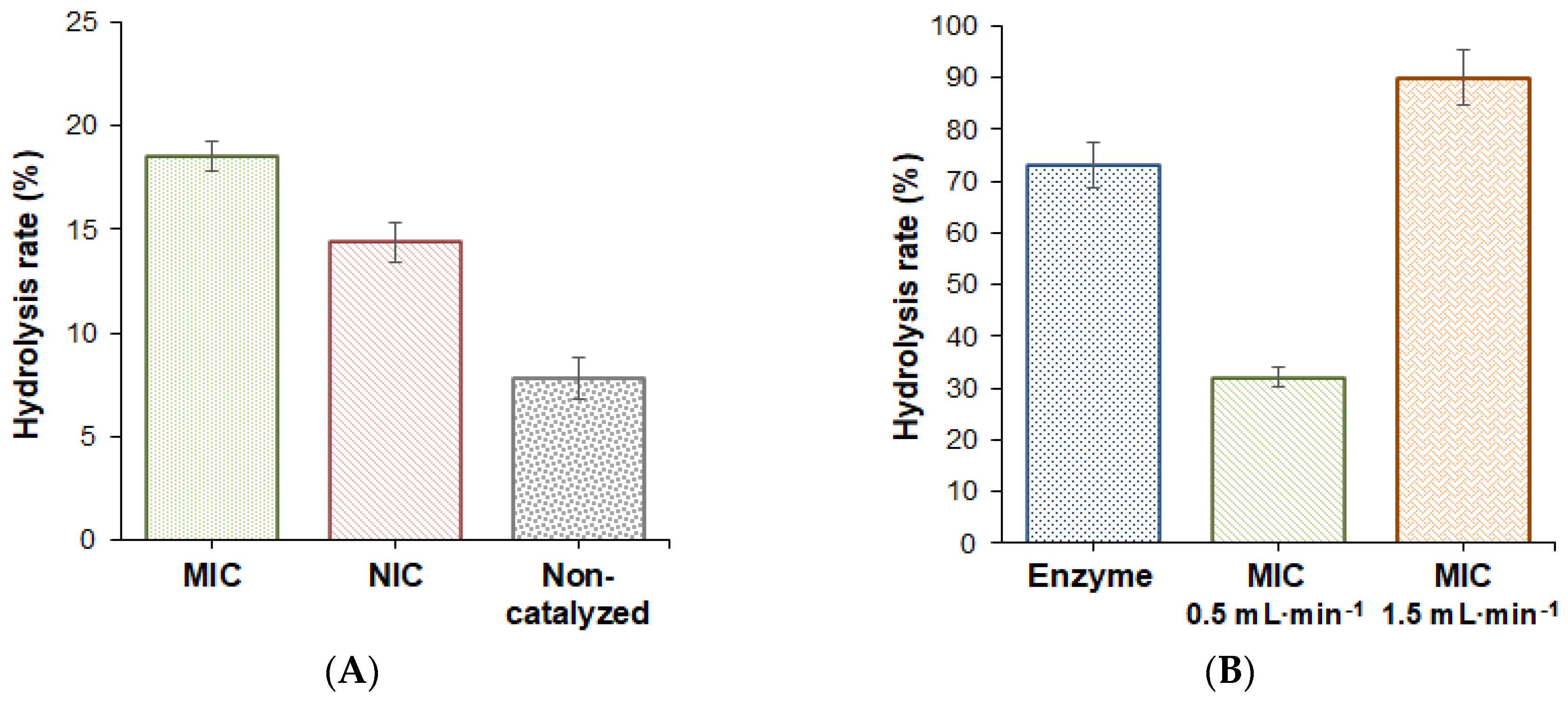
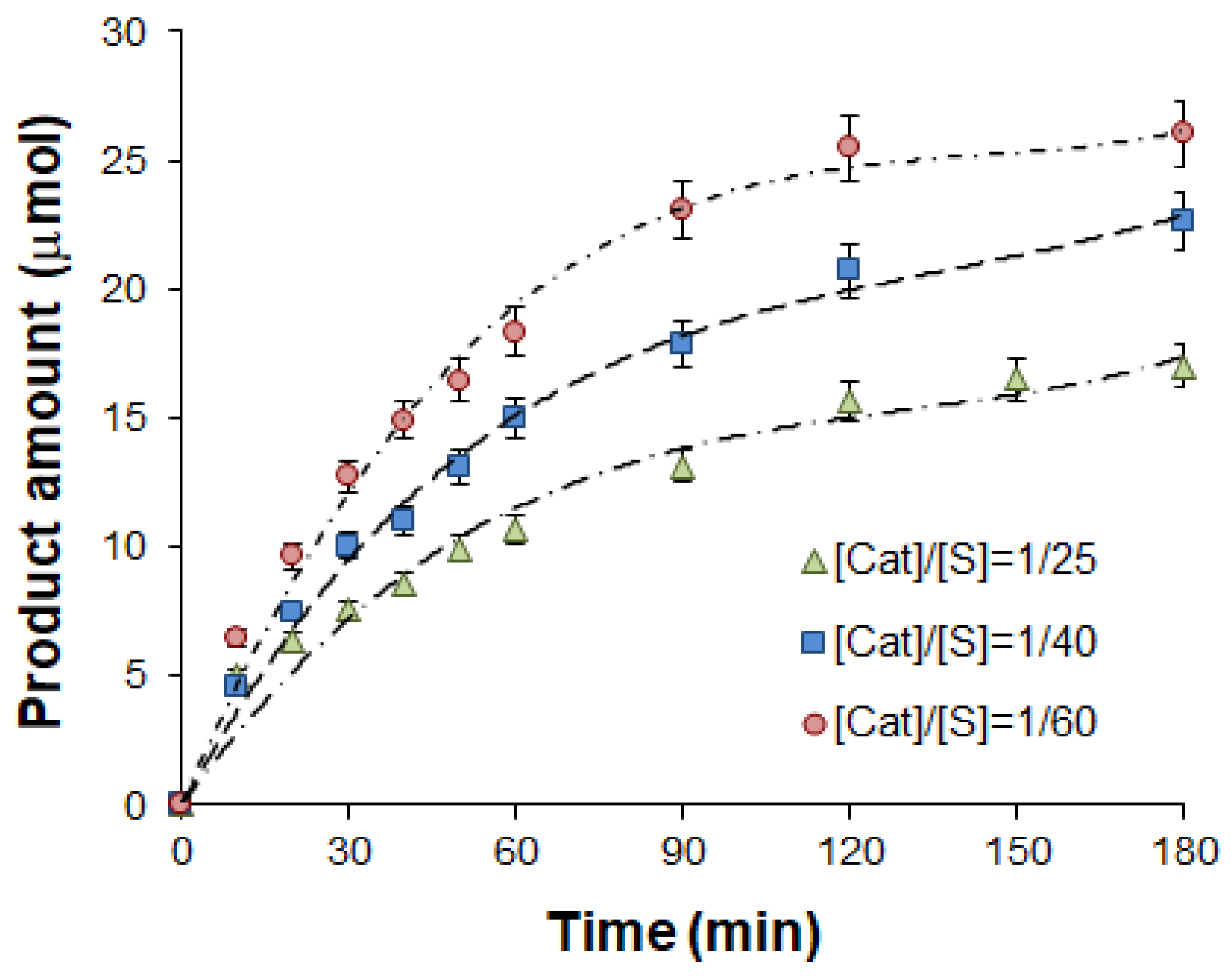
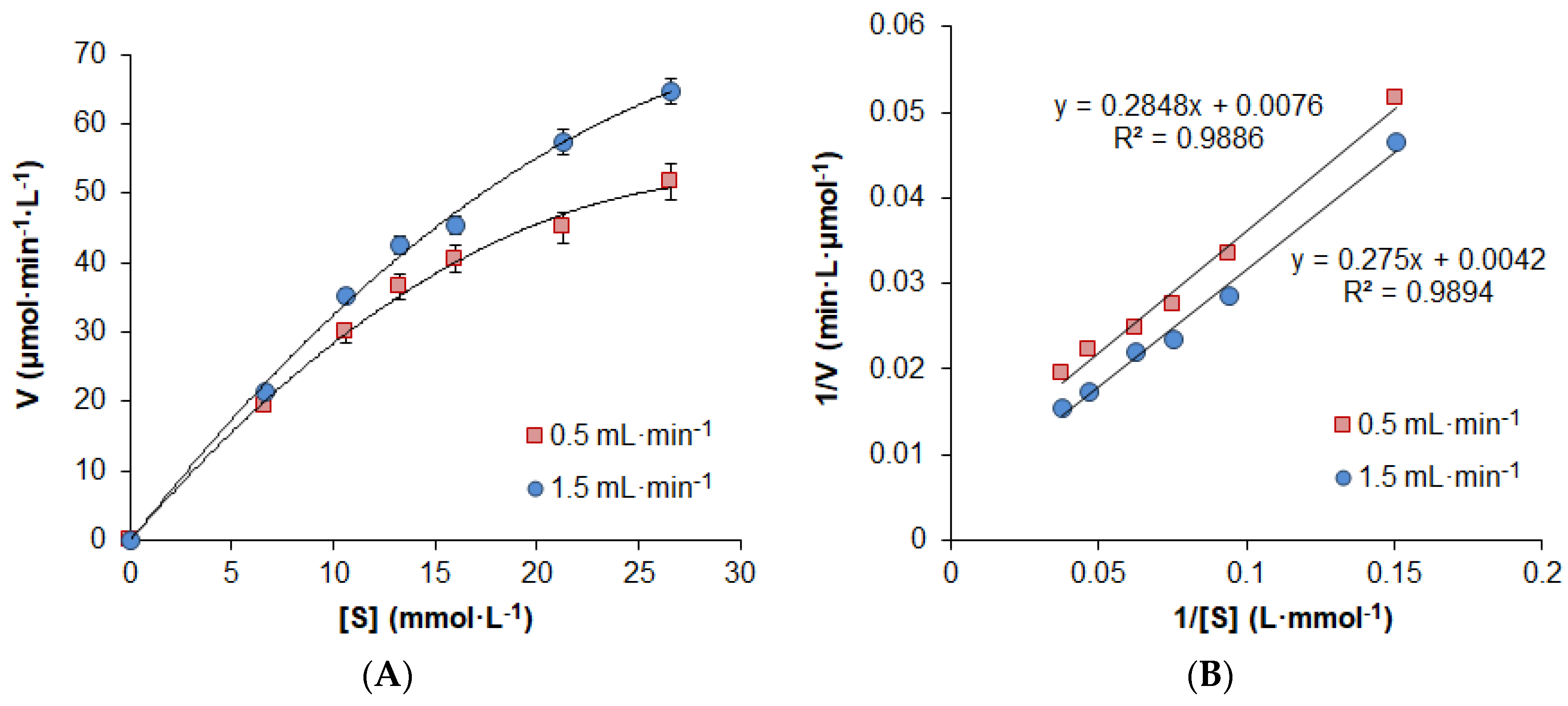
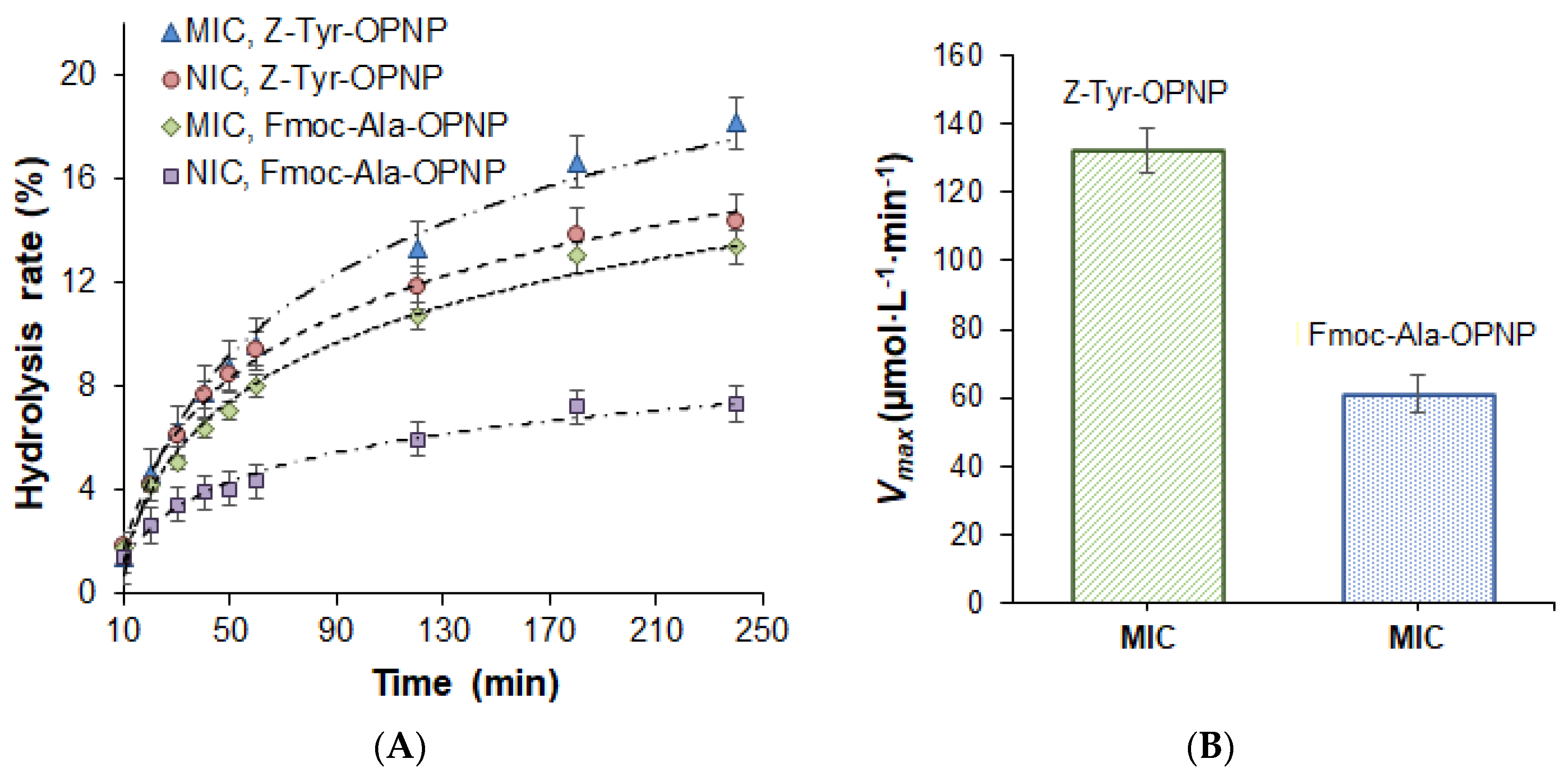
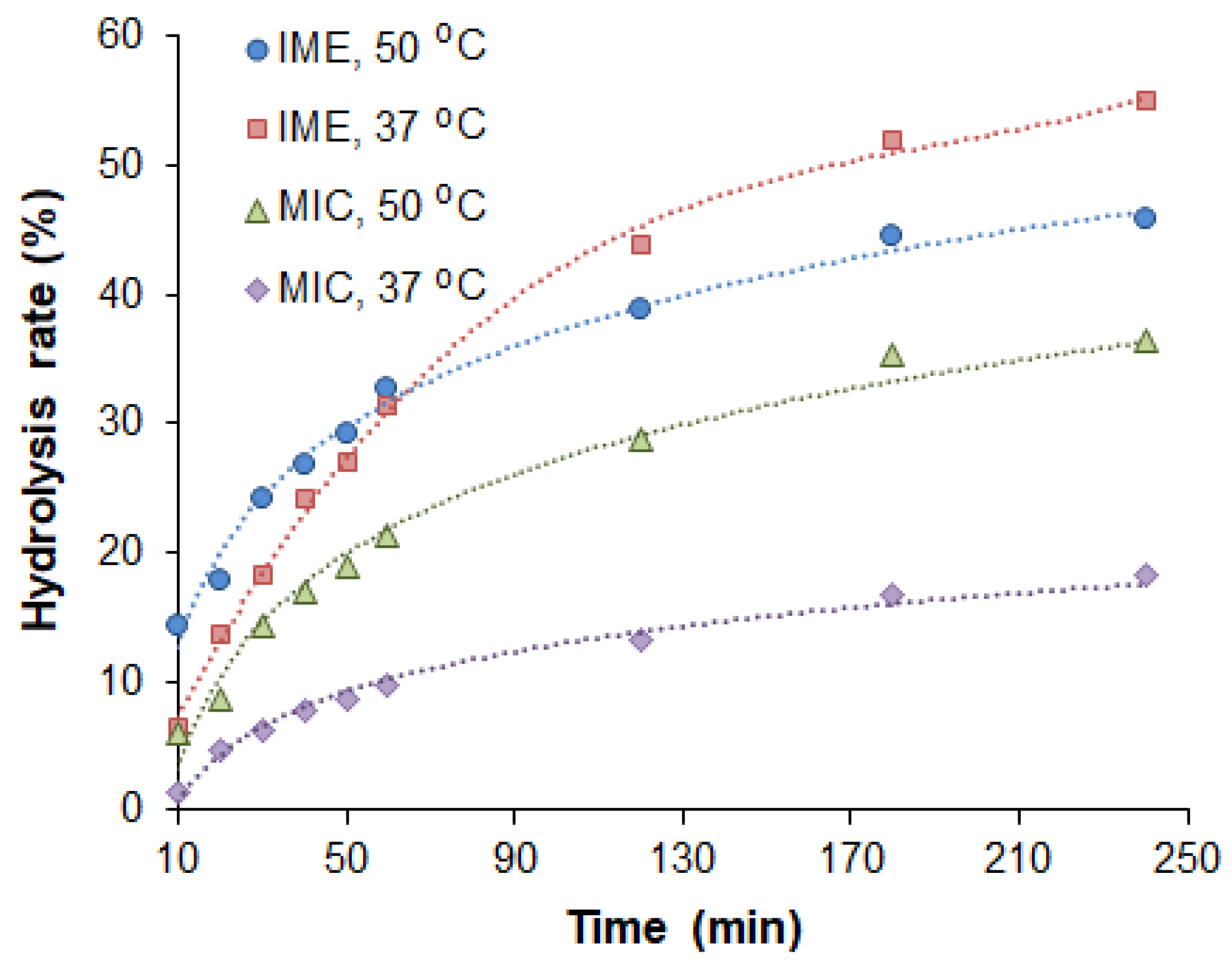
2.2. Study of Catalytical Properties
3. Materials and Methods
3.1. Chemicals and Supplements
3.2. Methods
3.2.1. Synthesis of Amino Acid-based Monomers (MA-His, MA-Ser, and MA-Asp)
3.2.2. Synthesis of Template and Substrates
3.2.3. Synthesis of Macroporous Mimic Monoliths
3.2.4. Characterization of Macroporous Monoliths
3.2.5. Chymotrypsin Immobilization
3.2.6. Evaluation of Catalytic Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Singh, R.; Kumar, M.; Mittal, A.; Mehta, P.K. Microbial enzymes: Industrial progress in 21st century. 3 Biotech 2016, 6, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Chapman, J.; Ismail, A.E.; Dinu, C.Z. Industrial Applications of Enzymes: Recent Advances, Techniques, and Outlooks. Catalysts 2018, 8, 238. [Google Scholar] [CrossRef] [Green Version]
- Alexander, C.; Davidson, L.; Hayes, W. Imprinted polymers: Artificial molecular recognition materials with applications in synthesis and catalysis. Tetrahedron 2003, 59, 2025–2057. [Google Scholar] [CrossRef]
- Mathew, D.; Thomas, B.; Devaky, K.S. Design, synthesis and characterization of enzyme-analogue-built polymer catalysts as artificial hydrolases. Artif. Cells Nanomed. Biotechnol. 2019, 47, 1149–1172. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Liu, C.; Pu, F.; Liu, Z.; Ren, J.; Qu, X. A GO–Se nanocomposite as an antioxidant nanozyme for cytoprotection. Chem. Commun. 2017, 53, 3082–3085. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Liu, X.; Chai, H.; Huang, Y. Recent advances in the construction and analytical applications of metal-organic frameworks-based nanozymes. TrAC Trends Anal. Chem. 2018, 105, 391–403. [Google Scholar] [CrossRef]
- Shi, C.; Li, Y.; Gu, N. Iron-Based Nanozymes in Disease Diagnosis and Treatment. ChemBioChem 2020, 21, 2722–2732. [Google Scholar] [CrossRef] [PubMed]
- Mathew, D.; Thomas, B.; Devaky, K.S. Geometrical effect of 3D-memory cavity on the imprinting efficiency of transition-state analogue-built artificial hydrolases. Polym. Bull. 2017, 75, 3883–3896. [Google Scholar] [CrossRef]
- Zeng, L.; Cui, H.; Chao, J.; Huang, K.; Wang, X.; Zhou, Y.; Jing, T. Colorimetric determination of tetrabromobisphenol A based on enzyme-mimicking activity and molecular recognition of metal-organic framework-based molecularly imprinted polymers. Microchim. Acta 2020, 187, 142. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Xu, Y.; Li, Z.; Yan, C.; Mei, K.; Ding, M.; Ding, S.; Guan, P.; Qian, L.; Du, C.; et al. Molecularly Imprinted Materials for Selective Biological Recognition. Macromol. Rapid Commun. 2019, 40, e1900096. [Google Scholar] [CrossRef]
- Cheng, Z.; Zhang, L.; Li, Y. Synthesis of an Enzyme-like Imprinted Polymer with the Substrate as the Template, and Its Catalytic Properties under Aqueous Conditions. Chem. Eur. J. 2004, 10, 3555–3561. [Google Scholar] [CrossRef]
- Chen, Z.; Hua, Z.; Wang, J.; Guan, Y.; Zhao, M.; Li, Y. Molecularly imprinted soluble nanogels as a peroxidase-like catalyst in the oxidation reaction of homovanillic acid under aqueous conditions. Appl. Catal. A Gen. 2007, 328, 252–258. [Google Scholar] [CrossRef]
- Li, J.; Zhu, M.; Wang, M.; Qi, W.; Su, R.; He, Z. Molecularly imprinted peptide-based enzyme mimics with enhanced activity and specificity. Soft Matter 2020, 16, 7033–7039. [Google Scholar] [CrossRef] [PubMed]
- Philip, C.; Devaky, K. Multiwalled carbon nanotubes with surface grafted transition state analogue imprints as chymotrypsin mimics for the hydrolysis of amino acid esters: Synthesis and kinetic studies. Mol. Catal. 2017, 436, 276–284. [Google Scholar] [CrossRef]
- Díaz-Díaz, G.; Antuña-Jiménez, D.; Blanco-López, M.C.; Lobo-Castañón, M.J.; Miranda-Ordieres, A.J.; Tuñón-Blanco, P. New materials for analytical biomimetic assays based on affinity and catalytic receptors prepared by molecular imprinting. TrAC Trends Anal. Chem. 2012, 33, 68–80. [Google Scholar] [CrossRef]
- Czulak, J.; Jakubiak-Marcinkowska, A.; Trochimczuk, A. Polymer Catalysts Imprinted with Metal Ions as Biomimics of Metalloenzymes. Adv. Mater. Sci. Eng. 2013, 2013, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Pan, J.; Qin, M.; Guo, T. Molecularly imprinted nanocapsule mimicking phosphotriesterase for the catalytic hydrolysis of organophosphorus pesticides. Eur. Polym. J. 2019, 110, 1–8. [Google Scholar] [CrossRef]
- Tadi, K.K.; Motghare, R.V.; Ganesh, V. Electrochemical detection of epinephrine using a biomimic made up of hemin modified molecularly imprinted microspheres. RSC Adv. 2015, 5, 99115–99124. [Google Scholar] [CrossRef]
- Lele, B.; Kulkarni, M.; Mashelkar, R. Molecularly imprinted polymer mimics of chymotrypsin. React. Funct. Polym. 1999, 39, 37–52. [Google Scholar] [CrossRef]
- Arrua, R.D.; Strumia, M.C.; Igarzabal, C.I.A. Macroporous Monolithic Polymers: Preparation and Applications. Materials 2009, 2, 2429–2466. [Google Scholar] [CrossRef] [Green Version]
- Svec, F. Porous polymer monoliths: Amazingly wide variety of techniques enabling their preparation. J. Chromatogr. A 2010, 1217, 902–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patrushev, Y.; Yudina, Y.; Sidelnikov, V. Monolithic rod columns for HPLC based on divinylbenzene-styrene copolymer with 1-vinylimidazole and 4-vinylpyridine. J. Liq. Chromatogr. Relat. Technol. 2018, 41, 458–466. [Google Scholar] [CrossRef]
- Andjelković, U.; Tufegdžić, S.; Popović, M. Use of monolithic supports for high-throughput protein and peptide separation in proteomics. Electrophoresis 2017, 38, 2851–2869. [Google Scholar] [CrossRef] [PubMed]
- Rathnasekara, R.; Khadka, S.; Jonnada, M.; El Rassi, Z. Polar and nonpolar organic polymer-based monolithic columns for capillary electrochromatography and high-performance liquid chromatography. Electrophoresis 2016, 38, 60–79. [Google Scholar] [CrossRef] [Green Version]
- Kurganov, A.A. Monolithic column in gas chromatography. Anal. Chim. Acta 2013, 775, 25–40. [Google Scholar] [CrossRef]
- Wang, R.; Li, W.; Chen, Z. Solid phase microextraction with poly(deep eutectic solvent) monolithic column online coupled to HPLC for determination of non-steroidal anti-inflammatory drugs. Anal. Chim. Acta 2018, 1018, 111–118. [Google Scholar] [CrossRef]
- Krenkova, J.; Lacher, N.A.; Svec, F. Highly Efficient Enzyme Reactors Containing Trypsin and Endoproteinase LysC Immobilized on Porous Polymer Monolith Coupled to MS Suitable for Analysis of Antibodies. Anal. Chem. 2009, 81, 2004–2012. [Google Scholar] [CrossRef]
- Liu, X.; Ouyang, C.; Zhao, R.; Shangguan, D.; Chen, Y.; Liu, G. Monolithic molecularly imprinted polymer for sulfamethoxazole and molecular recognition properties in aqueous mobile phase. Anal. Chim. Acta 2006, 571, 235–241. [Google Scholar] [CrossRef]
- Zhu, J.; Chen, D.; Ai, Y.; Dang, X.; Huang, J. A dummy molecularly imprinted monolith for selective solid-phase microextraction of vanillin and methyl vanillin prior to their determination by HPLC. Microchim. Acta 2017, 184, 1161–1167. [Google Scholar] [CrossRef]
- Antipchik, M.; Dzhuzha, A.; Sirotov, V.; Tennikova, T.; Korzhikova-Vlakh, E. Molecularly imprinted macroporous polymer monolithic layers for L-phenylalanine recognition in complex biological fluids. J. Appl. Polym. Sci. 2020. [Google Scholar] [CrossRef]
- Monolithic Materials: Preparation, Properties and Applications; Elsevier BV: Amsterdam, The Netherlands, 2003.
- Blow, D.M. Structure and mechanism of chymotrypsin. Acc. Chem. Res. 1976, 9, 145–152. [Google Scholar] [CrossRef]
- Gráf, L.A.; Szilágyi, L.A.; Venekei, I.A. Chymotrypsin. In Handbook of Proteolytic Enzym; Elsevier Ltd.: Oxford, UK, 2013; pp. 2626–2633. [Google Scholar]
- Vlakh, E.G.; Korzhikov, A.V.; Hubina, A.V.; Tennikova, T.B. Molecular imprinting: A tool of modern chemistry for the preparation of highly selective monolithic sorbents. Russ. Chem. Rev. 2015, 84, 952–980. [Google Scholar] [CrossRef]
- Xie, S.; Svec, F. Design of reactive porous polymer supports for high throughput bioreactors: Poly(2-vinyl-4,4-dimethylazlactone-co-acrylamide-co-ethylene dimethacrylate) monoliths. Biotechnol. Bioeng. 1999, 62, 30–35. [Google Scholar] [CrossRef]
- Ponomareva, E.A.; Volokitina, M.V.; Vinokhodov, D.O.; Korzhikova-Vlakh, E.; Tennikova, T. Biocatalytic reactors based on ribonuclease A immobilized on macroporous monolithic supports. Anal. Bioanal. Chem. 2012, 405, 2195–2206. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Biochemistry, 6th ed.; W.H. Freeman: New York, NY, USA, 2006; pp. 241–260. [Google Scholar]
- Volokitina, M.V.; Vlakh, E.G.; Platonova, G.A.; Vinokhodov, D.O.; Tennikova, T. Polymer monoliths as efficient solid phases for enzymatic polynucleotide degradation followed by fast HPLC analysis. J. Sep. Sci. 2013, 36, 2793–2805. [Google Scholar] [CrossRef]
- Enzyme Biocatalysis: Principles and Applications. Focus Catal. 2009, 2009, 8. [CrossRef]
- Bayramoglu, G.; Salih, B.; Arica, M.Y. Catalytic Activity of Immobilized Chymotrypsin on Hybrid Silica-Magnetic Biocompatible Particles and Its Application in Peptide Synthesis. Appl. Biochem. Biotechnol. 2019, 190, 1224–1241. [Google Scholar] [CrossRef]
- Yilmaz, V.; Yilmaz, H.; Arslan, Z.; Leszczynski, J. Novel Imprinted Polymer for the Preconcentration of Cadmium with Determination by Inductively Coupled Plasma Mass Spectrometry. Anal. Lett. 2016, 50, 482–499. [Google Scholar] [CrossRef] [Green Version]
- Vlakh, E.G.; Maksimova, E.F.; Tennikova, T.B. Monolithic polymeric sorbents for high-performance chromatography of synthetic polymers. Polym. Sci. Ser. B 2013, 55, 55–62. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Amount of Components, mmol | Grafting Time, h | Amount of Grafted MA-His **, μmol | |||||
|---|---|---|---|---|---|---|---|---|
| CoCl2 | MA-His | MAA | HEMA | EDMA | Per 100 mg of Copolymer | Per Column | ||
| 1 | 0.38 | 0.38 | 0.38 | 0.38 | 0.38 | 15 | 1.5 ± 0.1 | 4.6 |
| 2 * | 0.38 | 0.38 | 0.38 | 0.38 | 1.14 | 5 | – | − |
| 3 * | 0.38 | 0.38 | 0.38 | 0.38 | 1.14 | 8 | 2.1 ± 0.2 | 5.9 |
| Column * | Amount of Components, mmol | Amount of Catalytic Centers **, μmol | ||||||
|---|---|---|---|---|---|---|---|---|
| CoCl2 | Template | MA-His | MA-Asp | MA-Ser | EDMA | Per 100 mg of Copolymer | Per Column | |
| MIC | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.3 | 1.2 ± 0.3 | 2.6 |
| NIC | 0.1 | – | 0.1 | 0.1 | 0.1 | 0.3 | 0.7 ± 0.1 | 1.4 |
| Control | – | – | 0.1 | 0.1 | 0.1 | 0.3 | 0.3 ± 0.1 | 0.7 |
| Catalytic Column | Flow Rate, mL·min−1 | Lineweaver–Burk Method | Hanes Method | ||
|---|---|---|---|---|---|
| KM, mM | Vmax, µmol·L−1·min−1 | KM, mM | Vmax, µmol·L−1·min−1 | ||
| IME | 0.5 | 17 | 92 | 26 | 132 |
| MIC | 0.5 | 37 | 132 | 27 | 106 |
| NIC | 0.5 | 35 | 139 | 37 | 144 |
| MIC | 1.5 | 65 | 238 | 46 | 179 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stepanova, M.; Solomakha, O.; Ten, D.; Tennikova, T.; Korzhikova-Vlakh, E. Flow-Through Macroporous Polymer Monoliths Containing Artificial Catalytic Centers Mimicking Chymotrypsin Active Site. Catalysts 2020, 10, 1395. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10121395
Stepanova M, Solomakha O, Ten D, Tennikova T, Korzhikova-Vlakh E. Flow-Through Macroporous Polymer Monoliths Containing Artificial Catalytic Centers Mimicking Chymotrypsin Active Site. Catalysts. 2020; 10(12):1395. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10121395
Chicago/Turabian StyleStepanova, Mariia, Olga Solomakha, Daria Ten, Tatiana Tennikova, and Evgenia Korzhikova-Vlakh. 2020. "Flow-Through Macroporous Polymer Monoliths Containing Artificial Catalytic Centers Mimicking Chymotrypsin Active Site" Catalysts 10, no. 12: 1395. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10121395