Mechanistic Insights for Dry Reforming of Methane on Cu/Ni Bimetallic Catalysts: DFT-Assisted Microkinetic Analysis for Coke Resistance

 ,
, 
 ,
,
Abstract
:1. Introduction
2. Results and Discussion
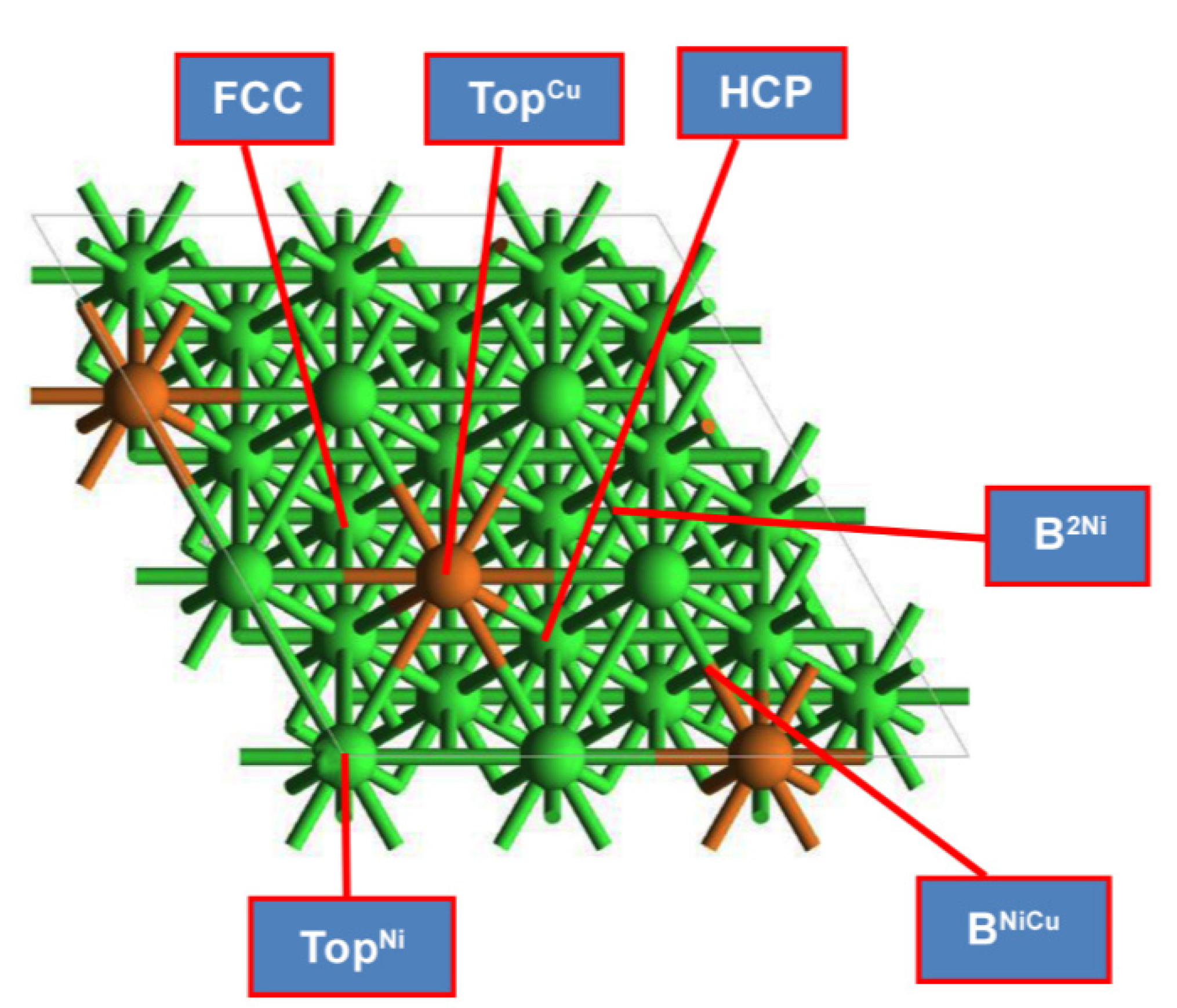
2.1. Adsorption Geometries and Energies on Ni2Cu Overlayer of Ni (111) Surface
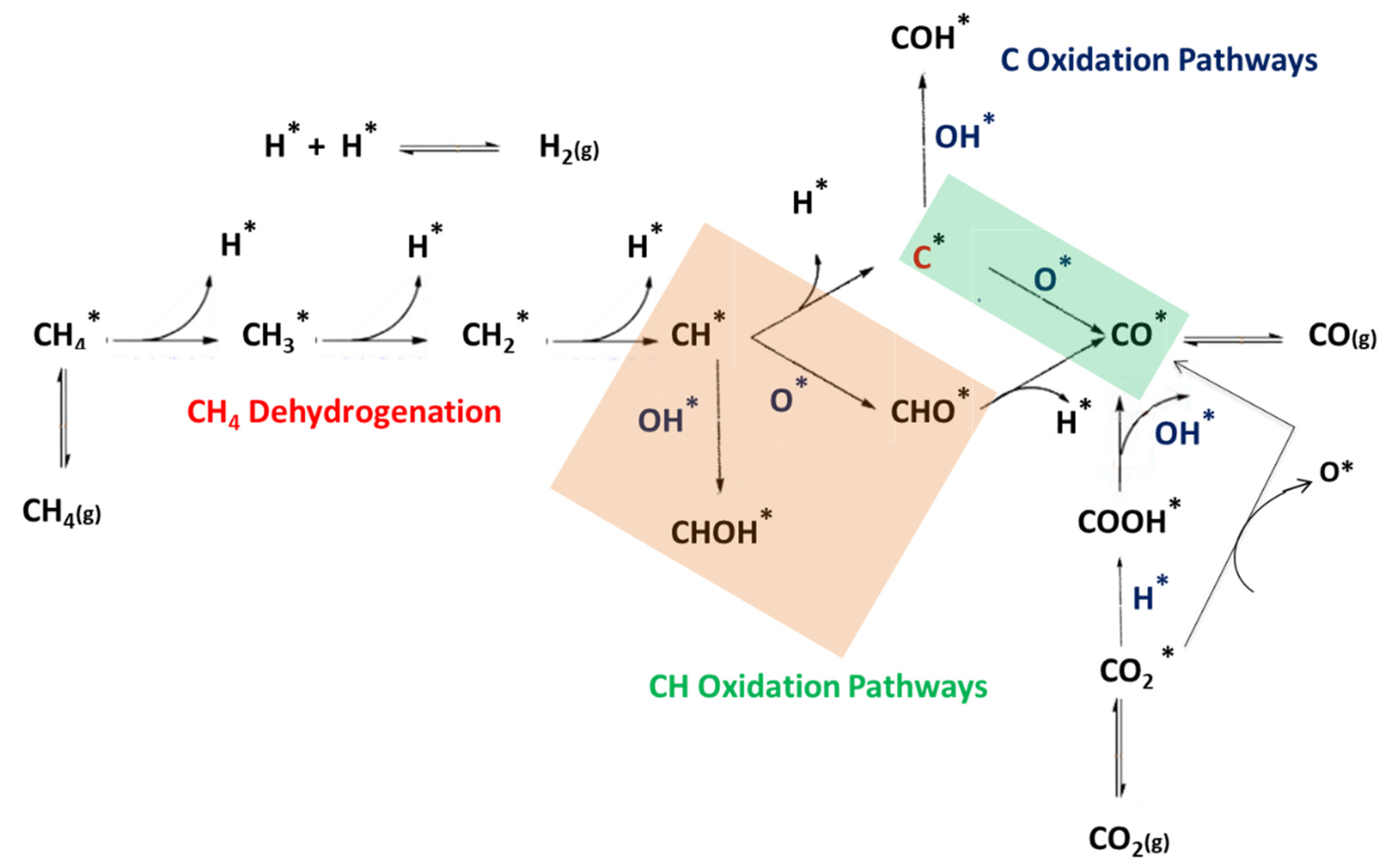
2.2. DRM Reaction Mechanism
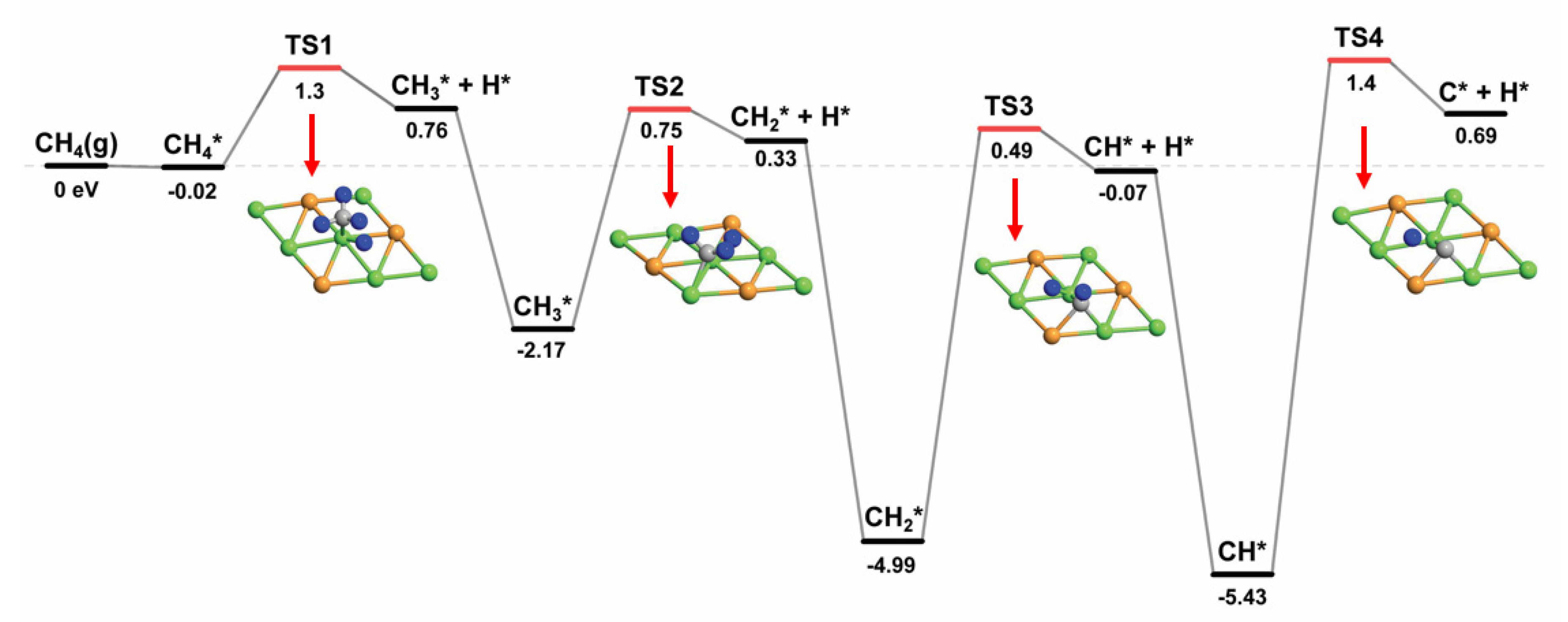
2.2.1. CHx Dissociation (x = 1–4)
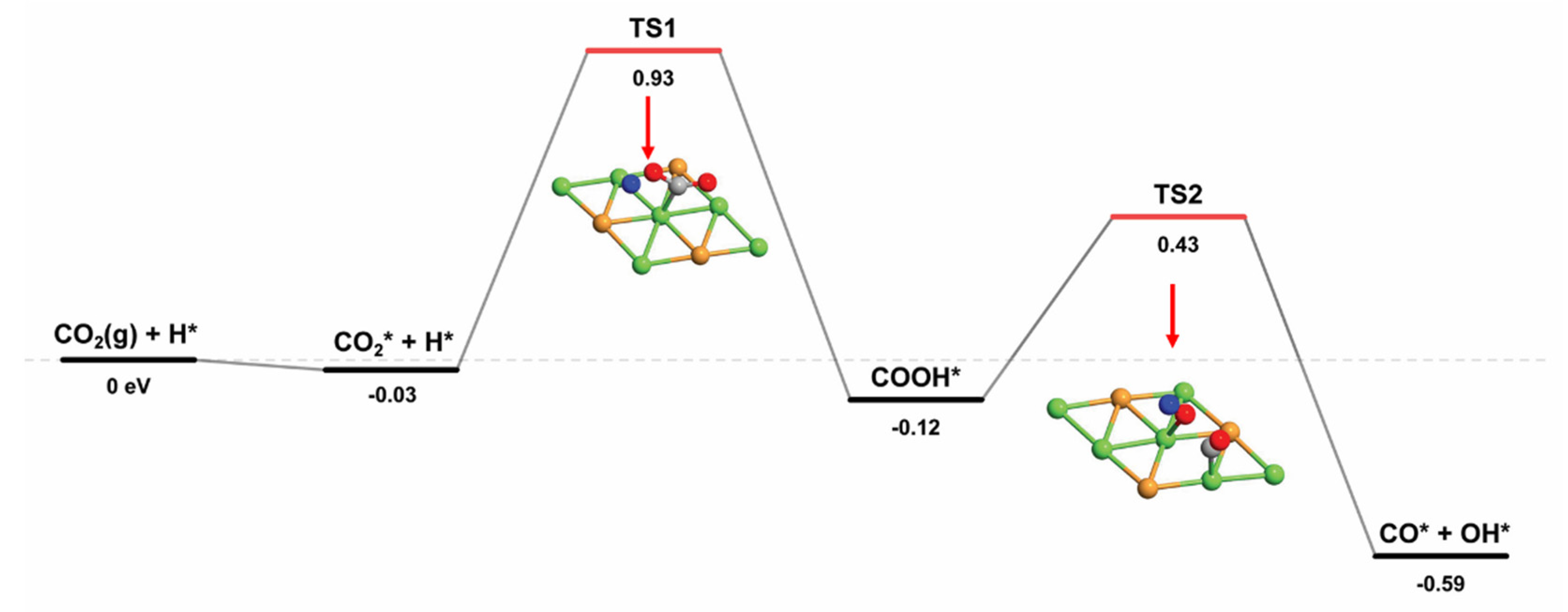
2.2.2. Two Reaction Pathways for CO2 Dissociation
2.2.3. Oxidation of C and CH
2.2.4. Carbon elimination by C + O and C + OH
2.2.5. CH + O and CH + OH Reactions
2.2.6. CHOH and COH Decomposition
2.2.7. H2 and H2O Formation
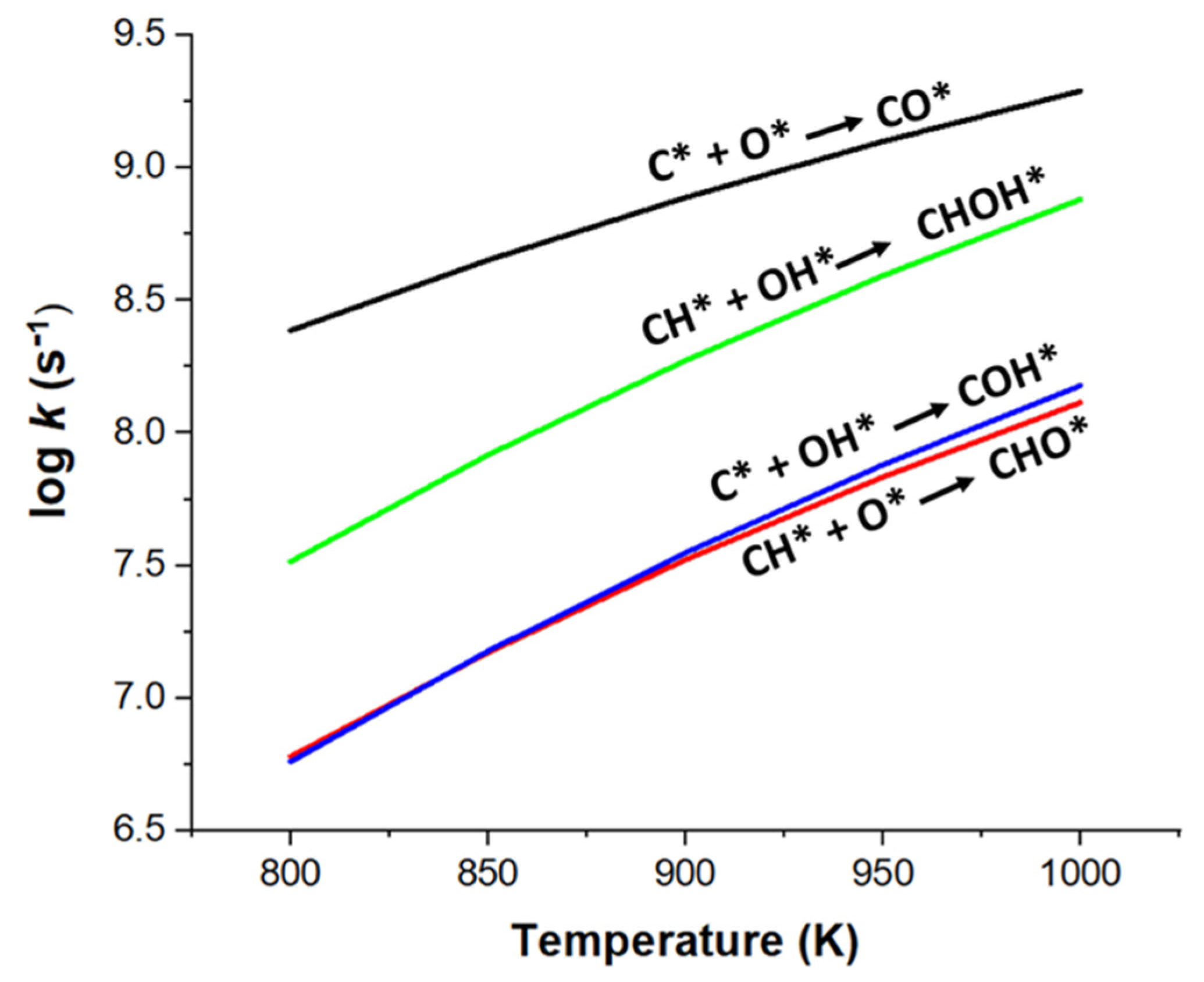
2.3. Effect of Temperature on Carbon Deposition Resistance
2.4. Dominant Reaction Pathways and Rate-Limiting Steps
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Uykun Mangaloğlu, D.; Baranak, M.; Ataç, Ö.; Atakül, H. Effect of the promoter presence in catalysts on the compositions of Fischer–Tropsch synthesis products. J. Ind. Eng. Chem. 2018, 66, 298–310. [Google Scholar] [CrossRef]
- Arora, S.; Prasad, R. An overview on dry reforming of methane: Strategies to reduce carbonaceous deactivation of catalysts. RSC Adv. 2016, 6, 108668–108688. [Google Scholar] [CrossRef]
- Challiwala, M.S.; Wilhite, B.A.; Ghouri, M.M.; Elbashir, N.O. Multidimensional modeling of a microfibrous entrapped cobalt catalyst Fischer-Tropsch reactor bed. AIChE J. 2018, 64, 1723–1731. [Google Scholar] [CrossRef]
- Gu, B.; Khodakov, A.Y.; Ordomsky, V.V. Selectivity shift from paraffins to α-olefins in low temperature Fischer–Tropsch synthesis in the presence of carboxylic acids. Chem. Commun. 2018, 54, 2345–2348. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Dhir, A.; Mohapatra, S.K.; Mahla, S.K. Dry reforming of methane using various catalysts in the process: Review. Biomass Convers. Biorefin. 2020, 10, 567–587. [Google Scholar] [CrossRef]
- Challiwala, M.S.; Afzal, S.; Choudhury, H.A.; Sengupa, D.; El-Halwagi, M.M.; Elbashir, N.O. Alternative via Pathways Dry Reforming for CO of 2 Utilization Methane. In Advances in Carbon Management Technologies Volume 1: Carbon Removal, Renewable and Nuclear Energy; CRC Press: Boca Raton, FL, USA, 2020; p. 253. Available online: https://www.taylorfrancis.com/books/9780429243608/chapters/10.1201/9780429243608-14 (accessed on 10 September 2020).
- Wei, J.; Iglesia, E. Isotopic and kinetic assessment of the mechanism of reactions of CH4 with CO2 or H2O to form synthesis gas and carbon on nickel catalysts. J. Catal. 2004, 224, 370–383. [Google Scholar] [CrossRef]
- Niu, J.; Ran, J.; Chen, D. Understanding the mechanism of CO2 reforming of methane to syngas on Ni@Pt surface compared with Ni(111) and Pt(111). Appl. Surf. Sci. 2020, 513, 145840. [Google Scholar] [CrossRef]
- Takami, D.; Yamamoto, A.; Yoshida, H. Dry reforming of methane over alumina-supported rhodium catalysts at low temperatures under visible and near-infrared light. Catal. Sci. Technol. 2020, 10, 5811–5814. [Google Scholar] [CrossRef]
- Zhu, Y.; Chen, K.; Barat, R.; Mitra, S. Dry reforming of methane over ruthenium/carbon nanotube catalyst. Chem. Eng. 2020, 4, 16. [Google Scholar] [CrossRef] [Green Version]
- Maina, S.C.P.; Ballarini, A.D.; Vilella, J.I.; de Miguel, S.R. Study of the performance and stability in the dry reforming of methane of doped alumina supported iridium catalysts. Catal. Today 2020, 344, 129–142. [Google Scholar] [CrossRef]
- Leba, A.; Yıldırım, R. Determining most effective structural form of nickel-cobalt catalysts for dry reforming of methane. Int. J. Hydro. Energy 2020, 45, 4268–4283. [Google Scholar] [CrossRef]
- Pakhare, D.; Spivey, J. A review of dry (CO2) reforming of methane over noble metal catalysts. Chem. Soc. Rev. 2014, 43, 7813–7837. [Google Scholar] [CrossRef]
- Kawi, S.; Kathiraser, Y.; Ni, J.; Oemar, U.; Li, Z.W.; Saw, E.T. Progress in synthesis of highly active and stable nickel-based catalysts for carbon dioxide reforming of methane. Chemsuschem. 2015, 8, 3556–3575. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yao, L.; Wang, S.H.; Mao, D.H.; Hu, C.W. Low-temperature catalytic CO2 dry reforming of methane on Ni-based catalysts: A review. Fuel Process. Technol. 2018, 169, 199–206. [Google Scholar] [CrossRef]
- Ren, J.; Qin, X.; Yang, J.Z.; Qin, Z.F.; Guo, H.L.; Lin, J.Y.; Li, Z. Methanation of carbon dioxide over Ni-M/ZrO2 (M = Fe, Co, Cu) catalysts: Effect of addition of a second metal. Fuel Process. Technol. 2015, 137, 204–211. [Google Scholar] [CrossRef]
- Bian, Z.F.; Das, S.; Wai, M.H.; Hongmanorom, P.; Kawi, S. A review on bimetallic nickel-based catalysts for CO2 reforming of methane. Chemphyschem 2017, 18, 3117–3134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.Y.; Fu, Z.M.; Yang, Z.X. The carbon-tolerance mechanism of Ni-based alloy with coinage metals. Phys. Lett. A 2013, 377, 2189–2194. [Google Scholar] [CrossRef]
- Xu, L.-L.; Wen, H.; Jin, X.; Bing, Q.-M.; Liu, J.-Y. DFT study on dry reforming of methane over Ni2Fe overlayer of Ni (1 1 1) surface. Appl. Surf. Sci. 2018, 443, 515–524. [Google Scholar] [CrossRef]
- Theofanidis, S.A.; Batchu, R.; Galvita, V.V.; Poelman, H.; Marin, G.B. Carbon gasification from Fe–Ni catalysts after methane dry reforming. Appl. Catal. B Environ. 2016, 185, 42–55. [Google Scholar] [CrossRef]
- Theofanidis, S.A.; Galvita, V.V.; Poelman, H.; Marin, G.B. Enhanced carbon-resistant dry reforming Fe–Ni catalyst: Role of Fe. ACS Catal. 2015, 5, 3028–3039. [Google Scholar] [CrossRef]
- Lo Faro, M.; Frontera, P.; Antonucci, P.; Arico, A.S. Ni-Cu based catalysts prepared by two different methods and their catalytic activity toward the ATR of methane. Chem. Eng. Res. Des. 2015, 93, 269–277. [Google Scholar] [CrossRef]
- Rahemi, N.; Haghighi, M.; Babaluo, A.A.; Allahyari, S.; Jafari, M.F. Syngas production from reforming of greenhouse gases CH4/CO2 over Ni–Cu/Al2O3 nanocatalyst: Impregnated vs. plasma-treated catalyst. Energ. Convers. Manag. 2014, 84, 50–59. [Google Scholar] [CrossRef]
- Wu, T.; Cai, W.Y.; Zhang, P.; Song, X.F.; Gao, L. Cu-Ni@ SiO2 alloy nanocomposites for methane dry reforming catalysis. RSC Adv. 2013, 3, 23976–23979. [Google Scholar] [CrossRef]
- Liu, H.Y.; Zhang, R.G.; Yan, R.X.; Li, J.R.; Wang, B.J.; Xie, K.C. Insight into CH4 dissociation on NiCu catalyst: A first-principles study. Appl. Surf. Sci. 2012, 258, 8177–8184. [Google Scholar] [CrossRef]
- An, W.; Zeng, X.C.; Turner, C.H. First-principles study of methane dehydrogenation on a bimetallic Cu/Ni (111) surface. J. Chem. Phys. 2009, 131, 174702. [Google Scholar] [CrossRef] [PubMed]
- Chatla, A.; Ghouri, M.M.; El Hassan, O.W.; Mohamed, N.; Prakash, A.V.; Elbashir, N.O. An experimental and first principles DFT investigation on the effect of Cu addition to Ni/Al2O3 catalyst for the dry reforming of methane. Appl. Catal. A-Gen. 2020, 602, 117699. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Cao, X.M.; Zhu, J.H.; Hu, P. Activity and coke formation of nickel and nickel carbide in dry reforming: A deactivation scheme from density functional theory. J. Catal. 2014, 311, 469–480. [Google Scholar] [CrossRef]
- Arevalo, R.L.; Aspera, S.M.; Escano, M.C.S.; Nakanishi, H.; Kasai, H. Tuning methane decomposition on stepped Ni surface: The role of subsurface atoms in catalyst design. Sci. Rep. 2017, 7, 13963. [Google Scholar] [CrossRef] [Green Version]
- Heiland, W. Charge-exchange processes and surface-chemistry. Surf. Sci. 1991, 251, 942–946. [Google Scholar] [CrossRef]
- Jang, W.J.; Shim, J.O.; Kim, H.M.; Yoo, S.Y.; Roh, H.S. A review on dry reforming of methane in aspect of catalytic properties. Catal. Today 2019, 324, 15–26. [Google Scholar] [CrossRef]
- Kim, S.M.; Abdala, P.M.; Margossian, T.; Hosseini, D.; Foppa, L.; Armutlulu, A.; van Beek, W.; Comas-Vives, A.; Copéret, C.; Müller, C. Cooperativity and dynamics increase the performance of NiFe dry reforming catalysts. J. Am. Chem. Soc. 2017, 139, 1937–1949. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Liu, Z.; Zhu, Y.-A.; Tong, G.; Zhang, J.; Engelbrekt, C.; Ulstrup, J.; Zhu, K.; Zhou, X. Tuning the composition of metastable CoxNiyMg100−x−y(OH)(OCH3) nanoplates for optimizing robust methane dry reforming catalyst. J. Catal. 2015, 330, 106–119. [Google Scholar] [CrossRef]
- Michaelides, A.; Hu, P. Methyl chemisorption on Ni (111) and C-H-M multicentre bonding: A density functional theory study. Surf. Sci. 1999, 437, 362–376. [Google Scholar] [CrossRef]
- Zhu, Y.-A.; Dai, Y.-C.; Chen, D.; Yuan, W.-K. First-principles calculations of CH4 dissociation on Ni (100) surface along different reaction pathways. J. Mol. Catal. A Chem. 2007, 264, 299–308. [Google Scholar] [CrossRef]
- Bradford, M.C.J.; Vannice, M.A. Catalytic reforming of methane with carbon dioxide over nickel catalysts II. Reaction kinetics. Appl. Catal. A-Gen. 1996, 142, 97–122. [Google Scholar] [CrossRef]
- Maslov, M.M.; Openov, L.A.; Podlivaev, A.I. On the vineyard formula for the pre-exponential factor in the Arrhenius law. Phys. Solid State 2014, 56, 1239–1244. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Jørgensen, M. Microkinetic Modeling of Nanoparticl Catalysis Using Density Functional Theory. Ph.D. Thesis, Chalmers University of Technology, Gothenburg, Sweden, 2017. [Google Scholar]
- Jørgensen, M.; Grönbeck, H. First-principles microkinetic modeling of methane oxidation over Pd (100) and Pd (111). ACS Catal. 2016, 6, 6730–6738. [Google Scholar] [CrossRef]
- Ray, D.; Ghosh, S.; Tiwari, A.K. Controlling heterogeneous catalysis of water dissociation using Cu-Ni bimetallic alloy surfaces: A quantum dynamics study. J. Phys. Chem. A 2018, 122, 5698–5709. [Google Scholar] [CrossRef]
- Zhang, R.G.; Guo, X.Q.; Wang, B.J.; Ling, L.M. Insight Into the effect of CuNi (111) and FeNi (111) surface structure and second metal composition on surface carbon elimination by O or OH: A comparison study with Ni (111) surface. J. Phys. Chem. C 2015, 119, 14135–14144. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Hammer, B.; Hansen, L.B.; Norskov, J.K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 1999, 59, 7413–7421. [Google Scholar] [CrossRef] [Green Version]
- Hammer, B.; Norskov, J.K. Theoretical surface science and catalysis—Calculations and concepts. Adv. Catal. 2000, 45, 71–129. [Google Scholar] [CrossRef]
- Methfessel, M.; Paxton, A.T. High-precision sampling for brillouin-zone integration in metals. Phys. Rev. B 1989, 40, 3616–3621. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.A.; Chen, D.; Zhou, X.G.; Yuan, W.K. DFT studies of dry reforming of methane on Ni catalyst. Catal. Today 2009, 148, 260–267. [Google Scholar] [CrossRef]
- Henkelman, G.; Jonsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.H.; Kang, U.; Park, H.; Abdel-Wahab, A.; Han, D.S. Computational density functional theory study on the selective conversion of CO2 to formate on homogeneously and heterogeneously mixed CuFeO2 and CuO surfaces. Catal. Today 2019, 335, 345–353. [Google Scholar] [CrossRef]
- Henkelman, G.; Jonsson, H. A dimer method for finding saddle points on high dimensional potential surfaces using only first derivatives. J. Chem. Phys. 1999, 111, 7010–7022. [Google Scholar] [CrossRef]
- Yoon, S.H.; Yu, C.; Han, A.; Park, H.; Elbashir, N.; Han, D.S. Computational characterization of nitrogen-doped carbon nanotube functionalized by Fe adatom and Fe substituent for oxygen reduction reaction. Appl. Surf. Sci. 2019, 485, 342–352. [Google Scholar] [CrossRef]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. Theoretical calculations of CH4 and H2 associative desorption from Ni (111): Could subsurface hydrogen play an important role? J. Chem. Phys. 2006, 124, 044706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, C.; Zhu, Y.A.; Yang, M.L.; Sui, Z.J.; Zhou, X.G.; Chen, D. Density functional theory-assisted microkinetic analysis of methane dry reforming on Ni catalyst. Ind. Eng. Chem. Res. 2015, 54, 5901–5913. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Eads (eV) | Favored Adsorption Site | |
|---|---|---|---|
| 1 | CH4* | −0.02 | N/A |
| 2 | CH3* | −2.17 | FCC |
| 3 | CH2* | −4.99 | FCC |
| 4 | CH* | −5.43 | FCC |
| 5 | C* | −7.04 | HCP |
| 6 | O* | −6.34 | FCC |
| 7 | OH* | −3.28 | B2Ni |
| 8 | CO* | −1.52 | B2Ni |
| 9 | COH* | −4.07 | B2Ni |
| 10 | CHOH* | −2.40 | B2Ni |
| 11 | CO2* | −0.03 | N/A |
| 12 | H* | −3.60 | FCC |
| 13 | H2* | −0.06 | N/A |
| 14 | CH3O* | −2.43 | B2Ni |
| 15 | CH2O | −0.04 | N/A |
| 16 | CH2OH* | −1.87 | TopNi |
| 17 | CH3OH* | −0.05 | N/A |
| 18 | CHO*a | −2.05 | TopNi |
| 19 | COH*a | −4.07 | B2Ni |
| 20 | H2O* | −0.06 | N/A |
| 21 | COOH* | −2.28 | TopNi |
| Reaction | Ea,f+ (eV) | Hf+ (eV) | Ea,r+ (eV) |
|---|---|---|---|
| CH4* → CH3* + H* | 1.30 | 0.78 | 0.52 |
| CH3* → CH2* + H* | 0.75 | 0.33 | 0.42 |
| CH2* → CH* + H* | 0.49 | −0.06 | 0.55 |
| CH* → C* + H* | 1.40 | 0.69 | 0.71 |
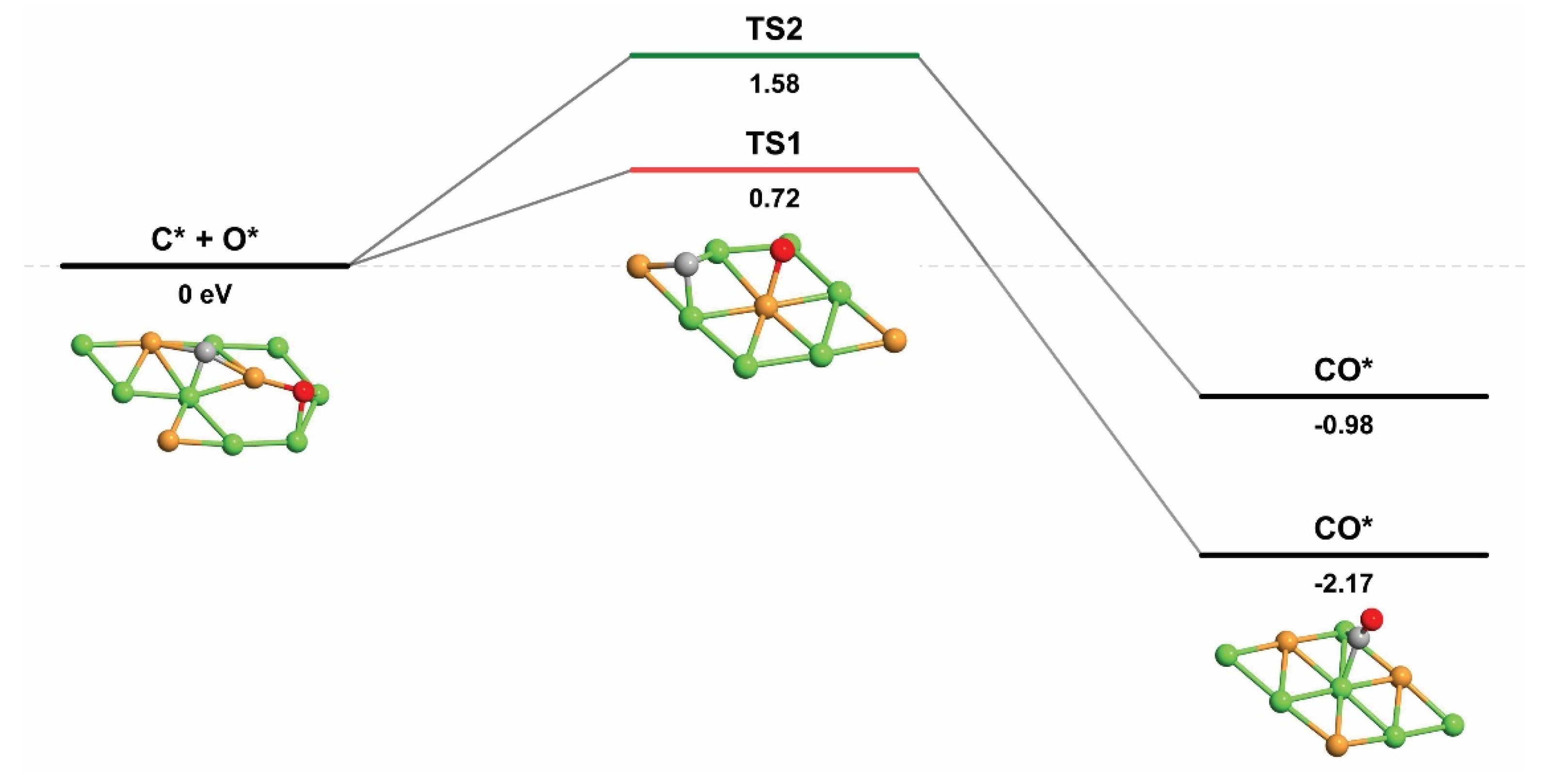
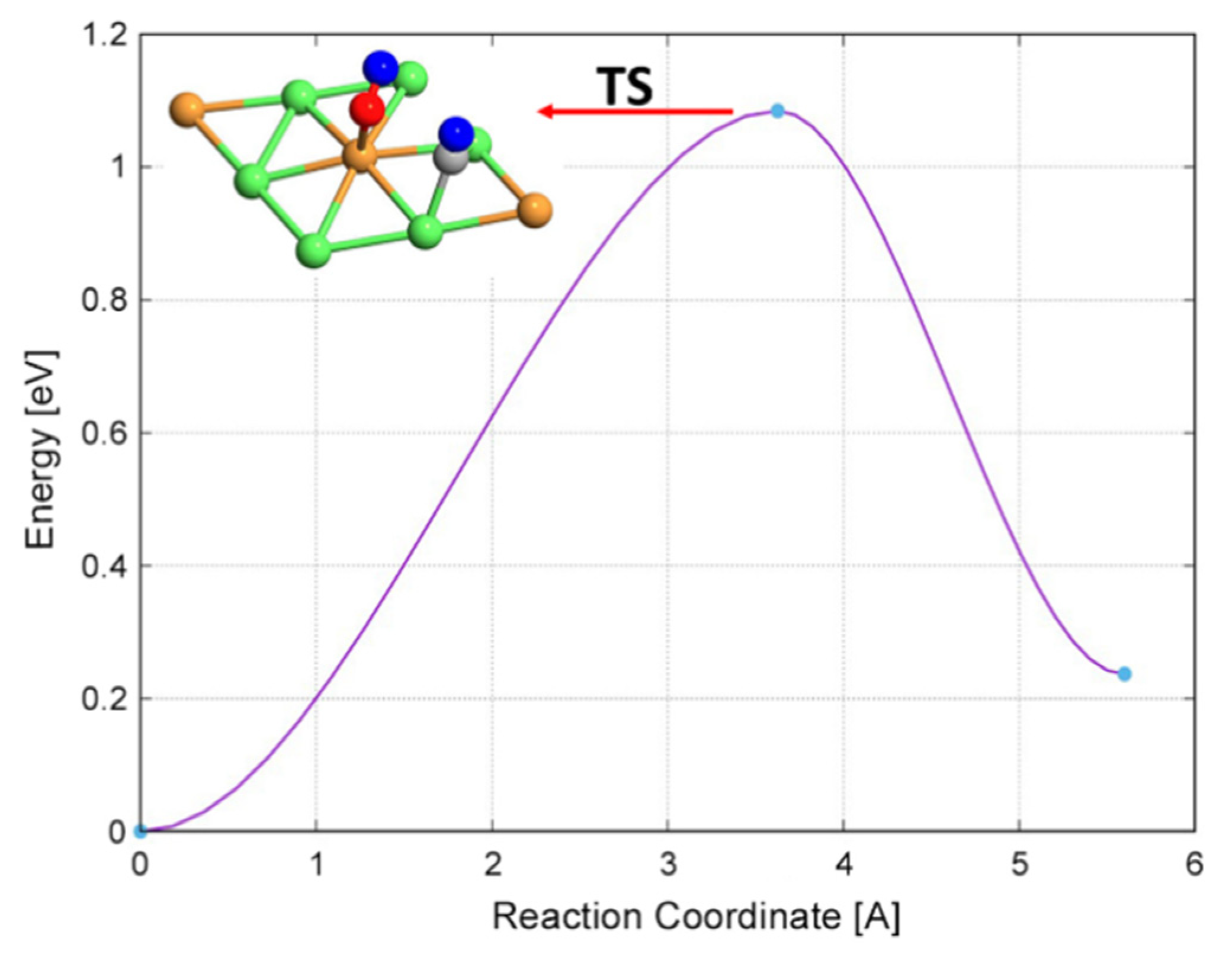
| C* + O* → CO* | 0.72 | −2.17 | 2.89 |
| CH* + O* → CHO* | 1.06 | −0.30 | 1.36 |
| CHO* → CO* + H* | 0.18 | −1.17 | 1.35 |
| C* + OH* → COH* | 1.13 | −0.95 | 2.07 |
| H* + H* → H2 | 0.73 | 0.57 | 0.16 |
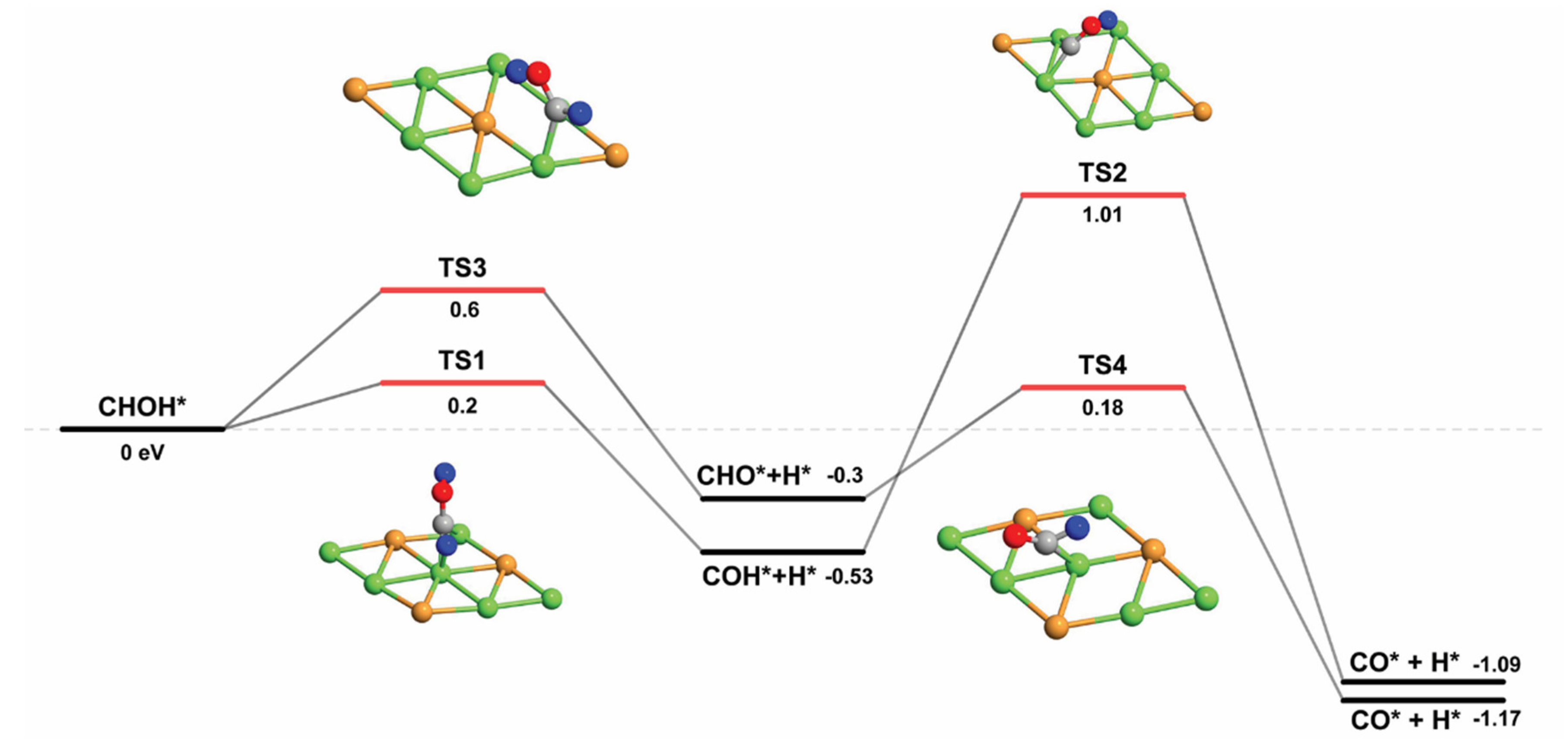
| CH* + OH* → CHOH* | 1.08 | 0.24 | 0.84 |
| CHOH* → CHO* + H* | 0.60 | −0.31 | 0.91 |
| CO2* + H* → COOH* | 0.93 | −0.12 | 1.05 |
| COOH* → CO* + OH* | 0.43 | −0.59 | 1.02 |
| O* + H* → OH* | 0.94 | −0.24 | 1.18 |
| H* + OH* → H2O* | 0.91 | −0.15 | 1.06 |
| CO2* → CO* + O* | 1.69 | 0.48 | 1.21 |
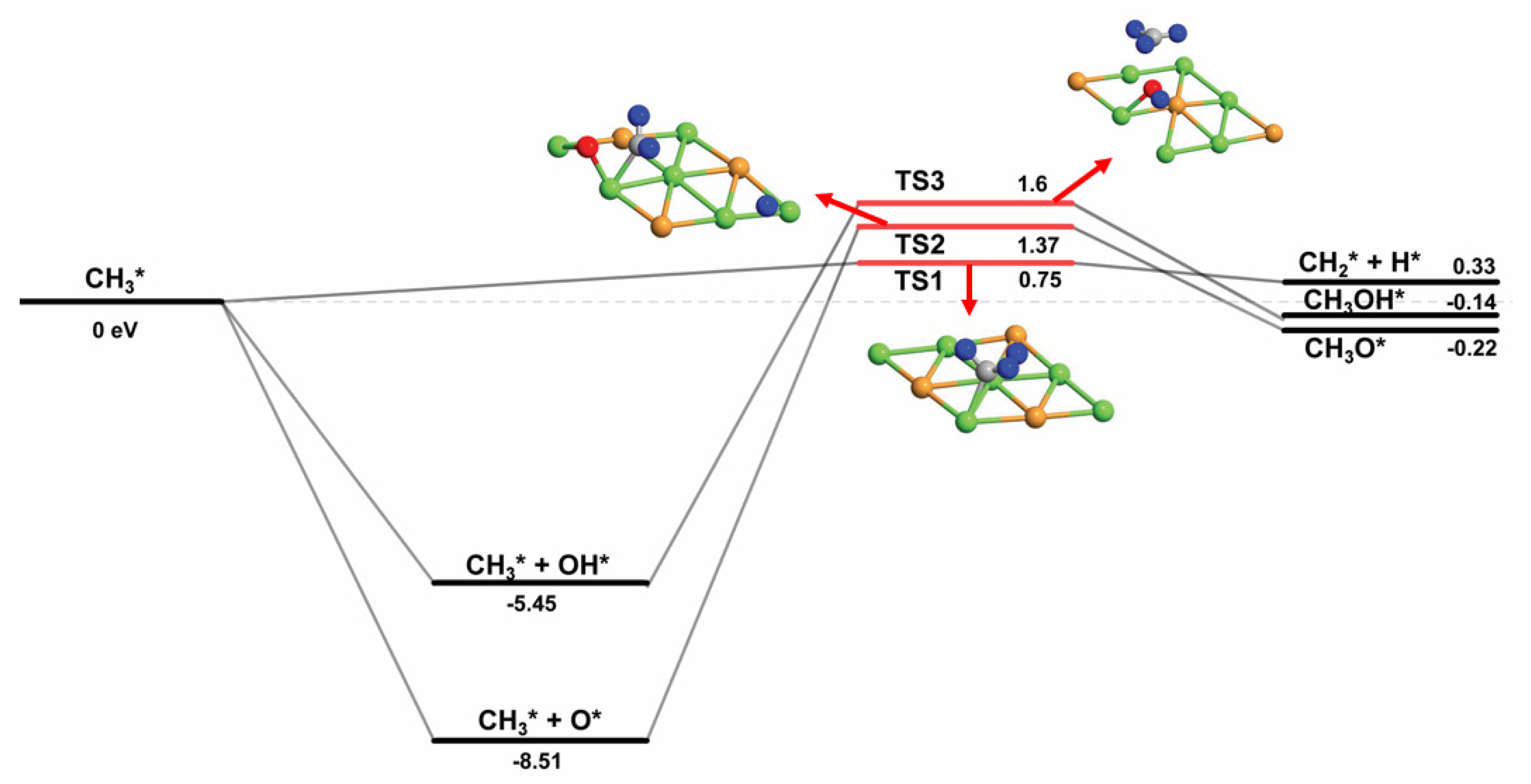
| CH3* + O* → CH3O* | 1.37 | −0.22 | 1.59 |
| CH2* + O* → CH2O* | 0.76 | −0.43 | 1.19 |
| CHOH* → COH* + H* | 0.20 | −0.53 | 0.73 |
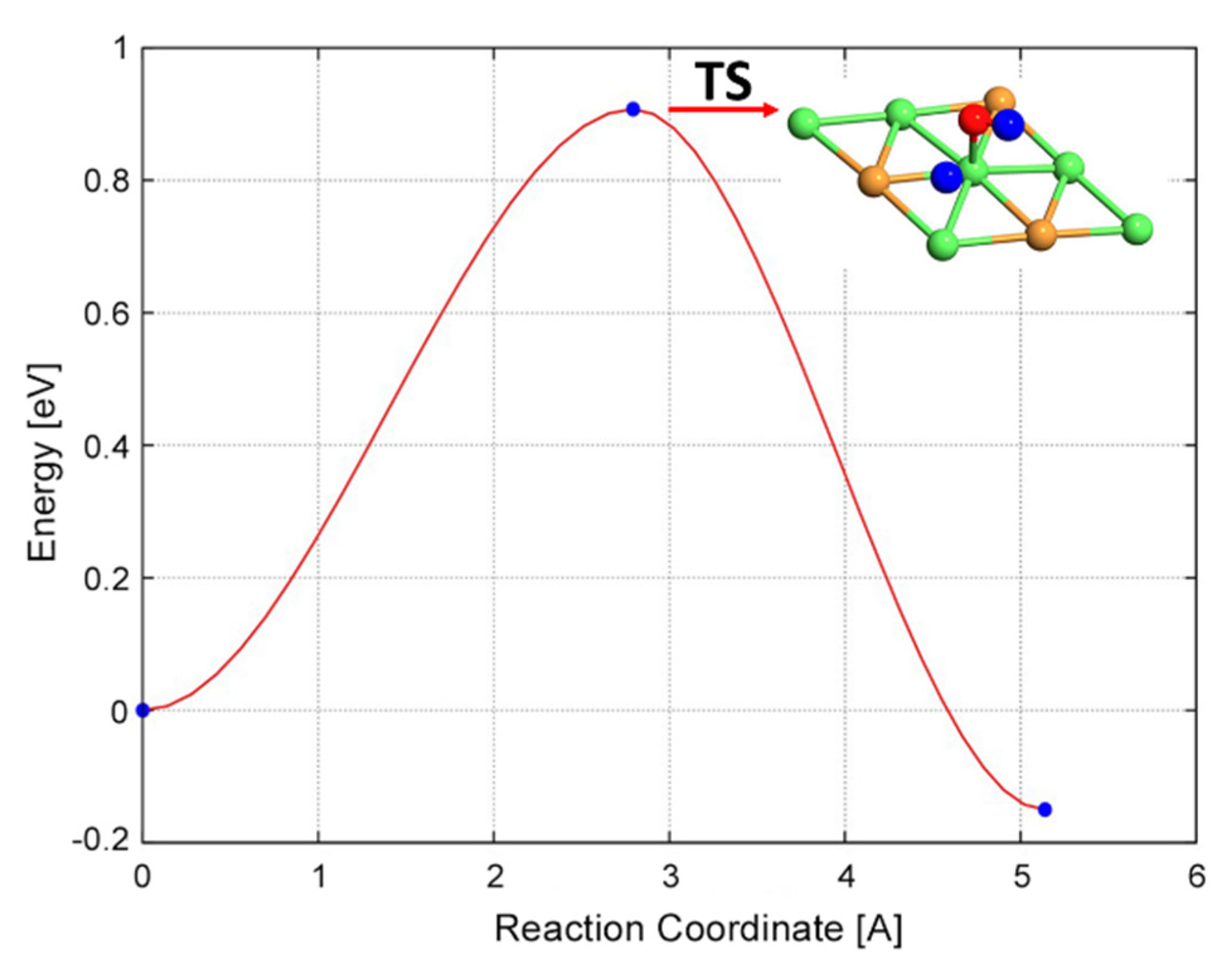
| COH* → CO* + H* | 1.01 | −1.09 | 2.10 |
| CH3* + OH* → CH3OH* | 1.60 | −0.14 | 1.74 |
| CH2* + OH* → CH2OH* | 0.78 | −0.10 | 0.88 |
| CH3O* → CH2O* + H* | 0.96 | 0.57 | 0.39 |
| CH2OH* → CH2O* + H* | 0.75 | −0.07 | 0.82 |
| CH2OH* → CHOH* + H* | 0.92 | 0.42 | 0.50 |
| CH3OH* → CH3O* + H* | 4.29 | 0.01 | 4.28 |
| CH3OH* → CH2OH* + H* | 2.46 | 1.39 | 1.10 |
| Reactions | 800 K | 850 K | 900 K | 950 K | 1000 K |
|---|---|---|---|---|---|
| CO* → C* + O* | 4.36 × 10−7 | 5.11 × 10−6 | 4.56 × 10−5 | 3.23 × 10−4 | 1.88 × 10−3 |
| CH* → C* + H* | 1.18 × 105 | 3.90 × 105 | 1.13 × 106 | 2.92 × 106 | 6.88 × 106 |
| C* + O* → CO* | 2.43 × 108 | 4.50 × 108 | 7.74 × 108 | 1.25 × 109 | 1.95 × 109 |
| C* + OH* → COH* | 5.75 × 106 | 1.50 × 107 | 3.53 × 107 | 7.59 × 107 | 1.51 × 108 |
| CH* + O* → CHO* | 6.01 × 106 | 1.49 × 107 | 3.32 × 107 | 6.83 × 108 | 1.31 × 108 |
| CH* + OH* → CHOH* | 3.28 × 107 | 8.27 × 107 | 1.88 × 108 | 3.93 × 108 | 7.62 × 108 |
| kCH/kC(O) | 0.02 | 0.03 | 0.04 | 0.05 | 0.07 |
| kCH/kC(OH) | 5.70 | 5.50 | 5.33 | 5.18 | 5.05 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Omran, A.; Yoon, S.H.; Khan, M.; Ghouri, M.; Chatla, A.; Elbashir, N. Mechanistic Insights for Dry Reforming of Methane on Cu/Ni Bimetallic Catalysts: DFT-Assisted Microkinetic Analysis for Coke Resistance. Catalysts 2020, 10, 1043. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10091043
Omran A, Yoon SH, Khan M, Ghouri M, Chatla A, Elbashir N. Mechanistic Insights for Dry Reforming of Methane on Cu/Ni Bimetallic Catalysts: DFT-Assisted Microkinetic Analysis for Coke Resistance. Catalysts. 2020; 10(9):1043. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10091043
Chicago/Turabian StyleOmran, Ahmed, Sun Hee Yoon, Murtaza Khan, Minhaj Ghouri, Anjaneyulu Chatla, and Nimir Elbashir. 2020. "Mechanistic Insights for Dry Reforming of Methane on Cu/Ni Bimetallic Catalysts: DFT-Assisted Microkinetic Analysis for Coke Resistance" Catalysts 10, no. 9: 1043. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10091043