Butane Isomerization as a Diagnostic Tool in the Rational Design of Solid Acid Catalysts

 , ,
, ,  ,
,
Abstract
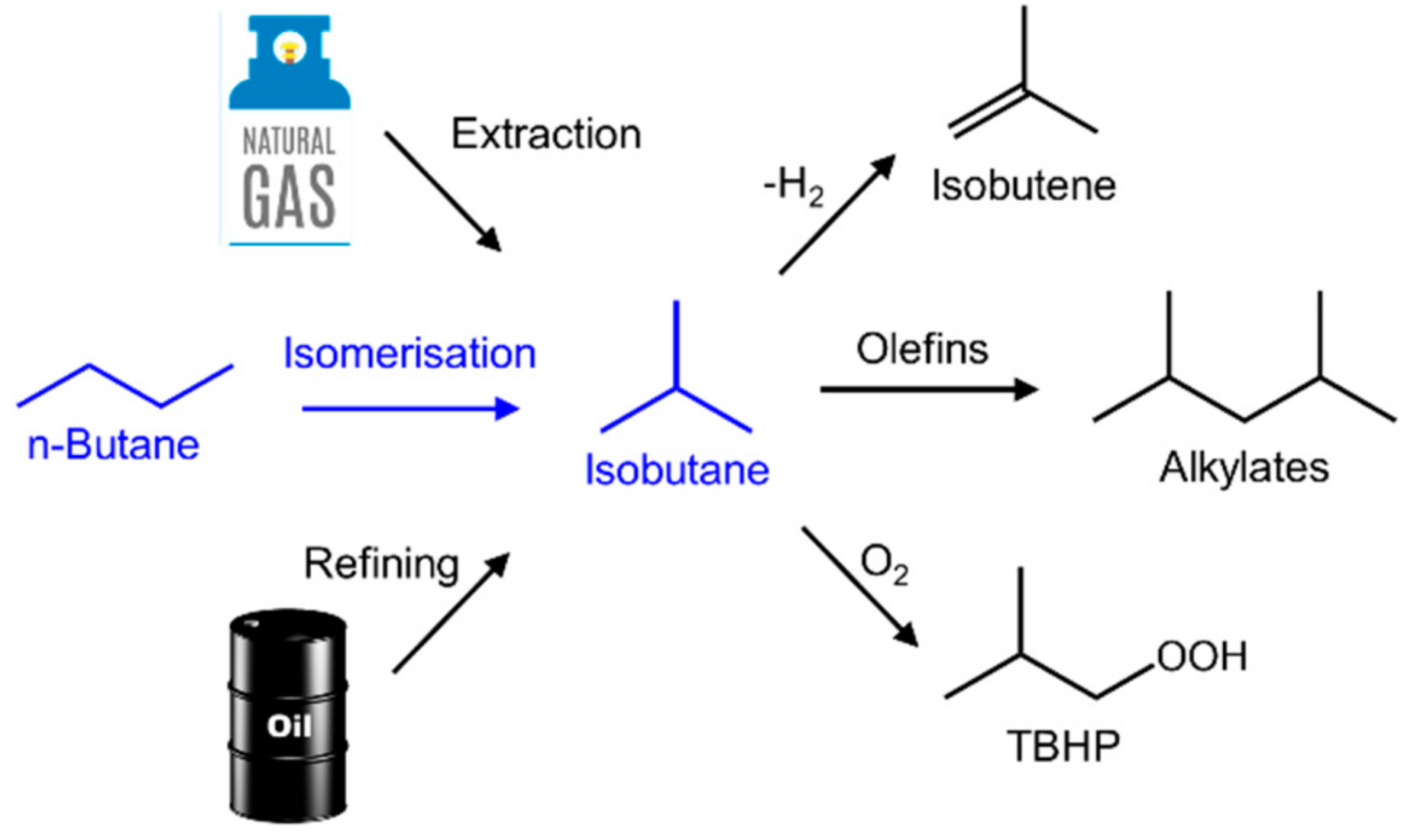
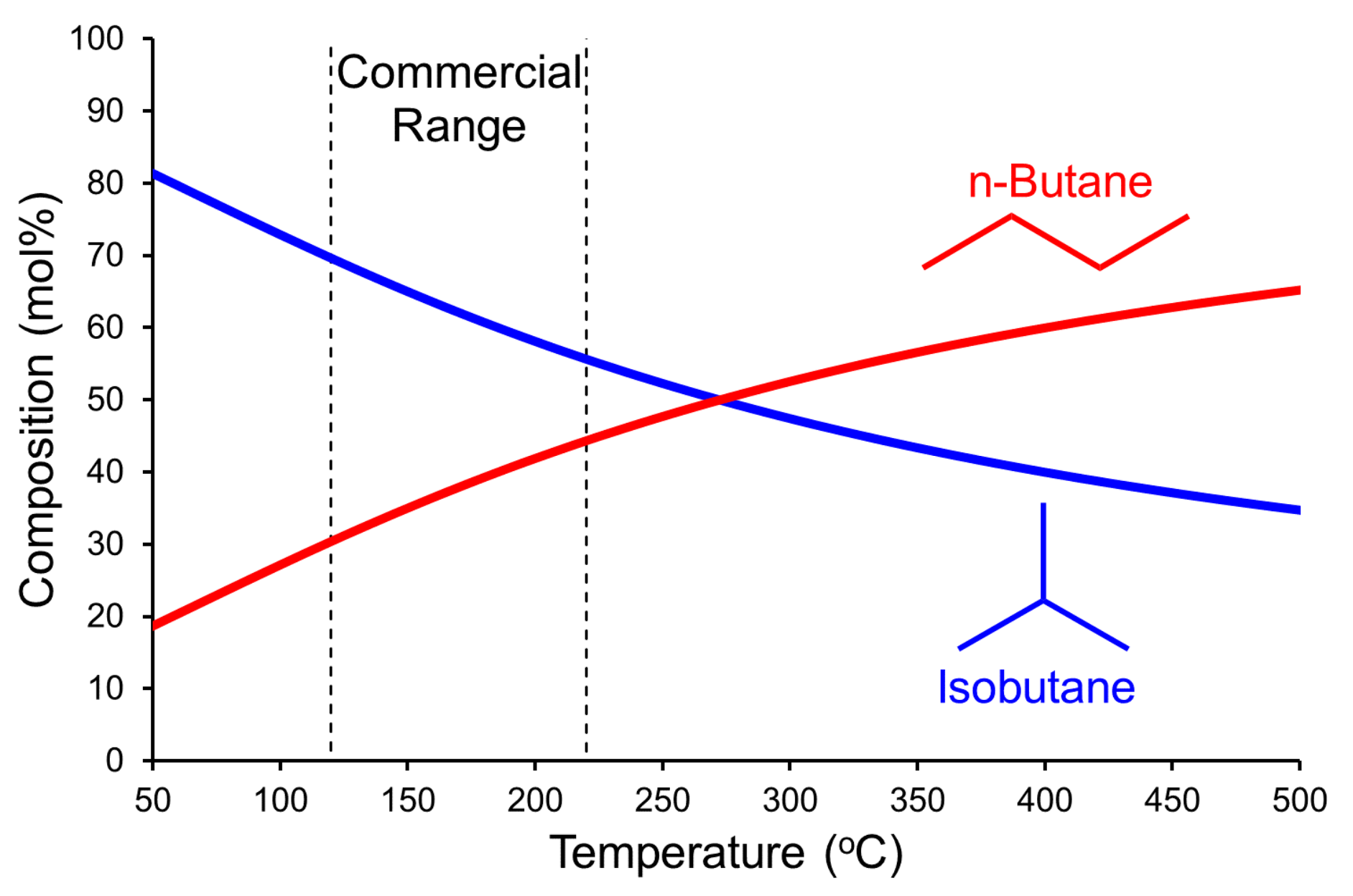
:1. Commercial and Fundamental Aspects of Butane Isomerization
2. Tailoring Microporous Species for Butane Isomerization
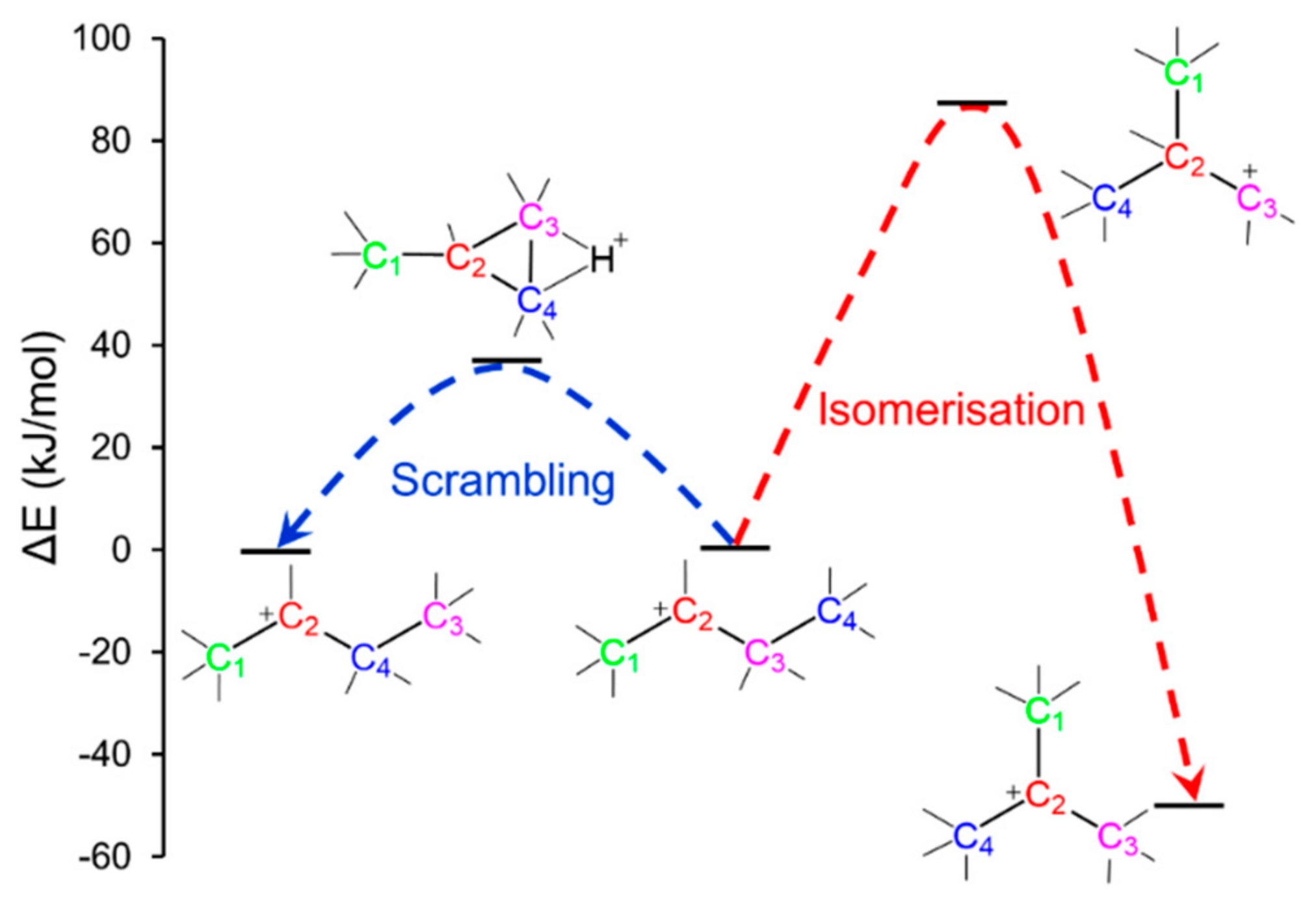
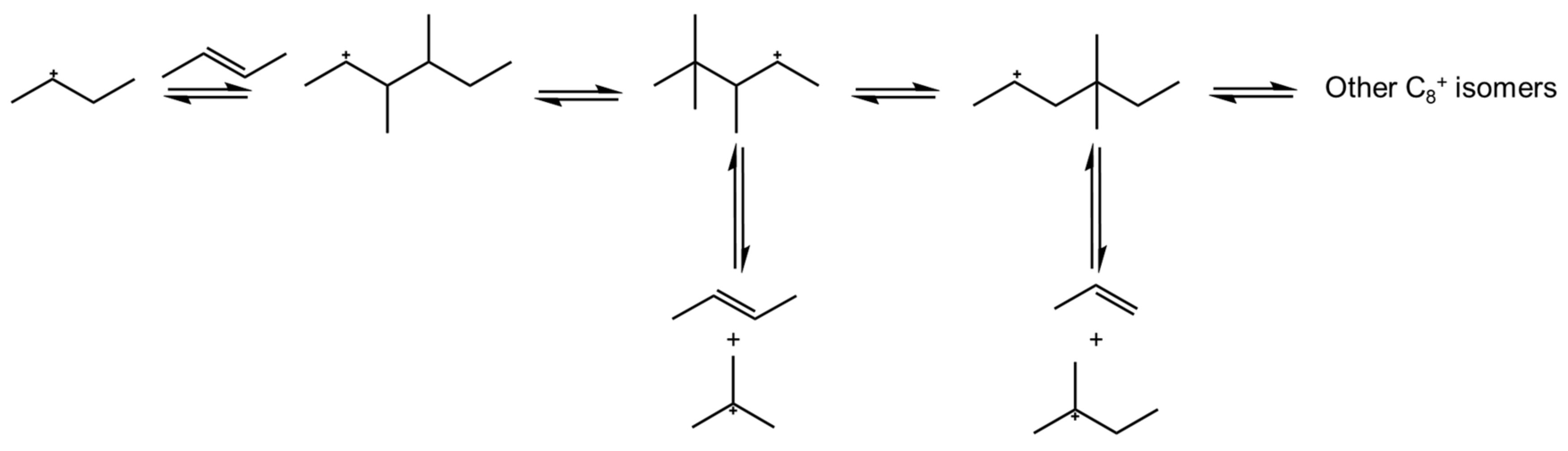
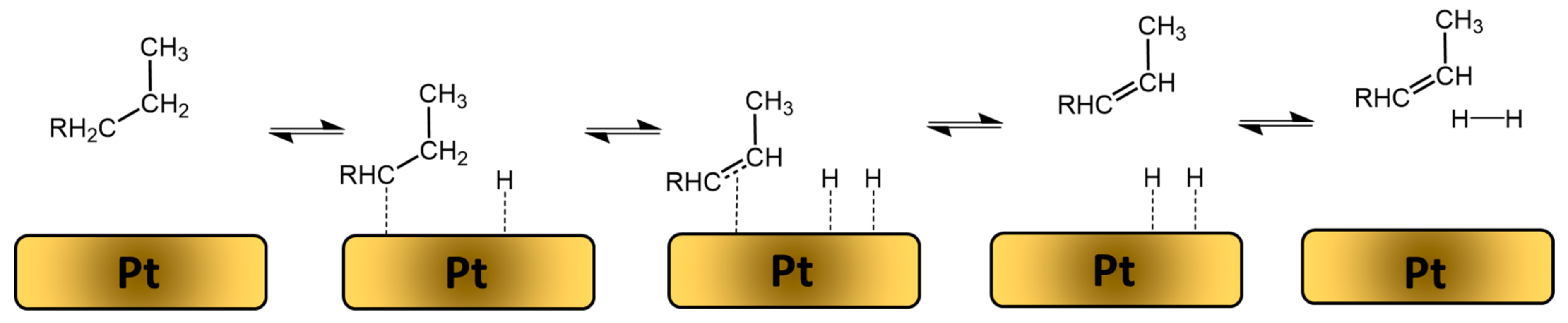
2.1. Main Principles
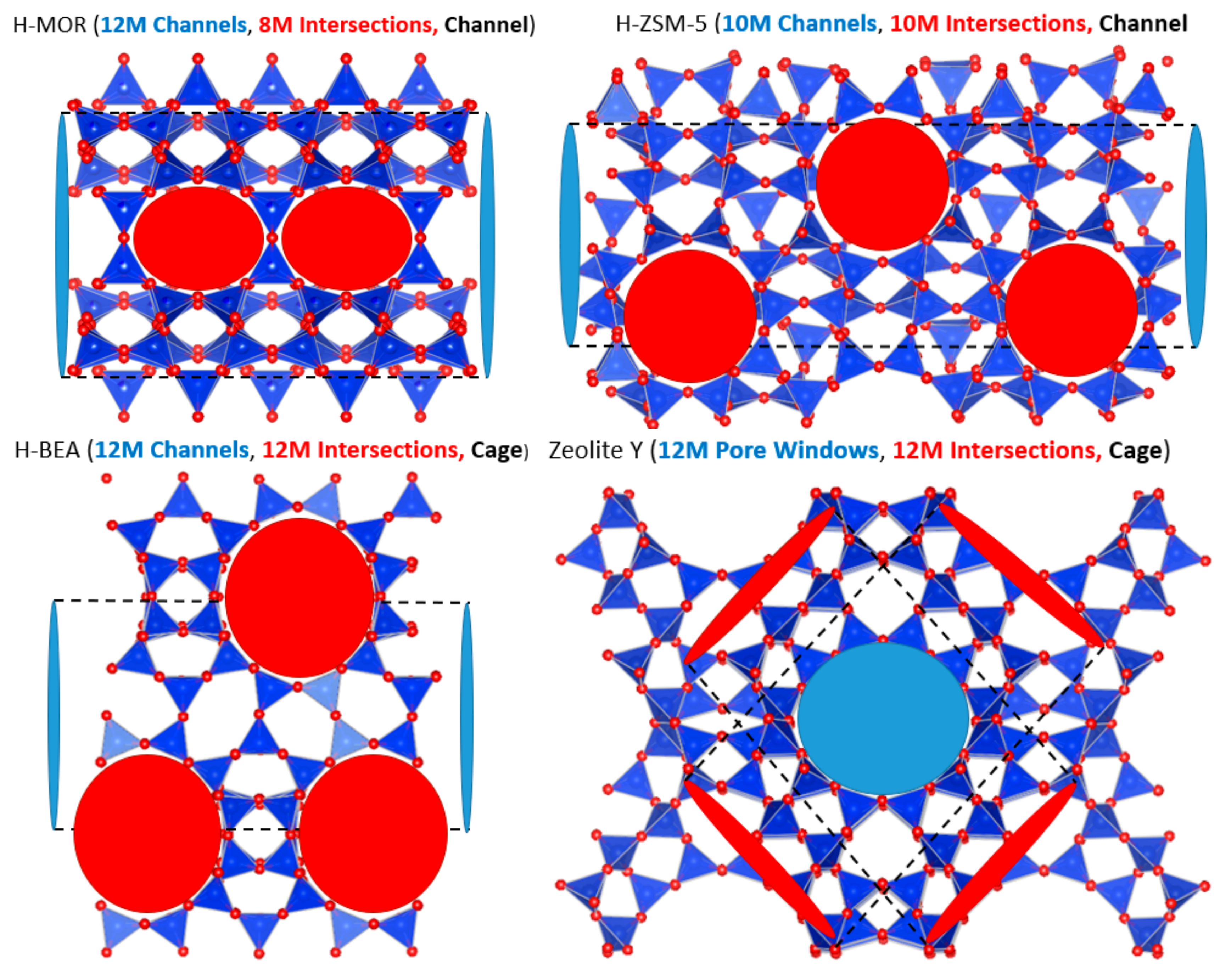
2.2. Influence of Framework Topology
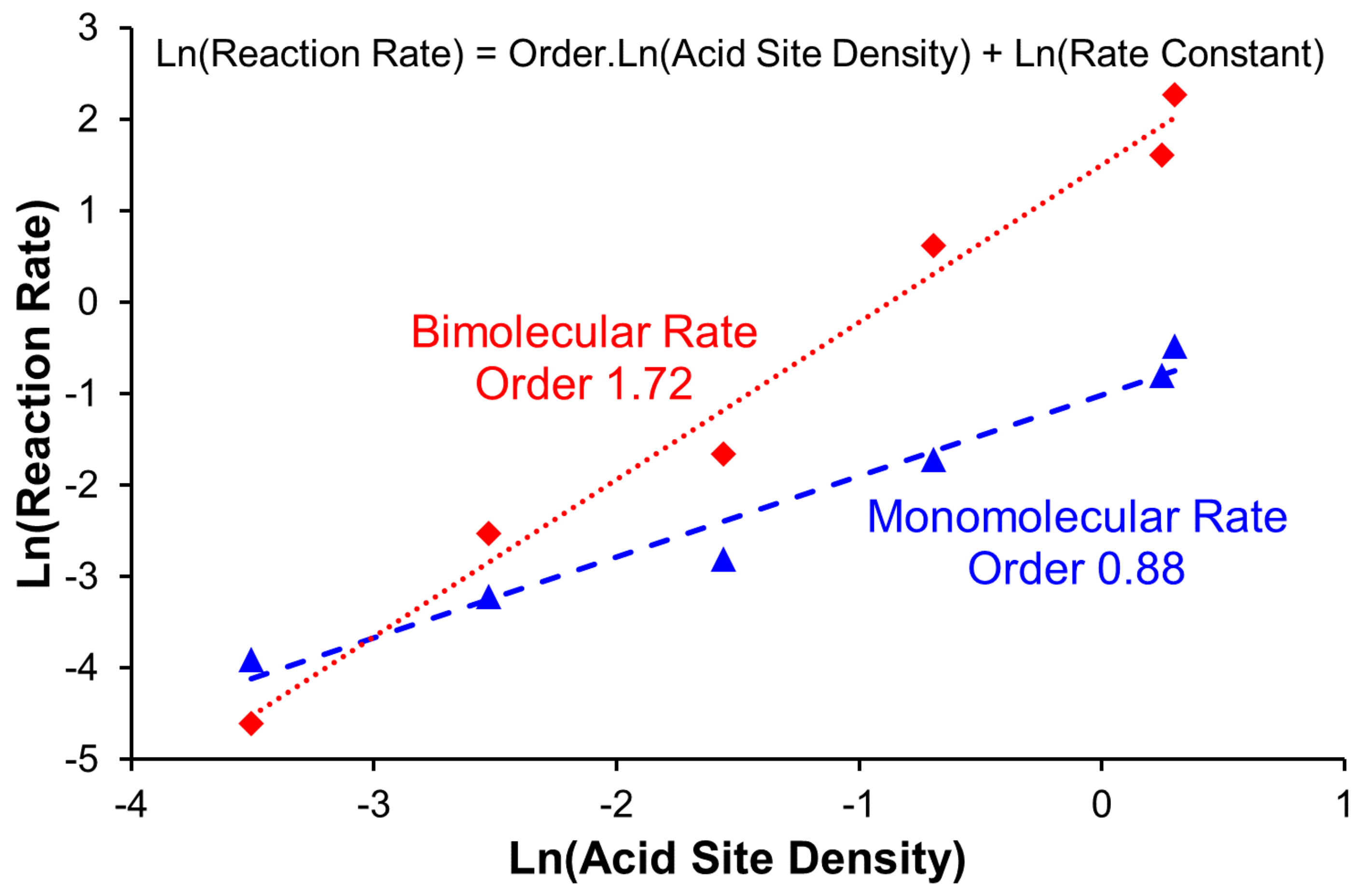
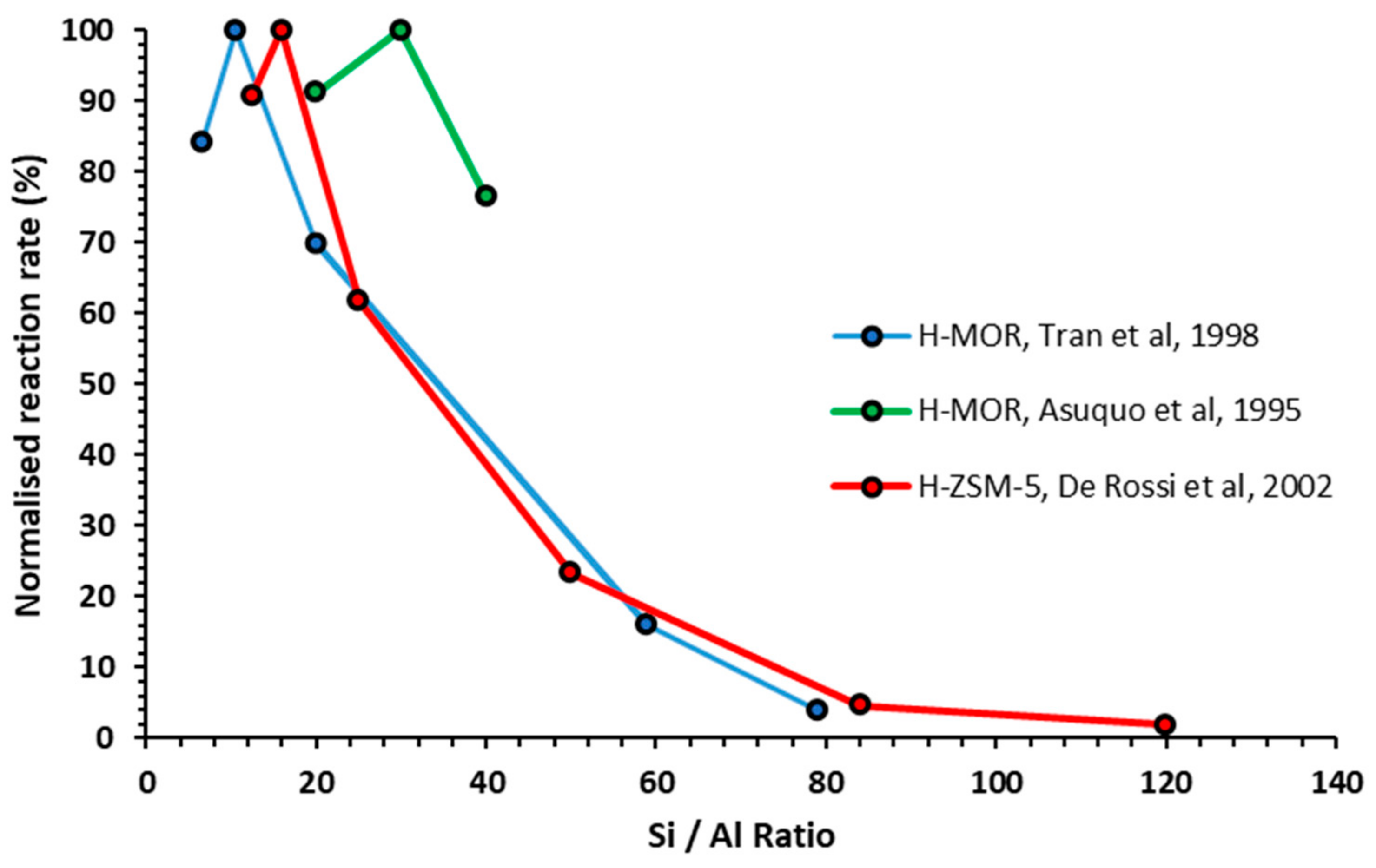
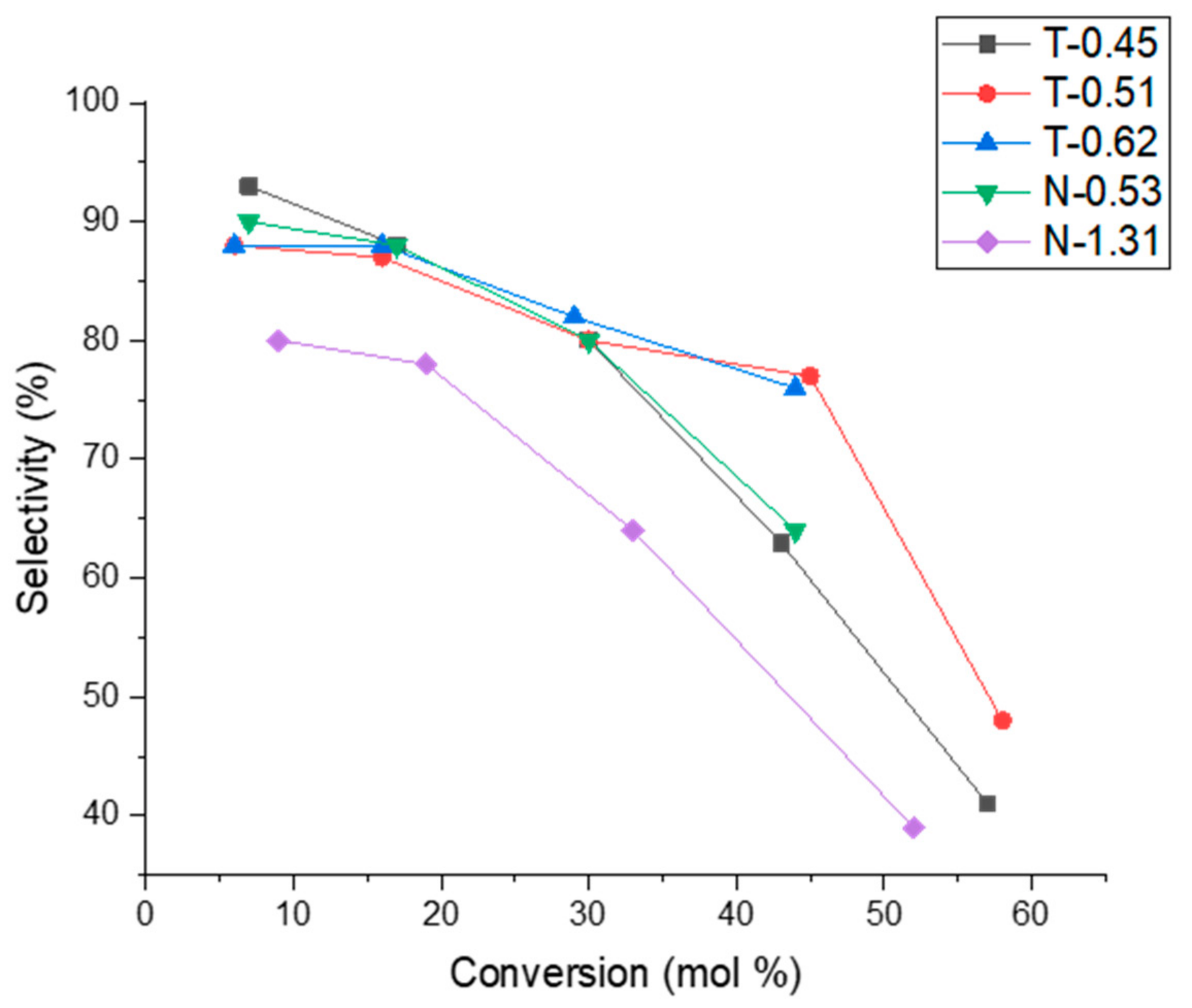
2.3. Influence of Acid Site Density
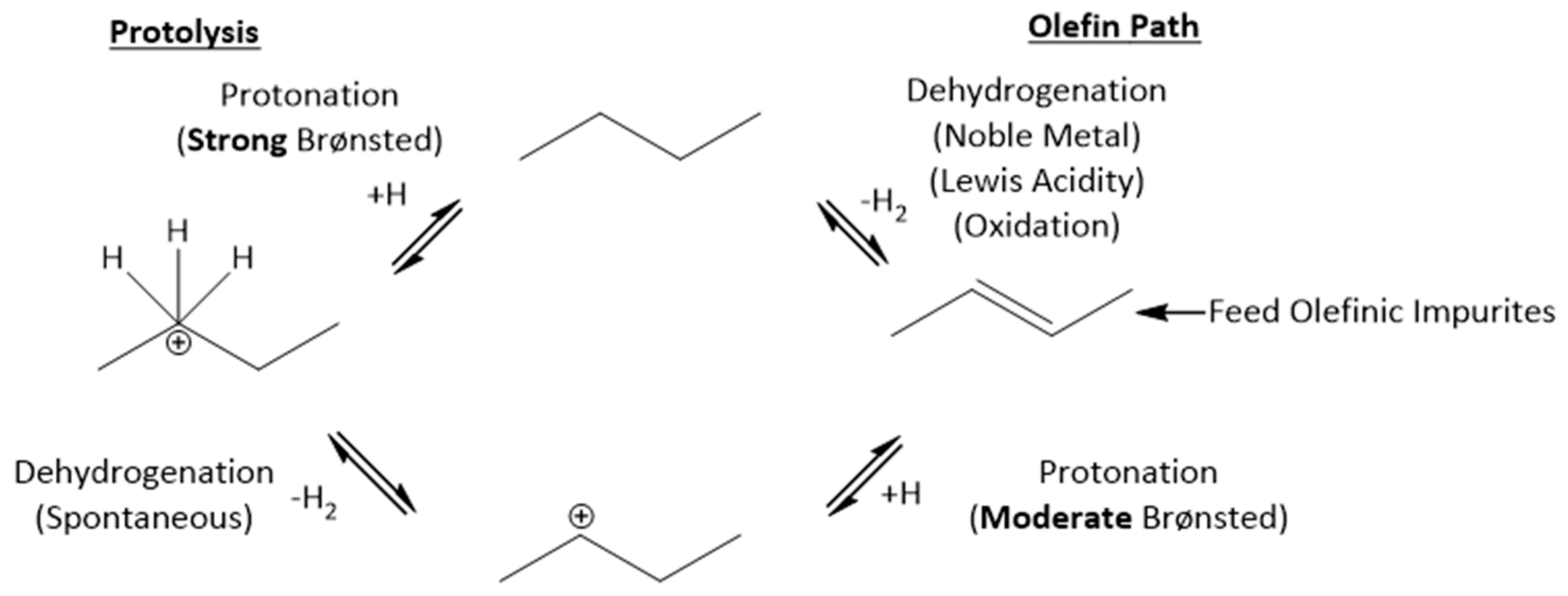
2.4. Influence of Acidic Site Strength and Olefin Levels
2.5. Influence of Mesoporosity in Zeolites
3. Solid-Acid Catalysts for Butane Isomerization
3.1. Friedel-Crafts Catalysts
3.2. Bifunctional Catalysts
3.3. Heteropoly Acids
3.4. Tungsten Oxide Supported on Zirocnia
3.5. Sulfated Zirconia
3.5.1. Main Principles
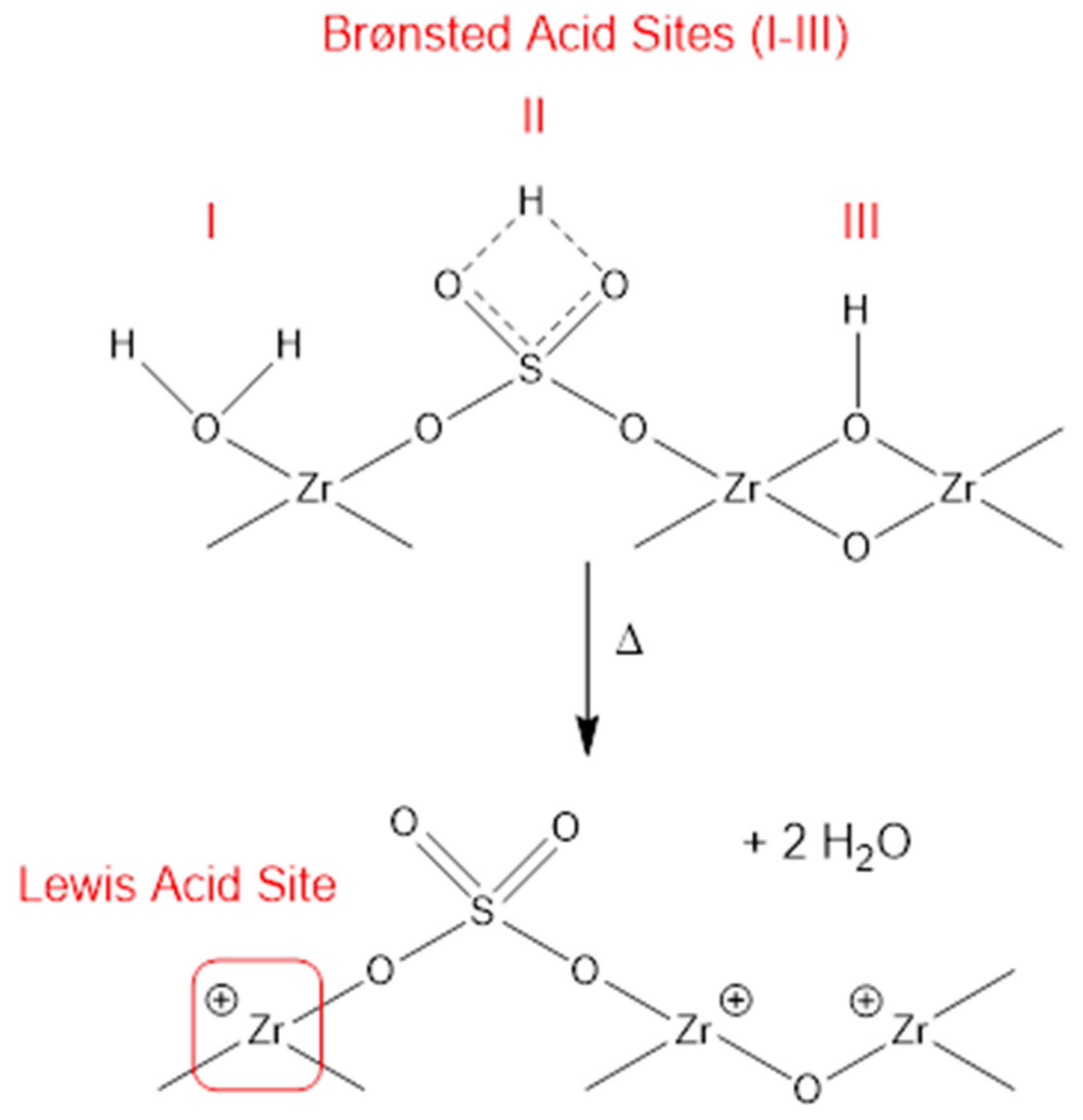
3.5.2. Active Sites in Sulfated Zirconia
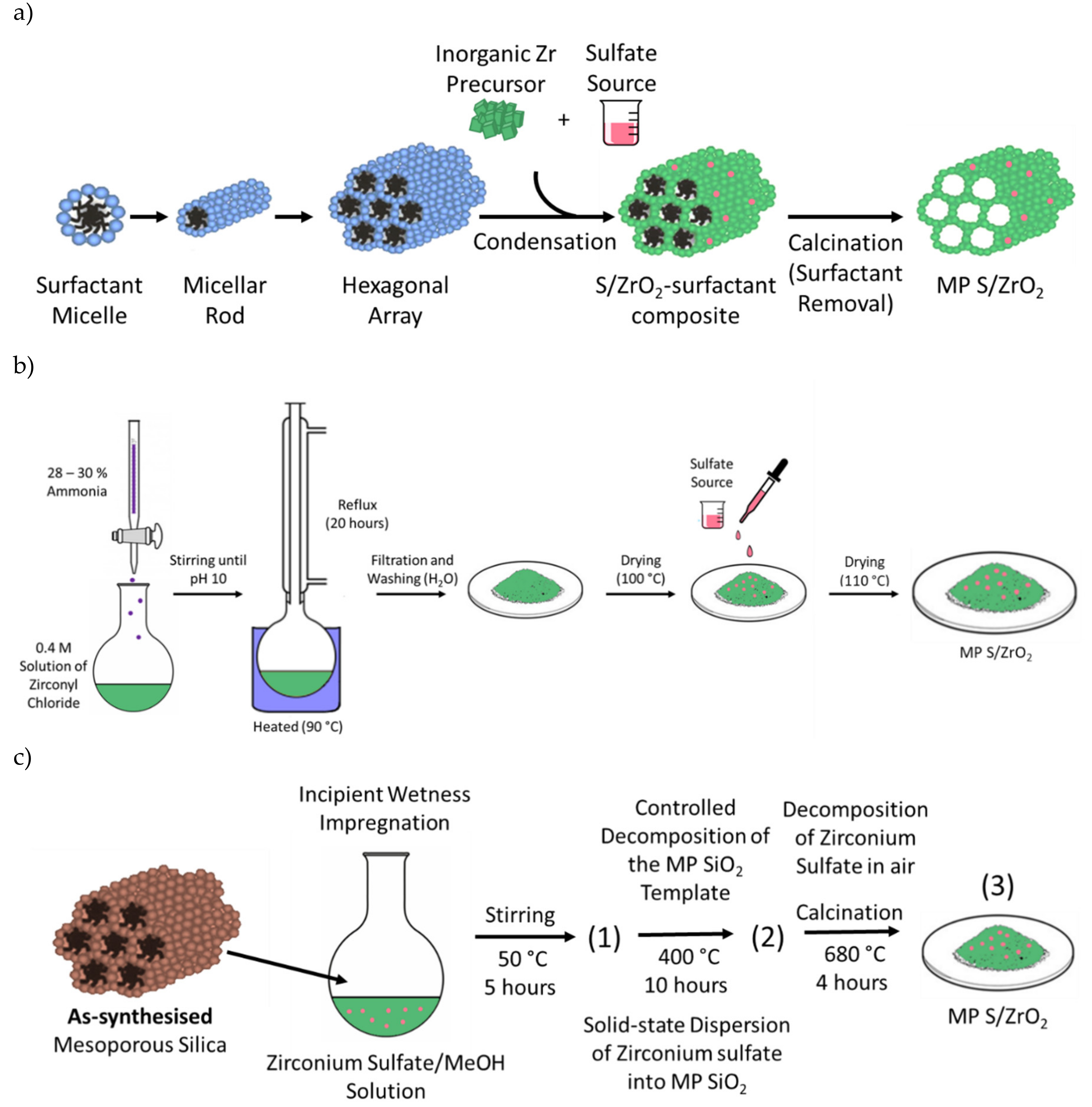
3.5.3. Incorporating Mesoporosity into Sulfated Zirconia
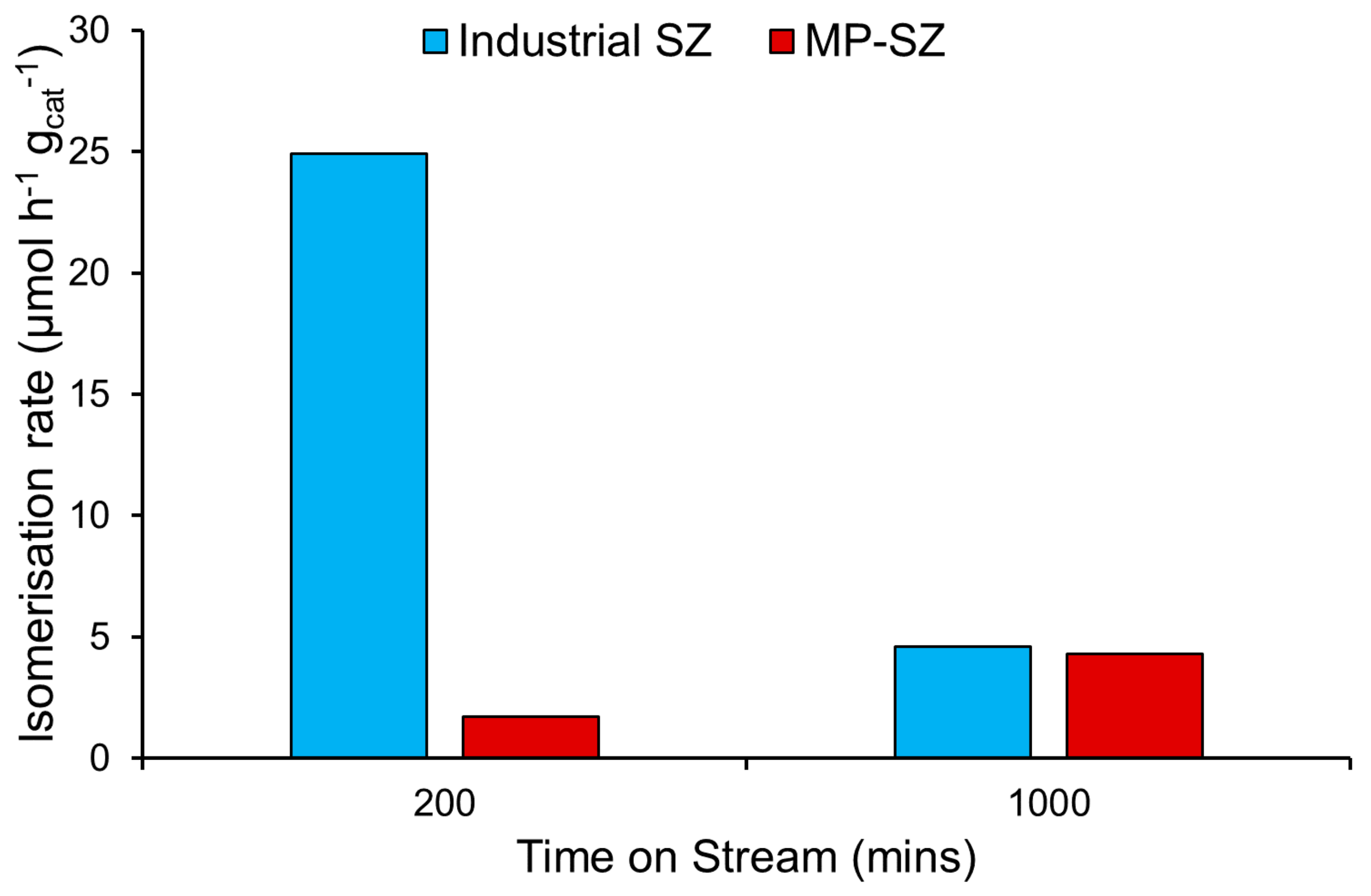
3.5.4. The Consequences of Mesoporosity on Butane Isomerization for Sulfated Zirconia Systems
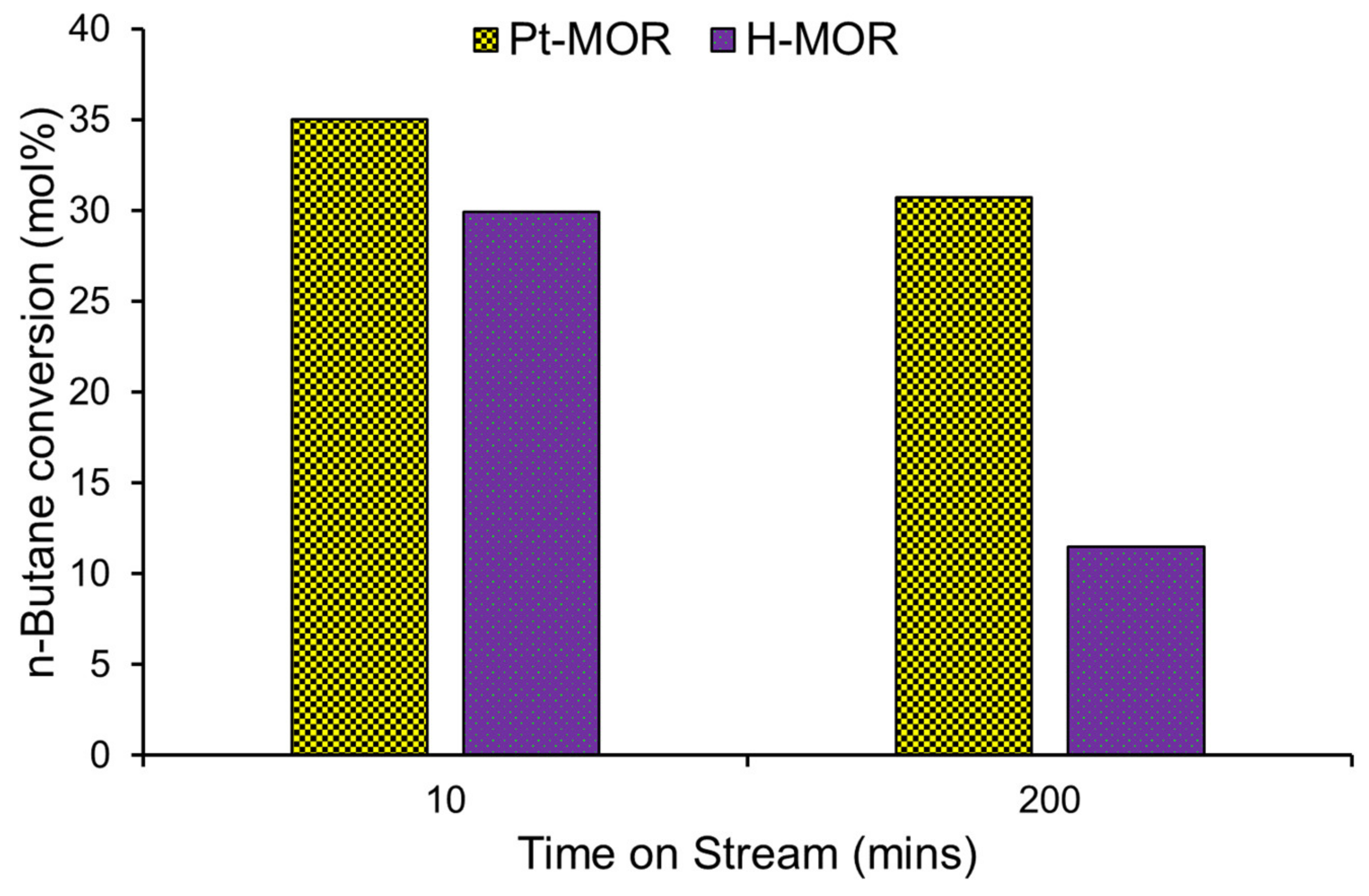
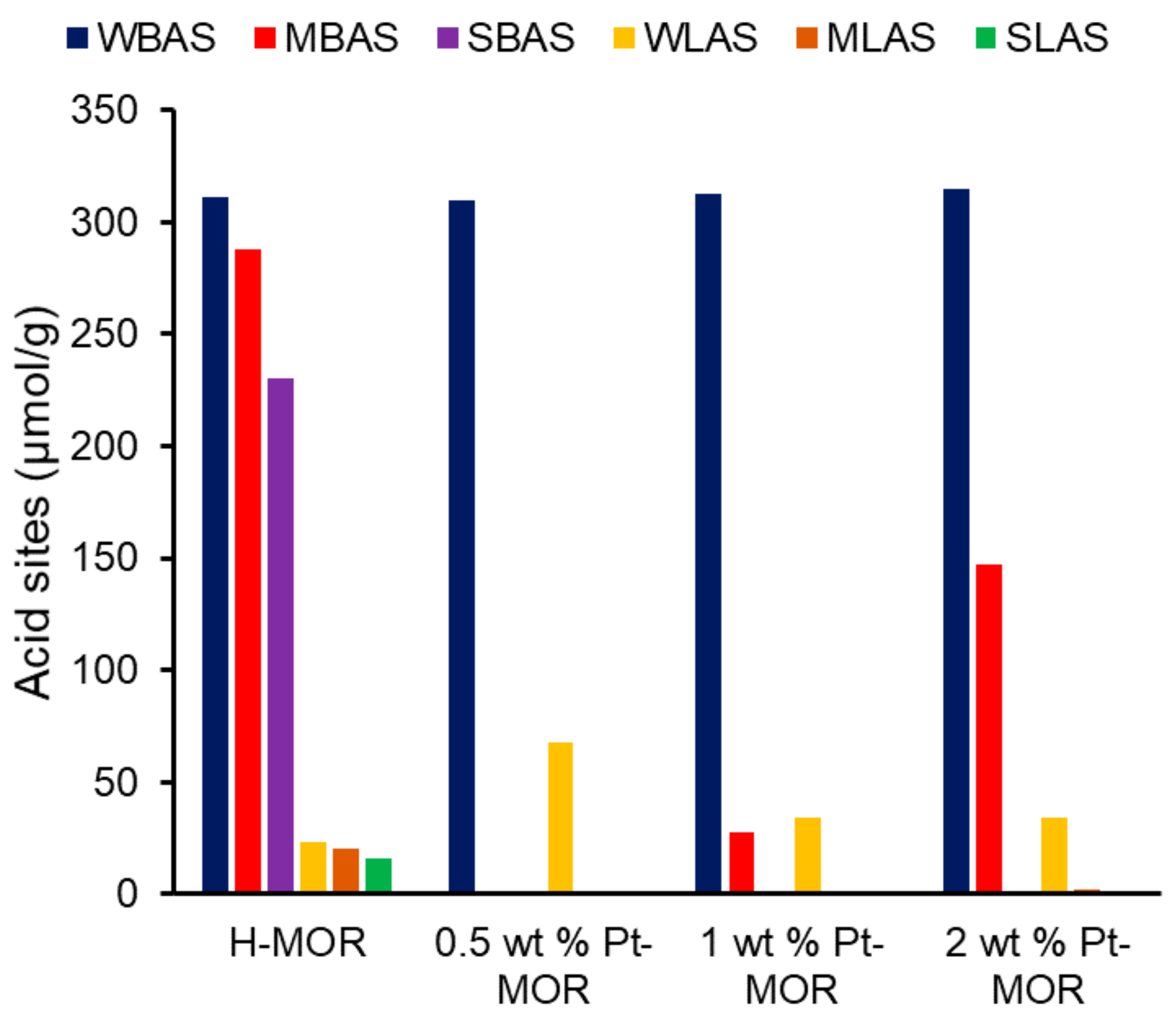
4. Modifying Acidity with Metal Dopants
4.1. The Importance of Dopant-Support Combinations
4.2. The Influence of Dopant Loading
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- LPG Production and Distribution. Available online: https://www.wlpga.org/about-lpg/production-distribution/ (accessed on 25 June 2020).
- Statistical Review of Global LPG. Available online: https://www.argusmedia.com/-/media/Files/white-papers/statistical-review-of-global-lpg-2016.ashx/ (accessed on 25 June 2020).
- Amer, M.; Wojcik, E.Z.; Sun, C.; Hoeven, R.; Hughes, J.M.X.; Faulkner, M.; Yunus, I.S.; Tait, S.; Johannissen, L.O.; Hardman, S.J.O.; et al. Low carbon strategies for sustainable bio-alkane gas production and renewable energy. Energy Environ. Sci. 2020, 13, 1818–1831. [Google Scholar] [CrossRef]
- U.S. Energy Information Administration. Hydrocarbon Gas Liquids (HGL): Recent Market Trends and Issues; U.S. Department of Energy: Washington, DC, USA, 2014.
- Singbal, S.; Yang, S.; Zhang, N. Modelling and integration of process networks for C4 hydrocarbons. Comput. Chem. Eng. 2020, 140. [Google Scholar] [CrossRef]
- Butane Market Size, Share & Trends Analysis Report by Application (LPG (Residential/Commercial, Chemical/Petrochemical, Industrial, Auto Fuel, Refinery), Petrochemicals, Refineries), by Region, and Segment Forecasts, 2018–2025; Grand View Research: San Francisco, CA, USA, 2016.
- Isobutane Market to Reach USD 34.00 Billion by 2026, Reports and Data. Available online: https://www.globenewswire.com/news-release/2019/03/21/1758764/0/en/Isobutane-Market-To-Reach-USD-34–00-Billion-By-2026-Reports-And-Data.html (accessed on 25 June 2020).
- Hidalgo, J.; Zbuzek, M.; Černý, R.; Jíša, P. Current uses and trends in catalytic isomerization, alkylation and etherification processes to improve gasoline quality. Open Chem. 2014, 12, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Akhmadova, K.K.; Magomadova, M.K.; Syrkin, A.M.; Egutkin, N.L. History, Current State, and Prospects for Development of Isobutane Alkylation with Olefins. Theor. Found. Chem. Eng. 2019, 53, 643–655. [Google Scholar] [CrossRef]
- Sullivan, D.; Metro, S.; Pujadó, P.R. Isomerization in Petroleum Processing. In Handbook of Petroleum Processing; Springer: Cham, Switzerland, 2015; pp. 479–497. [Google Scholar]
- Echevskii, G.V.; Aksenov, D.G.; Kodenev, E.G.; Ovchinnikova, E.V.; Chumachenko, V.A. Activity of a Sulfated Zirconia Catalyst in Isomerization of n-Butane Fractions. Pet. Chem. 2020, 59, S101–S107. [Google Scholar] [CrossRef]
- Optimising Profits by Blending Butane. Available online: https://www.digitalrefining.com/article/1001142/optimising-profits-by-blending-butane-ti#.X2cRKYvTWUk/ (accessed on 25 June 2020).
- You Can Just Iso My Butane: Isobutane and Isomerization in the Shale Gas World. Available online: https://rbnenergy.com/you-can-just-iso-my-butane-isobutane-and-isomerization (accessed on 25 June 2020).
- Butane Isomerization. Available online: https://www.enterpriseproducts.com/operations/petrochemical-refined-products-services/butane-isomerization (accessed on 25 June 2020).
- Evering, B.L. Commercial Isomerization. Adv. Catal. 1954, 6, 197–239. [Google Scholar]
- Honeywell UOP Catalysts: Empowering Hydrocarbon Industry. Available online: https://www.refiningandpetrochemicalsme.com/petrochemicals/24952-honeywell-uop-catalysts-empowering-hydrocarbon-industry (accessed on 25 June 2020).
- Klerk, A. Zeolites as Catalysts for Fuels Refining after Indirect Liquefaction Processes. Molecules 2018, 23, 115. [Google Scholar] [CrossRef] [Green Version]
- Fan, D.; Tian, P.; Xu, S.; Wang, D.; Yang, Y.; Li, J.; Wang, Q.; Yang, M.; Liu, Z. SAPO-34 templated by dipropylamine and diisopropylamine: Synthesis and catalytic performance in the methanol to olefin (MTO) reaction. New J. Chem. 2016, 40, 4236–4244. [Google Scholar] [CrossRef]
- Wang, P.; Yue, Y.; Wang, T.; Bao, X. Alkane isomerization over sulfated zirconia solid acid system. Int. J. Energy Res. 2020, 44, 3270–3294. [Google Scholar] [CrossRef]
- Yan, G.X.; Wang, A.; Wachs, I.E.; Baltrusaitis, J. Critical review on the active site structure of sulfated zirconia catalysts and prospects in fuel production. Appl. Catal. A Gen. 2019, 572, 210–225. [Google Scholar] [CrossRef]
- Sander, B.; Thelen, M.; Kraushaar-Czarnetzki, B. Non-corrosive and chlorine-free isomerisation process under supercritical conditions. Catal. Today 2002, 75, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Urzhuntsev, G.A.; Ovchinnikova, E.V.; Chumachenko, V.A.; Yashnik, S.A.; Zaikovsky, V.I.; Echevsky, G.V. Isomerization of n-butane over Pd–SO4/ZrO2 catalyst: Prospects for commercial application. Chem. Eng. J. 2014, 238, 148–156. [Google Scholar] [CrossRef]
- Bloch, H.P. Compressors and Modern Process Applications; John Wiley & Sons: Hoboken, NJ, USA, 2006. [Google Scholar]
- Pines, H.; Kvetinskas, B.; Kassel, L.S.; Ipatieff, V.N. Determination of Equilibrium Constants for Butanes and Pentanes. J. Am. Chem. Soc. 1945, 67, 631–637. [Google Scholar] [CrossRef]
- Surla, K.; Vleeming, H.; Guillaume, D.; Galtier, P. A single events kinetic model: N-butane isomerization. Chem. Eng. Sci. 2004, 59, 4773–4779. [Google Scholar] [CrossRef]
- Cañizares, P.; de Lucas, A.; Dorado, F.; Pérez, D. Effect of zeolite pore geometry on isomerization of n-butane. Appl. Catal. A Gen. 2000, 190, 233–239. [Google Scholar] [CrossRef]
- Suzuki, T.; Okuhara, T. Mechanism of Skeletal Isomerization ofn-Butane Using 1,4-13C2-n-Butane on Solid Strong Acids. Chem. Lett. 2000, 29, 470–471. [Google Scholar] [CrossRef]
- Asuquo, R.A.; Edermirth, G.; Lercher, J.A. n-Butane Isomerization over Acidic Mordenite. J. Catal. 1995, 155, 376–382. [Google Scholar] [CrossRef] [Green Version]
- Lohitharn, N.; Lotero, E.; Goodwinjr, J. A comprehensive mechanistic pathway for n-butane isomerization on sulfated zirconia. J. Catal. 2006, 241, 328–341. [Google Scholar] [CrossRef]
- Boronat, M.; Corma, A. Are carbenium and carbonium ions reaction intermediates in zeolite-catalyzed reactions? Appl. Catal. A Gen. 2008, 336, 2–10. [Google Scholar] [CrossRef]
- Boronat, M.; Viruela, P.; Corma, A. Theoretical Study on the Mechanism of the Superacid-Catalyzed Unimolecular Isomerization ofn-Butane and 1-Butene. J. Phys. Chem. 1996, 100, 633–637. [Google Scholar] [CrossRef]
- Caeiro, G.; Carvalho, R.H.; Wang, X.; Lemos, M.A.N.D.A.; Lemos, F.; Guisnet, M.; Ramôa Ribeiro, F. Activation of C2–C4 alkanes over acid and bifunctional zeolite catalysts. J. Mol. Catal. A Chem. 2006, 255, 131–158. [Google Scholar] [CrossRef]
- Ono, Y. A survey of the mechanism in catalytic isomerization of alkanes. Catal. Today 2003, 81, 3–16. [Google Scholar] [CrossRef]
- Guisnet, M.; Pinard, L. Characterization of acid-base catalysts through model reactions. Catal. Rev. Sci. Eng. 2018, 60, 337–436. [Google Scholar] [CrossRef]
- Jain, A.V.; Pradhan, N.C.; Dalai, A.K.; Bakhshi, N.N. Studies on Chlorided Pt/Al2O3 Catalysts: Preparation, Characterization and n-Butane Isomerization Activity. Catal. Lett. 2003, 86, 221–227. [Google Scholar] [CrossRef]
- Kumar, N.; Villegas, J.I.; Salmi, T.; Murzin, D.Y.; Heikkilä, T. Isomerization of n-butane to isobutane over Pt-SAPO-5, SAPO-5, Pt-H-mordenite and H-mordenite catalysts. Catal. Today 2005, 100, 355–361. [Google Scholar] [CrossRef]
- Na, K.; Iizaki, T.; Okuhara, T.; Misono, M. Molecular design of solid acid catalysts. Isomerization of n-butane catalyzed by acid cesium salts of 12-tungstophosphoric acid combined with platinum. J. Mol. Catal. A Chem. 1997, 115, 449–455. [Google Scholar] [CrossRef]
- Nieminen, V.; Kumar, N.; Salmi, T.; Murzin, D.Y. n-Butane isomerization over Pt–H–MCM-41. Catal. Commun. 2004, 5, 15–19. [Google Scholar] [CrossRef]
- Del Gallo, P.; Meunier, F.; Pham-Huu, C.; Crouzet, C.; Ledoux, M.J. Selectiven-Butane Isomerization over High Specific Surface Area MoO3-Carbon-Modified Catalyst. Ind. Eng. Chem. Res. 1997, 36, 4166–4175. [Google Scholar] [CrossRef]
- Yori, J.C.; Vera, C.R.; Parera, J.M. n-butane isomerization on tungsten oxide supported on zirconia. Appl. Catal. A Gen. 1997, 163, 165–175. [Google Scholar] [CrossRef]
- Smith, S.; Bhattacharyya, A. Catalytic Isomerization of Butane Using Ionic Liquids. WO2014210140A1, 31 December 2014. [Google Scholar]
- Chapman, S.; Potter, M.E.; Raja, R. The Molecular Design of Active Sites in Nanoporous Materials for Sustainable Catalysis. Molecules 2017, 22, 2127. [Google Scholar] [CrossRef] [Green Version]
- Raja, R.; Potter, M.E.; Newland, S.H. Predictive design of engineered multifunctional solid catalysts. Chem. Commun. 2014, 50, 5940–5957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, B.; Bordiga, S.; Prins, R.; van Bokhoven, J.A. Effect of framework Si/Al ratio and extra-framework aluminum on the catalytic activity of Y zeolite. Appl. Catal. A Gen. 2007, 333, 245–253. [Google Scholar] [CrossRef]
- Hunger, B.; Heuchel, M.; Clark, L.A.; Snurr, R.Q. Characterization of Acidic OH Groups in Zeolites of Different Types: An Interpretation of NH3-TPD Results in the Light of Confinement Effects. J. Phys. Chem. B 2002, 106, 3882–3889. [Google Scholar] [CrossRef]
- Camiloti, A.M.; Jahn, S.L.; Velasco, N.D.; Moura, L.F.; Cardoso, D. Acidity of Beta zeolite determined by TPD of ammonia and ethylbenzene disproportionation. Appl. Catal. A Gen. 1999, 182, 107–113. [Google Scholar] [CrossRef]
- Suzuki, K.; Aoyagi, Y.; Katada, N.; Choi, M.; Ryoo, R.; Niwa, M. Acidity and catalytic activity of mesoporous ZSM-5 in comparison with zeolite ZSM-5, Al-MCM-41 and silica–alumina. Catal. Today 2008, 132, 38–45. [Google Scholar] [CrossRef]
- Farcasiu, D.; Li, J.Q.; Cameron, S. Preparation of sulfated zirconia catalysts with improved control of sulfur content II. Effect of sulfur content on physical properties and catalytic activity. Appl. Catal. A Gen. 1997, 154, 173–184. [Google Scholar] [CrossRef]
- Chen, F.R.; Coudurier, G.; Joly, J.F.; Vedrine, J.C. Superacid and Catalytic Properties of Sulfated Zirconia. J. Catal 1993, 143, 616–626. [Google Scholar] [CrossRef]
- Katada, N.; Endo, J.-I.; Notsu, K.-I.; Yasunobu, N.; Naito, N.; Niwa, M. Superacidity and Catalytic Activity of Sulfated Zirconia. J. Phys. Chem. B 2000, 104, 10321–10328. [Google Scholar] [CrossRef]
- Tran, M.T.; Gnep, N.S.; Szabo, G.; Guisnet, M. Comparative study of the transformation of n-butane, n-hexane and n-heptane over H-MOR zeolites with various Si/Al ratios. Appl. Catal. A Gen. 1998, 170, 49–58. [Google Scholar] [CrossRef]
- Tran, M.T.; Gnep, N.S.; Szabo, G.; Guisnet, M. Isomerization ofn-Butane over H-Mordenites under Nitren and Hydrogen: Influence of the Acid Site Density. J. Catal. 1998, 174, 185–190. [Google Scholar] [CrossRef]
- De Rossi, S.; Moretti, G.; Ferraris, G.; Gazzoli, D. Butane isomerization on several H-zeolite catalysts. In Impact of Zeolites and other Porous Materials on the new Technologies at the Beginning of the New Millennium. In Proceedings of the 2nd International FEZA (Federation of the European Zeolite Associations) Conference, Taormina, Italy, 1–5 September 2002; pp. 715–722. [Google Scholar]
- Karger, J.; Petzold, M.; Pfeifer, H.; Ernst, S.; Weitkamp, J. Single-file diffusion and reaction in zeolites. J. Catal. 1992, 136, 283–299. [Google Scholar] [CrossRef]
- Cañizares, P.; de Lucas, A.; Dorado, F. n-Butane isomerization over H-mordenite: Role of the monomolecular mechanism. Appl. Catal. A Gen. 2000, 196, 225–231. [Google Scholar] [CrossRef]
- Liu, H.; Lei, G.D.; Sachtler, W.M.H. Alkane isomerization over solid acid catalysts Effects of one-dimensional micropores. Appl. Catal. A Gen. 1996, 137, 167–177. [Google Scholar] [CrossRef]
- Adeeva, V.; Liu, H.Y.; Xu, B.Q.; Sachtler, W.M.H. Alkane isomerization over sulfated zirconia and other solid acids. Top. Catal. 1998, 6, 61–76. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, P.; Yang, C.; Li, C. A Comparative Study of n-Butane Isomerization over H-Beta and H-ZSM-5 Zeolites at Low Temperatures: Effects of Acid Properties and Pore Structures. Catal. Lett. 2019, 149, 1017–1025. [Google Scholar] [CrossRef]
- Thybaut, J.W.; Marin, G.B. Multiscale Aspects in Hydrocracking: From Reaction Mechanism Over Catalysts to Kinetics and Industrial Application. Adv. Catal. 2016, 59, 109–238. [Google Scholar]
- Wang, P.; Zhang, W.; Zhang, Q.; Xu, Z.; Yang, C.; Li, C. Comparative study of n-butane isomerization over SO42−/Al2O3-ZrO2 and HZSM-5 zeolites at low reaction temperatures. Appl. Catal. A Gen. 2018, 550, 98–104. [Google Scholar] [CrossRef]
- Adeeva, V.; Lei, G.D.; Sachtler, W.M.H. Competitive mechanisms ofn-butane isomerization on sulfated zirconia catalysts. Catal. Lett. 1995, 33, 135–143. [Google Scholar] [CrossRef]
- Adeeva, V.; Sachtler, W.M.H. Mechanism of butane isomerization over industrial isomerization catalysts. Appl. Catal. A Gen. 1997, 163, 237–243. [Google Scholar] [CrossRef]
- Wang, P.Z.; Wang, S.Q.; Yue, Y.Y.; Wang, T.H.; Bao, X.J. Effects of acidity and topology of zeolites on the n-alkane conversion at low reaction temperatures. Microporous Mesoporous Mater. 2020, 292, 109748. [Google Scholar] [CrossRef]
- Koradia, P. Optimization of SiO2/Al2O3 mole ratio of mordenite for n-pentane isomerization. J. Catal. 1980, 66, 290–293. [Google Scholar] [CrossRef]
- Guisnet, M.; Fouche, V.; Belloum, M.; Bournonville, J.P.; Travers, C. Isomerization of n-hexane on platinum dealuminated mordenite catalysts I. Influence of the silicon-to-aluminium ratio of the zeolite. Appl. Catal. 1991, 71, 283–293. [Google Scholar] [CrossRef]
- Namba, S. Catalytic application of hydrophobic properties of high-silica zeolites I. Hydrolysis of ethyl acetate in aqueous solution. J. Catal. 1981, 72, 16–20. [Google Scholar] [CrossRef]
- Fajula, F. Hydration of n-butenes using zeolite catalysts. Influence of the aluminium content on activity. J. Catal. 1984, 89, 60–68. [Google Scholar] [CrossRef]
- Kurniawan, T.; Muraza, O.; Bakare, I.A.; Sanhoob, M.A.; Al-Amer, A.M. Isomerization of n-Butane over Cost-Effective Mordenite Catalysts Fabricated via Recrystallization of Natural Zeolites. Ind. Eng. Chem. Res. 2018, 57, 1894–1902. [Google Scholar] [CrossRef]
- Kumar, N.; Nieminen, V.; Demirkan, K.; Salmi, T.; Yu Murzin, D.; Laine, E. Effect of synthesis time and mode of stirring on physico-chemical and catalytic properties of ZSM-5 zeolite catalysts. Appl. Catal. A Gen. 2002, 235, 113–123. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, M.; Zhang, W.; Yang, C.; Li, C. Consequence of heterogeneity of active sites for reactivity mechanism of n -butane isomerization over SO42−/ZrO2-Al2O3 catalyst. Appl. Catal. A Gen. 2017, 542, 311–316. [Google Scholar] [CrossRef]
- Thomas, J.M.; Raja, R.; Lewis, D.W. Single-site heterogeneous catalysts. Angew. Chem. Int. Ed. Engl. 2005, 44, 6456–6482. [Google Scholar] [CrossRef] [Green Version]
- Potter, M.E.; Paterson, A.J.; Raja, R. Transition Metal versus Heavy Metal Synergy in Selective Catalytic Oxidations. Acs. Catal. 2012, 2, 2446–2451. [Google Scholar] [CrossRef]
- Sastre, G.; Lewis, D.W.; Catlow, C.R.A. Modeling of silicon substitution in SAPO-5 and SAPO-34 molecular sieves. J. Phys. Chem. B 1997, 101, 5249–5262. [Google Scholar] [CrossRef]
- Oliveira, A.C.; Essayem, N.; Tuel, A.; Clacens, J.-M.; Tâarit, Y.B. Comparative study of transformation of linear alkanes over modified mordenites and sulphated zirconia catalysts: Influence of the zeolite acidity on the performance of n-butane isomerization. J. Mol. Catal. A Chem. 2008, 293, 31–38. [Google Scholar] [CrossRef]
- Wang, J.-H.; Mou, C.-Y. Catalytic behavior of nanostructured sulfated zirconia promoted by alumina: Butane isomerization. Catal. Today 2008, 131, 162–172. [Google Scholar] [CrossRef]
- Wulfers, M.J.; Jentoft, F.C. Mechanism of n-butane skeletal isomerization on H-mordenite and Pt/H-mordenite. J. Catal. 2015, 330, 507–519. [Google Scholar] [CrossRef]
- Wulfers, M.J.; Tzolova-Müller, G.; Villegas, J.I.; Murzin, D.Y.; Jentoft, F.C. Evolution of carbonaceous deposits on H-mordenite and Pt-doped H-mordenite during n-butane conversion. J. Catal. 2012, 296, 132–142. [Google Scholar] [CrossRef]
- Fogash, K.B.; Hong, Z.; Kobe, J.M.; Dumesic, J.A. Deactivation of sulfated-zirconia and H-mordenite catalysts during n-butane and isobutane isomerization. Appl. Catal. A Gen. 1998, 172, 107–116. [Google Scholar] [CrossRef]
- Engelhardt, J. Reaction of Butanes on Na,H–Y Zeolites and H-Mordenites. J. Catal. 1996, 164, 449–458. [Google Scholar] [CrossRef]
- Hong, Z.; Fogash, K.B.; Dumesic, J.A. Reaction kinetic behavior of sulfated-zirconia catalysts for butane isomerization. Catal. Today 1999, 51, 269–288. [Google Scholar] [CrossRef]
- Asuquo, R.A.; Eder-Mirth, G.; Seshan, K.; Pieterse, J.A.Z.; Lercher, J.A. Improving the Stability of H–Mordenite forn-Butane Isomerization. J. Catal. 1997, 168, 292–300. [Google Scholar] [CrossRef] [Green Version]
- Baburek, E. Isomerization of n-butane over acid zeolites Role of Broensted and Lewis acid sites. Appl. Catal. A Gen. 1999, 185, 123–130. [Google Scholar] [CrossRef]
- Hartmann, M.; Kevan, L. Transition-metal ions in aluminophosphate and silicoaluminophosphate molecular sieves: Location, interaction with adsorbates and catalytic properties. Chem. Rev. 1999, 99, 635–664. [Google Scholar] [CrossRef] [PubMed]
- Sankar, G.; Raja, R.; Thomas, J.M. Redox solid catalysts for the selective oxidation of cyclohexane in air. Catal. Lett. 1998, 55, 15–23. [Google Scholar] [CrossRef]
- Barrett, P.A.; Sankar, G.; Catlow, C.R.A.; Thomas, J.M. X-ray Absorption Spectroscopic Study of Brønsted, Lewis, and Redox Centers in Cobalt-Substituted Aluminum Phosphate Catalysts. J. Phys. Chem. 1996, 100, 8977–8985. [Google Scholar] [CrossRef]
- Mohammed, K.M.H.; Chutia, A.; Callison, J.; Wells, P.P.; Gibson, E.K.; Beale, A.M.; Catlow, C.R.A.; Raja, R. Design and control of Lewis acid sites in Sn-substituted microporous architectures. J. Mater. Chem. A 2016, 4, 5706–5712. [Google Scholar] [CrossRef] [Green Version]
- Verboekend, D.; Pérez-Ramírez, J. Design of hierarchical zeolite catalysts by desilication. Catal. Sci. Technol. 2011, 1, 879–890. [Google Scholar] [CrossRef] [Green Version]
- Cañizares, P.; Dorado, F.; Sánchez-Herrera, P. Hydroisomerization of n-butane over hybrid catalysts. Appl. Catal. A Gen. 2001, 217, 69–78. [Google Scholar] [CrossRef]
- Stöcker, M. Gas phase catalysis by zeolites. Microporous Mesoporous Mater. 2005, 82, 257–292. [Google Scholar] [CrossRef]
- Christensen, C.; Johannsen, K.; Tornqvist, E.; Schmidt, I.; Topsoe, H.; Christensen, C. Mesoporous zeolite single crystal catalysts: Diffusion and catalysis in hierarchical zeolites. Catal. Today 2007, 128, 117–122. [Google Scholar] [CrossRef]
- Liu, J.; Li, Y.; Chen, Z.; Li, Z.; Yang, Q.; Hu, L.; Jiang, G.; Xu, C.; Wang, Y.; Zhao, Z. Hierarchical ZSM-5 Zeolites with Tunable Sizes of Building Blocks for Efficient Catalytic Cracking of i-Butane. Ind. Eng. Chem. Res. 2018, 57, 10327–10335. [Google Scholar] [CrossRef]
- Modhera, B.K.; Chakraborty, M.; Bajaj, H.C.; Parikh, P.A. Influences of Mesoporosity Generation in ZSM-5 and Zeolite Beta on Catalytic Performance During n-Hexane Isomerization. Catal. Lett. 2011, 141, 1182–1190. [Google Scholar] [CrossRef]
- McKetta, J.J.; Cunningham, W.A. Encyclopedia of Chemical Processing and Design; M. Dekker: New York, NY, USA, 1988. [Google Scholar]
- Perry, S.F. Isomerization. Ind. Eng. Chem. 1948, 40, 1624–1627. [Google Scholar] [CrossRef]
- McKetta, J.J., Jr. Chemical Processing Handbook; CRC Press: Boca Raton, FL, USA, 1993. [Google Scholar]
- Guisnet, M. Transformation of propane, n-butane and n-hexane over H3PW12O40 and cesium salts. Comparison to sulfated zirconia and mordenite catalysts. Top. Catal. 2000, 11/12, 247–254. [Google Scholar] [CrossRef]
- Na, K.; Okuhara, T.; Misono, M. Skeletal isomerization of n-butane over caesium hydrogen salts of 12-tungstophosphoric acid. J. Chem. Soc. Faraday Trans. 1995, 91, 367–373. [Google Scholar] [CrossRef]
- Ebitani, K. Skeletal isomerization of hydrocarbons over zirconium oxide promoted by Platinum and Sulfate Ion. J. Catal. 1991, 130, 257–267. [Google Scholar] [CrossRef]
- Hino, M.; Arata, K. Synthesis of solid superacid of tungsten oxide supported on zirconia and its catalytic action for reactions of butane and pentane. J. Chem. Soc. Chem. Commun. 1988, 18, 1259–1260. [Google Scholar] [CrossRef]
- Larsen, G.; Petkovic, L.M. Effect of preparation method and selective poisoning on the performance of platinum supported on tungstated zirconia catalysts for alkane isomerization. Appl. Catal. A Gen. 1996, 148, 155–166. [Google Scholar] [CrossRef]
- Kuba, S.; Lukinskas, P.; Grasselli, R.K.; Gates, B.C.; Knözinger, H. Structure and properties of tungstated zirconia catalysts for alkane conversion. J. Catal. 2003, 216, 353–361. [Google Scholar] [CrossRef]
- Holm, V.C.F.; Bailey, G.C. Sulfate-Treated Zirconia-Gel Catalyst. U.S. Patent 30325991962, 5 January 1962. [Google Scholar]
- Laizet, J.B.; Søiland, A.K.; Leglise, J.; Duchet, J.C. Top Catal. 2000, 10, 89–97. [CrossRef]
- Shi, G.; Yu, F.; Wang, Y.; Pan, D.; Wang, H.; Li, R. A novel one-pot synthesis of tetragonal sulfated zirconia catalyst with high activity for biodiesel production from the transesterification of soybean oil. Renew. Energy 2016, 92, 22–29. [Google Scholar] [CrossRef]
- Shao, Y.; Li, Y.; Sun, K.; Zhang, Z.; Tian, H.; Gao, G.; Li, Q.; Liu, Q.; Liu, Q.; Hu, X. Sulfated Zirconia with Different Crystal Phases for the Production of Ethyl Levulinate and 5-Hydroxymethylfurfural. Energy Technol. 2019, 8. [Google Scholar] [CrossRef]
- Hino, M.; Kobayashi, S.; Arata, K. Solid catalyst treated with anion. 2. Reactions of butane and isobutane catalyzed by zirconium oxide treated with sulfate ion. Solid superacid catalyst. J. Am. Chem. Soc. 1979, 101, 6439–6441. [Google Scholar] [CrossRef]
- Song, X.; Sayari, A. Sulfated Zirconia-Based Strong Solid-Acid Catalysts: Recent Progress. Catal. Rev. 1996, 38, 329–412. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Tanabe, K.; Yao Chin, K. Preparation and characterization of ZrO2 and SO42−-promoted ZrO2. Mater. Chem. Phys. 1987, 16, 67–77. [Google Scholar] [CrossRef]
- Ishida, T.; Yamaguchi, T.; Tanabe, K. Acid Property of Sulfur-Promoted Zirconium Oxide on Silica as Solid Superacid. Chem. Lett. 1988, 17, 1869–1872. [Google Scholar] [CrossRef] [Green Version]
- Arata, K.; Hino, M. Preparation of superacids by metal oxides and their catalytic action. Mater. Chem. Phys. 1990, 26, 213–237. [Google Scholar] [CrossRef]
- Arata, K. Solid Superacids; Academic Press: Hakodate, Japan, 1990; Volume 37, pp. 165–211. [Google Scholar]
- Morterra, C.; Cerrato, G.; Pinna, F.; Signoretto, M. Crystal Phase, Spectral Features, and Catalytic Activity of Sulfate-Doped Zirconia Systems. J. Catal. 1995, 157, 109–123. [Google Scholar] [CrossRef]
- Song, S.X.; Kydd, R.A. Activation of sulfated zirconia catalysts Effect of water content on their activity in n-butane isomerization. J. Chem. Soc. Faraday Trans. 1998, 94, 1333–1338. [Google Scholar] [CrossRef]
- Yaluris, G.; Larson, R.B.; Kobe, J.M.; González, M.R.; Fogash, K.B.; Dumesic, J.A. Selective Poisoning and Deactivation of Acid Sites on Sulfated Zirconia Catalysts forn-Butane Isomerization. J. Catal 1996, 158, 336–342. [Google Scholar] [CrossRef]
- Huang, Y.-Y.; Zhao, B.-Y.; Xie, Y.-C. Modification of sulfated zirconia by tungsten oxide: Acidity enhancement and structural characterization. Appl. Catal. A Gen. 1998, 171, 75–83. [Google Scholar] [CrossRef]
- Triwahyono, S.; Abdullah, Z.; Jalil, A.A. The Effect of Sulfate Ion on the Isomerization of n-Butane to iso-Butane. J. Nat. Gas. Chem. 2006, 15, 247–252. [Google Scholar] [CrossRef]
- Sun, Q.; Hu, K.; Leng, K.; Yi, X.; Aguila, B.; Sun, Y.; Zheng, A.; Meng, X.; Ma, S.; Xiao, F.-S. A porous Brønsted superacid as an efficient and durable solid catalyst. J. Mater. Chem. A 2018, 6, 18712–18719. [Google Scholar] [CrossRef]
- Olah, G.A.; Prakash, K.S.; Sommer, J. Superacids; John Wiley: New York, NY, USA, 1985. [Google Scholar] [CrossRef]
- Davis, B.H.; Keogh, R.A.; Srinivasan, R. Sulfated zirconia as a hydrocarbon conversion catalyst. Catal. Today 1994, 20, 219–256. [Google Scholar] [CrossRef]
- Umansky, B. On the strength of solid acids. J. Catal. 1991, 127, 128–140. [Google Scholar] [CrossRef]
- Kustov, L.M.; Kazansky, V.B.; Figueras, F.; Tichit, D. Investigation of the Acidic Properties of ZrO2 Modified by SO2−4 Anions. J. Catal. 1994, 150, 143–149. [Google Scholar] [CrossRef]
- Adeeva, V.; Dehaan, J.W.; Janchen, J.; Lei, G.D.; Schunemann, V.; Vandeven, L.J.M.; Sachtler, W.M.H.; Vansanten, R.A. Acid Sites in Sulfated and Metal-Promoted Zirconium Dioxide Catalysts. J. Catal. 1995, 151, 364–372. [Google Scholar] [CrossRef] [Green Version]
- Babou, F.; Bigot, B.; Coudurier, G.; Sautet, P.; Védrine, J.C. 4.23 Sulfated Zirconia for n-Butane Isomerization Experimental and Theoretical Approaches. In Acid-Base Catalysis II, Proceedings of the International Symposium on Acid-Base Catalysis II, Sapporo, Japan, 2–3 December 1993; Elsevier Science Ltd.: Amsterdam, The Netherlands, 1994; pp. 519–529. [Google Scholar]
- Comelli, R.A.; Vera, C.R.; Parera, J.M. Influence of ZrO2 Crystalline Structure and Sulfate Ion Concentration on the Catalytic Activity of SO2−4- ZrO2. J. Catal. 1995, 151, 96–101. [Google Scholar] [CrossRef]
- Liu, H.; Adeeva, V.; Lei, G.D.; Sachtler, W.M.H. Butane isomerization over platinum promoted sulfated zirconia catalysts. J. Mol. Catal. A Chem. 1995, 100, 35–48. [Google Scholar] [CrossRef]
- Liu, H.; Lei, G.D.; Sachtler, W.M.H. Pentane and butane isomerization over platinum promoted sulfated zirconia catalysts. Appl. Catal. A Gen. 1996, 146, 165–180. [Google Scholar] [CrossRef]
- Risch, M.; Wolf, E.E. n-Butane and n-pentane isomerization over mesoporous and conventional sulfated zirconia catalysts. Catal. Today 2000, 62, 255–268. [Google Scholar] [CrossRef]
- Hino, M.; Arata, K. Synthesis of solid superacid catalyst with acid strength of H0 ⩽–16.04. J. Chem. Soc. Chem. Commun. 1980, 18, 851–852. [Google Scholar] [CrossRef]
- Xu, B.-Q.; Sachtler, W.M.H. Isomerization ofn-Butane over Deuterated Sulfated Zirconia. J. Catal. 1997, 165, 231–240. [Google Scholar] [CrossRef]
- Signoretto, M.; Pinna, F.; Strukul, G.; Chies, P.; Cerrato, G.; Di Ciero, S.; Morterra, C. Platinum-Promoted and Unpromoted Sulfated Zirconia Catalysts Prepared by a One-Step Aerogel Procedure. J. Catal 1997, 167, 522–532. [Google Scholar] [CrossRef]
- Pinna, F.; Signoretto, M.; Strukul, G.; Cerrato, G.; Morterra, C. Isomerization of n-butane on sulfated zirconia: Evidence for the dominant role of Lewis acidity on the catalytic activity. Catal. Lett. 1994, 26, 339–344. [Google Scholar] [CrossRef]
- Azambre, B.; Zenboury, L.; Weber, J.V.; Burg, P. Surface characterization of acidic ceria–zirconia prepared by direct sulfation. Appl. Surf. Sci. 2010, 256, 4570–4581. [Google Scholar] [CrossRef]
- Na, K.; Choi, M.; Ryoo, R. Recent advances in the synthesis of hierarchically nanoporous zeolites. Microporous Mesoporous Mater. 2013, 166, 3–19. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, N.; Xi, D.; Yang, M.; Yu, J. Organosilane surfactant-directed synthesis of hierarchical porous SAPO-34 catalysts with excellent MTO performance. Chem. Commun. 2014, 50, 6502–6505. [Google Scholar] [CrossRef]
- Huang, Y.-Y.; McCarthy, T.J.; Sachtler, W.M.H. Preparation and catalytic testing of mesoporous sulfated zirconium dioxide with partially tetragonal wall structure. Appl. Catal. A Gen. 1996, 148, 135–154. [Google Scholar] [CrossRef]
- Kresge, C.T.; Vartuli, J.C.; Roth, W.J.; Leonowicz, M.E. The discovery of ExxonMobil’s M41S family of mesoporous molecular sieves. In Mesoporous Crystals and Related Nano-Structured Materials, Proceedings of the Meeting on Mesoporous Crystals and Related Nano-Structured Materials, Stockholm, Sweden, 1–5 June 2004; Elsevier: Amsterdam, The Netherlands, 2004; pp. 53–72. [Google Scholar]
- Antonelli, D.M.; Ying, J.Y. Synthesis of Hexagonally Packed Mesoporous TiO2 by a Modified Sol–Gel Method. Angew. Chem. Int. Ed. Engl. 1995, 34, 2014–2017. [Google Scholar] [CrossRef]
- Huo, Q.; Margolese, D.I.; Ciesla, U.; Feng, P.; Gier, T.E.; Sieger, P.; Leon, R.; Petroff, P.M.; Schüth, F.; Stucky, G.D. Generalized synthesis of periodic surfactant/inorganic composite materials. Nature 1994, 368, 317–321. [Google Scholar] [CrossRef]
- Wirnsberger, G.; Gatterer, K.; Fritzer, H.P.; Grogger, W.; Pillep, B.; Behrens, P.; Hansen, M.F.; Koch, C.B. Mesostructured Iron Oxyhydroxides. 1. Synthesis, Local Structure, and Magnetism. Chem. Mater. 2001, 13, 1453–1466. [Google Scholar] [CrossRef]
- Kimura, T.; Sugahara, Y.; Kuroda, K. Synthesis of a Hexagonal Mesostructured Aluminophosphate. Chem. Lett. 1997, 26, 983–984. [Google Scholar] [CrossRef]
- Monnier, A.; Schuth, F.; Huo, Q.; Kumar, D.; Margolese, D.; Maxwell, R.S.; Stucky, G.D.; Krishnamurty, M.; Petroff, P.; Firouzi, A.; et al. Cooperative formation of inorganic-organic interfaces in the synthesis of silicate mesostructures. Science 1993, 261, 1299–1303. [Google Scholar] [CrossRef] [PubMed]
- Kresge, C.T.; Leonowicz, M.E.; Roth, W.J.; Vartuli, J.C.; Beck, J.S. Ordered mesoporous molecular sieves synthesized by a liquid-crystal template mechanism. Nature 1992, 359, 710–712. [Google Scholar] [CrossRef]
- Yadav, G.D.; Murkute, A.D. Preparation of a novel catalyst UDCaT-5: Enhancement in activity of acid-treated zirconia—Effect of treatment with chlorosulfonic acid vis-à-vis sulfuric acid. J. Catal. 2004, 224, 218–223. [Google Scholar] [CrossRef]
- Osatiashtiani, A.; Lee, A.F.; Granollers, M.; Brown, D.R.; Olivi, L.; Morales, G.; Melero, J.A.; Wilson, K. Hydrothermally Stable, Conformal, Sulfated Zirconia Monolayer Catalysts for Glucose Conversion to 5-HMF. Acs. Catal. 2015, 5, 4345–4352. [Google Scholar] [CrossRef]
- Feng, Y.; Zuo, M.; Wang, T.; Jia, W.; Zhao, X.; Zeng, X.; Sun, Y.; Tang, X.; Lei, T.; Lin, L. Efficient synthesis of glucose into 5-hydroxymethylfurfural with SO42−/ZrO2 modified H+ zeolites in different solvent systems. J. Taiwan Inst. Chem. Eng. 2019, 96, 431–438. [Google Scholar] [CrossRef]
- Yan, H.; Yang, Y.; Tong, D.; Xiang, X.; Hu, C. Catalytic conversion of glucose to 5-hydroxymethylfurfural over SO42−/ZrO2 and SO42−/ZrO2–Al2O3 solid acid catalysts. Catal. Commun. 2009, 10, 1558–1563. [Google Scholar] [CrossRef]
- Sudhakar Reddy, J.; Sayari, A. Nanoporous zirconium oxide prepared using the supramolecular templating approach. Catal. Lett. 1996, 38, 219–223. [Google Scholar] [CrossRef]
- Ciesla, U.; Schacht, S.; Stucky, G.D.; Unger, K.K.; Schüth, F. Formation of a Porous Zirconium Oxo Phosphate with a High Surface Area by a Surfactant-Assisted Synthesis. Angew. Chem. Int. Ed. Engl. 1996, 35, 541–543. [Google Scholar] [CrossRef]
- Romannikov, V.N.; Fenelonov, V.B.; Paukshtis, E.A.; Derevyankin, A.Y.; Zaikovskii, V.I. Mesoporous basic zirconium sulfate: Structure, acidic properties and catalytic behaviour. Microporous Mesoporous Mater. 1998, 21, 411–419. [Google Scholar] [CrossRef]
- Ciesla, U.; Fröba, M.; Stucky, G.; Schüth, F. Highly Ordered Porous Zirconias from Surfactant-Controlled Syntheses: Zirconium Oxide−Sulfate and Zirconium Oxo Phosphate. Chem. Mater. 1999, 11, 227–234. [Google Scholar] [CrossRef]
- Schüth, F.; Ciesla, U.; Schacht, S.; Thieme, M.; Huo, Q.; Stucky, G. Ordered mesoporous silicas and zirconias: Control on length scales between nanometer and micrometer. Mater. Res. Bull. 1999, 34, 483–494. [Google Scholar] [CrossRef]
- Lindén, M.; Blanchard, J.; Schacht, S.; Schunk, S.A.; Schüth, F. Phase Behavior and Wall Formation in Zr(SO4)2/CTABr and TiOSO4/CTABr Mesophases. Chem. Mater. 1999, 11, 3002–3008. [Google Scholar] [CrossRef]
- Mamak, M.; Coombs, N.; Ozin, G. Mesoporous Yttria-Zirconia and Metal-Yttria-Zirconia Solid Solutions for Fuel Cells. Adv. Mater. 2000, 12, 198–202. [Google Scholar] [CrossRef]
- Chen, H.-R.; Shi, J.-L.; Yu, J.; Wang, L.-Z.; Yan, D.-S. Synthesis of titanium-doped ordered porous zirconium oxide with high-surface-area. Microporous Mesoporous Mater. 2000, 39, 171–176. [Google Scholar] [CrossRef]
- Pârvulescu, V.I.; Pârvulescu, V.; Endruschat, U.; Lehmann, C.W.; Grange, P.; Poncelet, G.; Bönnemann, H. Preparation and characterization of mesoporous zirconium oxide. Part 2. Microporous Mesoporous Mater. 2001, 44–45, 221–226. [Google Scholar]
- Wong, M.S.; Antonelli, D.M.; Ying, J.Y. Synthesis and characterization of phosphated mesoporous zirconium oxide. Nanostructured Mater. 1997, 9, 165–168. [Google Scholar] [CrossRef]
- Wong, M.S.; Ying, J.Y. Amphiphilic Templating of Mesostructured Zirconium Oxide. Chem. Mater. 1998, 10, 2067–2077. [Google Scholar] [CrossRef]
- Antonelli, D.M. Synthesis and Mechanistic Studies of Sulfated Meso- and Microporous Zirconias with Chelating Carboxylate Surfactants. Adv. Mater. 1999, 11, 487–492. [Google Scholar] [CrossRef]
- Huang, Y.; Sachtler, W.M.H. Preparation of mesostructured lamellar zirconia. Chem. Commun. 1997, 13, 1181–1182. [Google Scholar] [CrossRef]
- Pacheco, G.; Zhao, E.; Diaz Valdes, E.; Garcia, A.; Fripiat, J.J. Microporous zirconia from anionic and neutral surfactants. Microporous Mesoporous Mater. 1999, 32, 175–188. [Google Scholar] [CrossRef]
- Zhao, E.; Hardcastle, S.E.; Pacheco, G.; Garcia, A.; Blumenfeld, A.L.; Fripiat, J.J. Aluminum-doped mesoporous zirconia obtained from anionic surfactants. Microporous Mesoporous Mater. 1999, 31, 9–21. [Google Scholar] [CrossRef]
- Pacheco, G.; Fripiat, J.J. Physical Chemistry of the Thermal Transformation of Mesoporous and Microporous Zirconia. J. Phys. Chem. B 2000, 104, 11906–11911. [Google Scholar] [CrossRef]
- Yang, X.; Jentoft, F.C.; Jentoft, R.E.; Girgsdies, F.; Ressler, T. Sulfated zirconia with ordered mesopores as an active catalyst for n-butane isomerization. Catal. Lett. 2002, 81, 25–31. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, Q.; Xu, M.; Wang, S.; You, J. In situ spectroscopic studies of decomposition of ZrSiO4 during alkali fusion process using various hydroxides. Rsc. Adv. 2015, 5, 11658–11666. [Google Scholar] [CrossRef]
- Stein, A.; Fendorf, M.; Jarvie, T.P.; Mueller, K.T.; Benesi, A.J.; Mallouk, T. E Salt-Gel Synthesis of Porous Transition-Metal Oxides. Chem. Mater. 1995, 7, 304–313. [Google Scholar] [CrossRef]
- Schüth, F. Surface Properties and Catalytic Performance of Novel Mesostructured Oxides. Ber. Der. Bunsenges. Phys. Chem. 1995, 99, 1306–1315. [Google Scholar] [CrossRef]
- Larsen, G.; Lotero, E.; Nabity, M.; Petkovic, L.M.; Shobe, D.S. Surfactant-Assisted Synthesis of Mesoporous Zirconia Powders with High Surface Areas. J. Catal. 1996, 164, 246–248. [Google Scholar] [CrossRef]
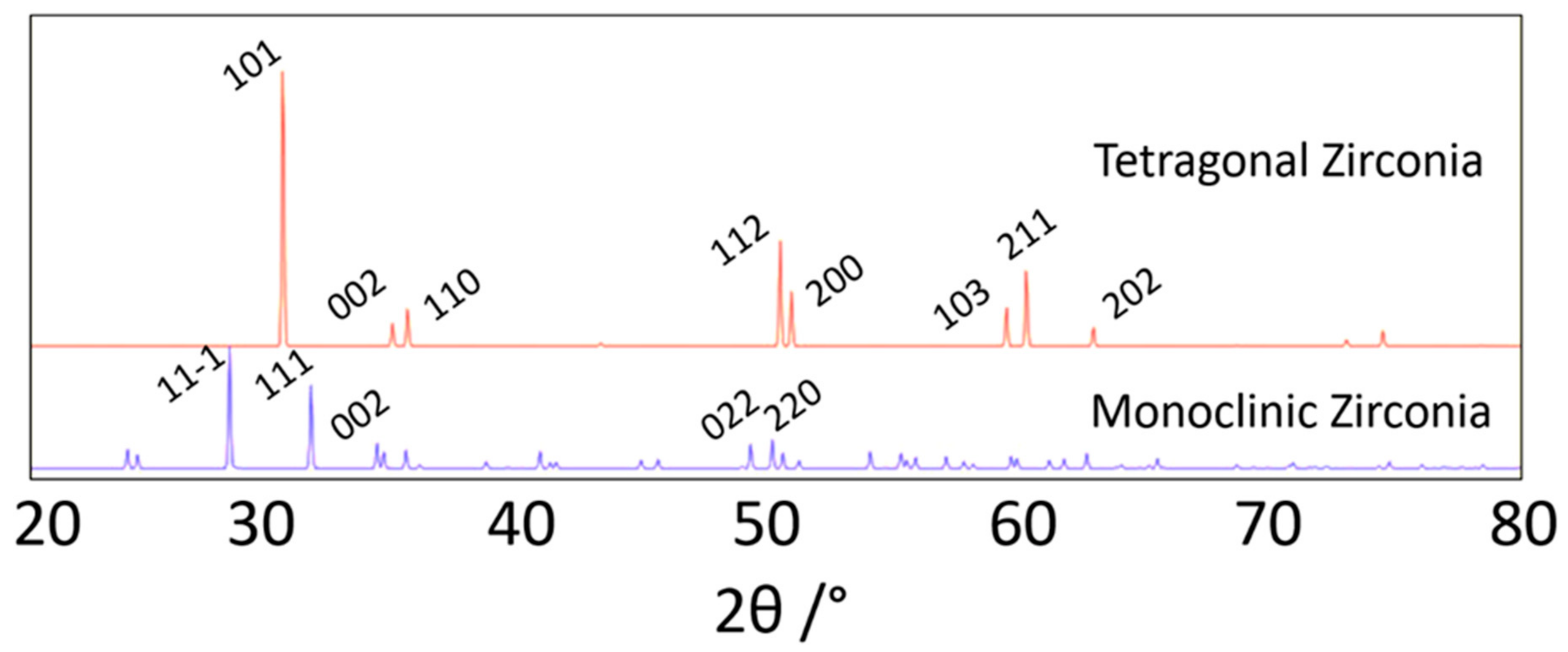
- Simha, N.K. Crystallography of the Tetragonal → Monoclinic Transformation in Zirconia. J. Phys. Iv. 2014, 5, C8-1121–C8-1126. [Google Scholar] [CrossRef]
- Livage, J.; Henry, M.; Sanchez, C. Sol-gel chemistry of transition metal oxides. Prog. Solid State Chem. 1988, 18, 259–341. [Google Scholar] [CrossRef]
- Debsikdar, J.C. Transparent zirconia gel-monolith from zirconium alkoxide. J. Non-Cryst. Solids 1986, 86, 231–240. [Google Scholar] [CrossRef]
- Sanchez, C.; Livage, J.; Henry, M.; Babonneau, F. Chemical modification of alkoxide precursors. J. Non-Cryst. Solids 1988, 100, 65–76. [Google Scholar] [CrossRef]
- Risch, M.; Wolf, E.E. Effect of the preparation of a mesoporous sulfated zirconia catalyst in n-butane isomerization activity. Appl. Catal. A Gen. 2001, 206, 283–293. [Google Scholar] [CrossRef]
- Knowles, J.A.; Hudson, M.J. Preparation and characterisation of mesoporous, high surface area zirconium(IV) oxides. J. Chem. Soc. Chem. Commun. 1995, 20, 2083–2084. [Google Scholar] [CrossRef]
- Hudson, M.J.; Knowles, J.A. Preparation and characterisation of mesoporous, high-surface-area zirconium(IV) oxide. J. Mater. Chem. 1996, 6, 89–95. [Google Scholar] [CrossRef]
- Corma, A.; Fornés, V.; Juan-Rajadell, M.I.; Nieto, J.M.L. Influence of preparation conditions on the structure and catalytic properties of SO42−/ZrO2 superacid catalysts. Appl. Catal. A Gen. 1994, 116, 151–163. [Google Scholar] [CrossRef]
- Grau, J.M.; Vera, C.R.; Parera, J.M. Alternatives for a better performance of Pt in SO2−4–ZrO2 catalysts for n-octane hydroisomerization-cracking. Selective adsorption of Pt over composites of SO2−4–ZrO2 mixed or supported onto Al2O3 and SiO2. Appl. Catal. A Gen. 1998, 172, 311–326. [Google Scholar] [CrossRef]
- Huang, Y.-Y.; Zhao, B.-Y.; Xie, Y.-C. A new method to prepare silica- or alumina-supported sulfated zirconia. Appl. Catal. A Gen. 1998, 173, 27–35. [Google Scholar] [CrossRef]
- Lei, T.; Xu, J.S.; Tang, Y.; Hua, W.M.; Gao, Z. New solid superacid catalysts for n-butane isomerization: γ-Al2O3 or SiO2 supported sulfated zirconia. Appl. Catal. A Gen. 2000, 192, 181–188. [Google Scholar] [CrossRef]
- Chen, C.-L.; Li, T.; Cheng, S.; Xu, N.; Mou, C.-Y. Catalytic Behavior of Alumina-Promoted Sulfated Zirconia Supported on Mesoporous Silica in Butane Isomerization. Catal. Lett. 2002, 78, 223–229. [Google Scholar] [CrossRef]
- Chen, C.-L.; Li, T.; Cheng, S.; Lin, H.-P.; Bhongale, C.J.; Mou, C.-Y. Direct impregnation method for preparing sulfated zirconia supported on mesoporous silica. Microporous Mesoporous Mater. 2001, 50, 201–208. [Google Scholar] [CrossRef]
- Babůrek, E.; Nováková, J. Effect of platinum in bifunctional isomerization of n-butane over acid zeolites. Appl. Catal. A Gen. 2000, 190, 241–251. [Google Scholar] [CrossRef]
- Villegas, J.; Kumar, N.; Heikkila, T.; Lehto, V.; Salmi, T.; Murzin, D. Isomerization of n-butane to isobutane over Pt-modified Beta and ZSM-5 zeolite catalysts: Catalyst deactivation and regeneration. Chem. Eng. J. 2006, 120, 83–89. [Google Scholar] [CrossRef]
- Chao, K.J.; Wu, H.C.; Leu, L.J. Skeletal Isomerization of n-Butane on Zeolites and Sulfated Zirconium Oxide Promoted by Platinum: Effect of Reaction Pressure. J. Catal. 1995, 157, 289–293. [Google Scholar] [CrossRef]
- Pirngruber, G.D.; Zinck-Stagno, O.P.E.; Seshan, K.; Lercher, J.A. The Effect of the Pore Structure of Medium-Pore Zeolites on the Dehydroisomerization of n-Butane: A Comparison of Pt–FER, Pt–TON, and Pt–ZSM5. J. Catal. 2000, 190, 374–386. [Google Scholar] [CrossRef]
- Villegas, J.I.; Kumar, N.; Salmi, T.; Murzin, D.Y.; Heikkilä, T.; Hudec, P.; Smiešková, A. Isomerization of n-butane over Pt-modified mordenite zeolite catalysts: Effect of Pt loadings and dealumination. In Molecular Sieves: From Basic Research to Industrial Applications, Proceedings of the 3rd International Zeolite Symposium (3rd FEZA), Prague, Czech Republic, 23–26 August 2005; Elsevier: Amsterdam, The Netherlands, 2005; pp. 1859–1866. [Google Scholar]
- Nieminen, V.; Kangas, M.; Salmi, T.; Murzin, D.Y. Kinetic Study ofn-Butane Isomerization over Pt−H-Mordenite. Ind. Eng. Chem. Res. 2005, 44, 471–484. [Google Scholar] [CrossRef]
- Yori, J.C.; Parera, J.M. n-Butane isomerization on metal-promoted sulfated zirconia. Appl. Catal. A Gen. 1996, 147, 145–157. [Google Scholar] [CrossRef]
- Cañizares, P.; De Lucas, A.; Valverde, J.L.; Dorado, F. n-Butane Hydroisomerization over Pt/HZSM-5 Catalysts. Ind. Eng. Chem. Res. 1997, 36, 4797–4808. [Google Scholar] [CrossRef]
- Moreno-Castilla, C.; Porcel-Jiménez, A.; Carrasco-Marín, F.; Utrera-Hidalgo, E. Pt/carbon catalysts: Effect of pretreatment on the dispersion and morphology of the Pt particles, on their capacity to chemisorb H2 and on the H2/n-C4H10 reaction. J. Mol. Catal. 1991, 66, 329–341. [Google Scholar] [CrossRef]
- Cañizares, P.; de Lucas, A.; Dorado, F.; Aguirre, J. n-Butane hydroisomerization over Pd/HZSM-5 catalysts. Palladium loaded by ion exchange. Microporous Mesoporous Mater. 2001, 42, 245–254. [Google Scholar] [CrossRef]
- Løften, T.; Gnep, N.S.; Guisnet, M.; Blekkan, E.A. Iron and manganese promoted sulfated zirconia: Acidic properties and n-butane isomerization activity. Catal. Today 2005, 100, 397–401. [Google Scholar] [CrossRef]
- Rahimi, N.; Karimzadeh, R. Kinetic modeling of catalytic cracking of C4 alkanes over La/HZSM-5 catalysts in light olefin production. J. Anal. Appl. Pyrolysis 2015, 115, 242–254. [Google Scholar] [CrossRef]
- Grau, J. Crystal phase dependent metal–support interactions in Pt/SO42−-ZrO2 catalysts for hydroconversion of n-alkanes. Appl. Catal. A Gen. 2004, 265, 141–152. [Google Scholar] [CrossRef]
- Hwang, C.-C.; Mou, C.-Y. Comparison of the promotion effects on sulfated mesoporous zirconia catalysts achieved by alumina and gallium. Appl. Catal. A Gen. 2009, 365, 173–179. [Google Scholar] [CrossRef]
- McIntosh, D.J.; Kydd, R.A.; Hill, J.M. Comparison of Cr, Mn, Fe, Co, and Ni as promoters for n-Butane conversion over sulfated zirconia. Chem. Eng. Commun. 2004, 191, 137–149. [Google Scholar] [CrossRef]
- Ahmed, M.A. Surface characterization and catalytic activity of sulfated-hafnia promoted zirconia catalysts for n-butane isomerization. Fuel Process. Technol. 2011, 92, 1121–1128. [Google Scholar] [CrossRef]
- Sattler, J.J.; Ruiz-Martinez, J.; Santillan-Jimenez, E.; Weckhuysen, B.M. Catalytic dehydrogenation of light alkanes on metals and metal oxides. Chem Rev. 2014, 114, 10613–10653. [Google Scholar] [CrossRef]
- Abu, I.I.; Das, D.D.; Mishra, H.K.; Dalai, A.K. Studies on platinum-promoted sulfated zirconia alumina: Effects of pretreatment environment and carrier gas on n-butane isomerization and benzene alkylation activities. J. Colloid Interface Sci. 2003, 267, 382–390. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Process | Butamer | C4 Isom | BIC | CFB-Isom | Lummus | Isomalk-3 |
|---|---|---|---|---|---|---|
| Company | UOP | BP | Boreskov Institute of Catalysis | UPC | Lummus | JSC SIE Neftheim |
| Catalyst | Pt/Al2O3-Cl | Pt/Al2O3-Cl | Pd/SO4.ZrO2 | SO4/ZrO2·Al2O3 | Al2O3-Cl | Pt/SO4·ZrO2 |
| Pressure/Bar | 25–32 | 14–28 | 23–25 | 1–1.5 | 4–10 | >15 |
| Temperature/°C | 160–220 | 150–200 | 120–160 | 160–200 | 100–160 | 180–200 |
| Feed additives | Chlorides | Chlorides | None | None | Chlorides | None |
| Butane/H2 mol ratio | 14–34 | Unknown | 7–20 | No H2 | Unknown | 10–17 |
| Minimum conversion/mol% | 50 | Unknown | 60 | 52 | 60 | 50 |
| Minimum selectivity/mol% | 98 | 95 | 90 | 72 | 99 | 90 |
| Caustic output | Yes | Yes | No | No | Yes | No |
| Sample | Surfactant | BET Surface Area (m2/g) | Pore Volume (cm3/g) | Pore Size (nm) | Ref |
|---|---|---|---|---|---|
| MP-SZ | C16H33N(CH3)3Br | 230 | 0.12 | 2–2.5 | [148] |
| MP-SZ | C18H37N(CH3)3Br | 320 | 0.15 | 2–2.5 | [148] |
| MP-SZ | C20H41N(CH3)3Br | 390 | 0.22 | 2–2.5 | [148] |
| MP-SZ | C16H33N(CH3)3Br | 531 | 0.28 | 3.3 | [149] |
| MP-SZ | C16H33N(CH3)3Br | 373 | 0.17 | 1.8 | [150] |
| MP-SZ | C16H33N(CH3)3Br | 202 | 0.11 | 2.2 | [163] |
| MP-SZ | C16H36NCl + acac | 347 | 0.32 | 1.8 | [135] |
| MP-YZ | C16H33N(CH3)3Br | 116 | 0.05 | 1.9 | [153] |
| MP-TiZ | C16H33N(CH3)3Br | 423 | 0.19 | 1.8 | [154] |
| MP-Z | C16H33N(CH3)3Br | 269 | n/A | 2.8 | [155] |
| MP-Z | C12H25PO42− | 320 | N/A | 2.5 | [156] |
| MP-Z | C4H9PO42− | 233 | 0.21 | 1.5 | [157] |
| MP-Z | C8H17PO42− | 313 | 0.23 | 1.9 | [157] |
| MP-Z | C12H25PO42− | 356 | 0.27 | 2.5 | [157] |
| MP-Z | C16H33PO42− | 361 | 0.33 | 2.6 | [157] |
| MP-Z | C5H11COOH | 403 | N/A | 1.7 | [158] |
| MP-Z | C10H21COOH | 621 | N/A | 3.0 | [158] |
| MP-Z | C14H29SO42−Na+ | N/A | N/A | N/A | [160] |
| MP-Z | Tergitol® 7 | 249 | 0.15 | 1.4 | [160] |
| MP-Z | C16H33NH2 | 347 | 0.31 | 1.9 | [135] |
| Synthetic Variables | Deposition Method | Metal | Loading (wt %) | Support | Ref |
|---|---|---|---|---|---|
| Support and loading | Ion exchange | Pt | 1.5 and 3 | MOR, ZSM-5 and BEA | [181] |
| Support | Wetness impregnation | Pt | 2 | SAPO-5 and MOR | [36] |
| Support | Ion exchange | Pt | 2 | Beta and ZSM-5 | [182] |
| Support | Incipient wetness | Pt | 0.33 | MOR | [76] |
| Support, temperature and pressure | Incipient wetness | Pt | 0.5 | SZ, ZSM-5, MOR and Beta | [183] |
| Temperature and loading | Wetness impregnation | Pt | 0.02, 0.25 and 1.3 | MOR | [81] |
| Support, Loading and metal dispersion | Wetness impregnation | Pt | 0.31-0.09 | FER, TON and ZSM-5 | [184] |
| Support, loading | Impregnation | Pt | 0.5,1 and 2 | MOR | [185] |
| Pressure and temperature | Impregnation | Pt | 0.5 | MOR | [186] |
| Metal type | Incipient wetness | Pt | 0.25 | SZ | [187] |
| Loading, deposition method and calcination conditions | Incipient Wetness/Ion exchange | Pt | 0.24–0.97 | ZSM-5 | [188] |
| WHSV and carrier gas | Ion exchange | Pt | 0.54 | MCM-41 | [38] |
| Support | Impregnation | Pt | 0.5 | SZ | [98] |
| Support and calcination conditions | Wetness impregnation | Pt | 1.0 | Carbon | [189] |
| Support and loading | Incipient wetness | Pd | 0.27–0.57 | ZSM-5 and MOR | [26] |
| Calcination conditions, acidity and metal precursor | Ion exchange | Pd | 0.53 | ZSM-5 | [190] |
| Loading | During synthesis | Fe and Mn | 2 (total) | SZ | [191] |
| Addition of Metal and diluent amount | Wet impregnation | La | 10 | ZSM-5 | [192] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Potter, M.E.; Le Brocq, J.J.M.; Oakley, A.E.; McShane, E.B.; Vandegehuchte, B.D.; Raja, R. Butane Isomerization as a Diagnostic Tool in the Rational Design of Solid Acid Catalysts. Catalysts 2020, 10, 1099. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10091099
Potter ME, Le Brocq JJM, Oakley AE, McShane EB, Vandegehuchte BD, Raja R. Butane Isomerization as a Diagnostic Tool in the Rational Design of Solid Acid Catalysts. Catalysts. 2020; 10(9):1099. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10091099
Chicago/Turabian StylePotter, Matthew E., Joshua J.M. Le Brocq, Alice E. Oakley, Evangeline B. McShane, Bart D. Vandegehuchte, and Robert Raja. 2020. "Butane Isomerization as a Diagnostic Tool in the Rational Design of Solid Acid Catalysts" Catalysts 10, no. 9: 1099. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10091099