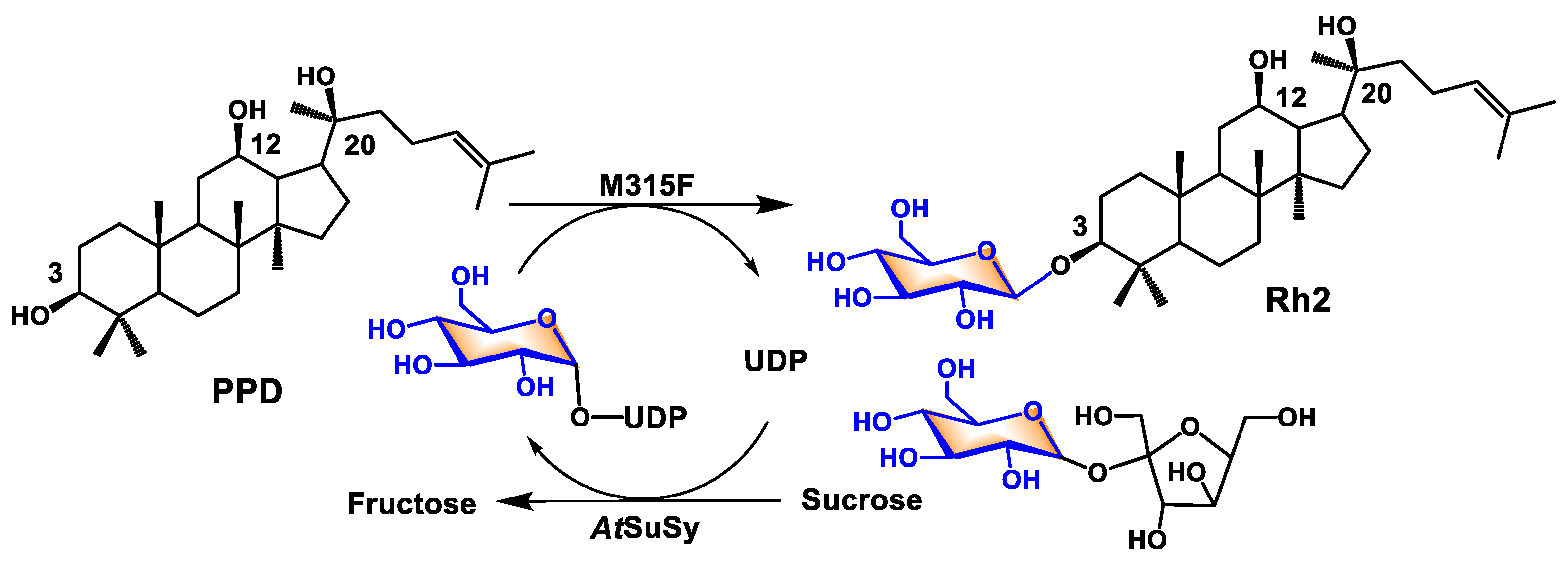
Biocatalysis for Rare Ginsenoside Rh2 Production in High Level with Co-Immobilized UDP-Glycosyltransferase Bs-YjiC Mutant and Sucrose Synthase AtSuSy


Abstract
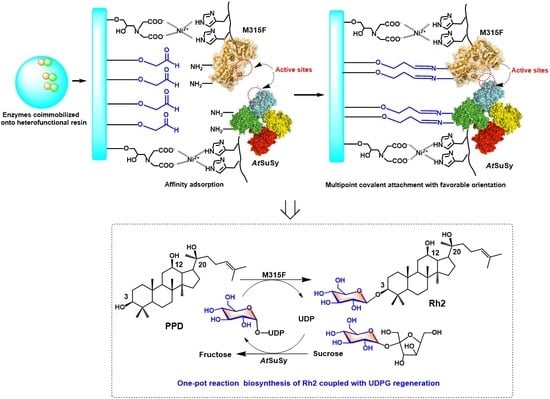
:
1. Introduction
2. Results and Discussion
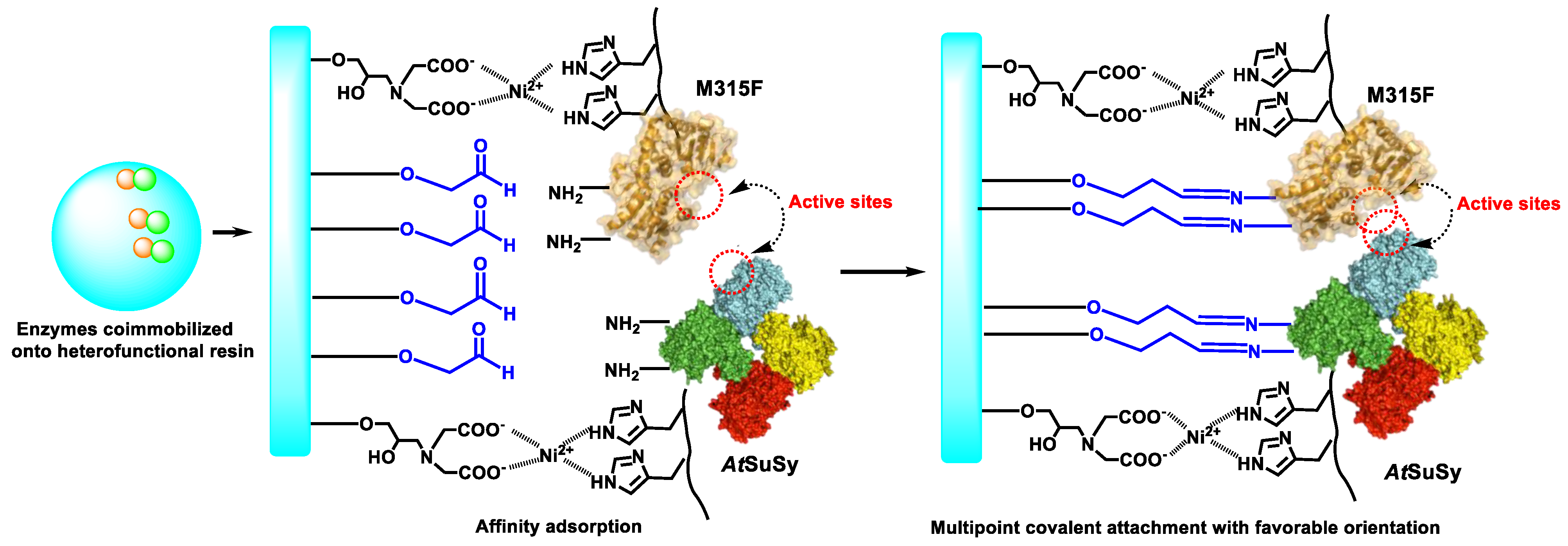
2.1. Co-Immobilization of Dual Enzyme onto Heterofunctional Carriers
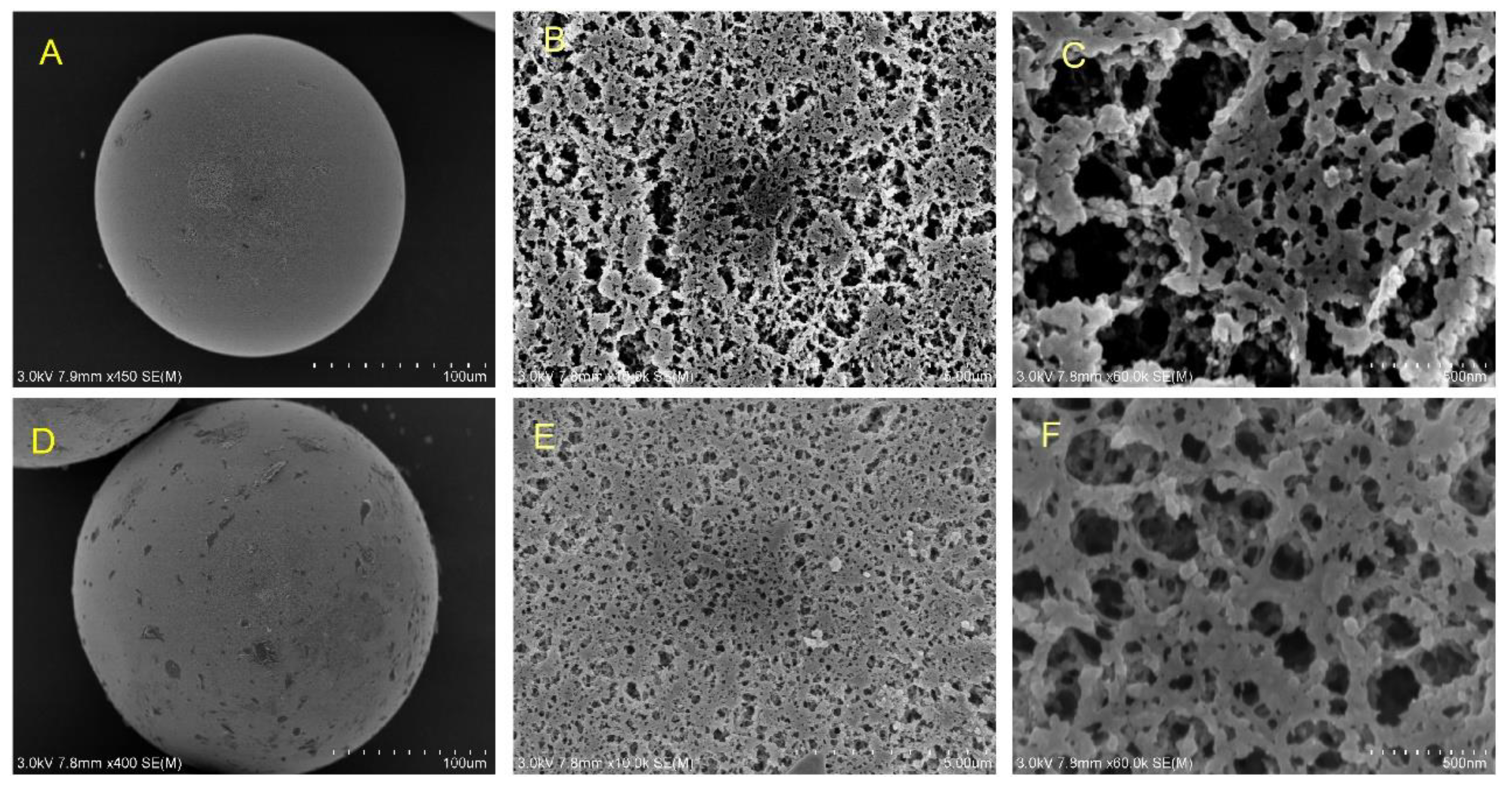
2.2. Physicochemical Properties of Heterofunctional Carriers
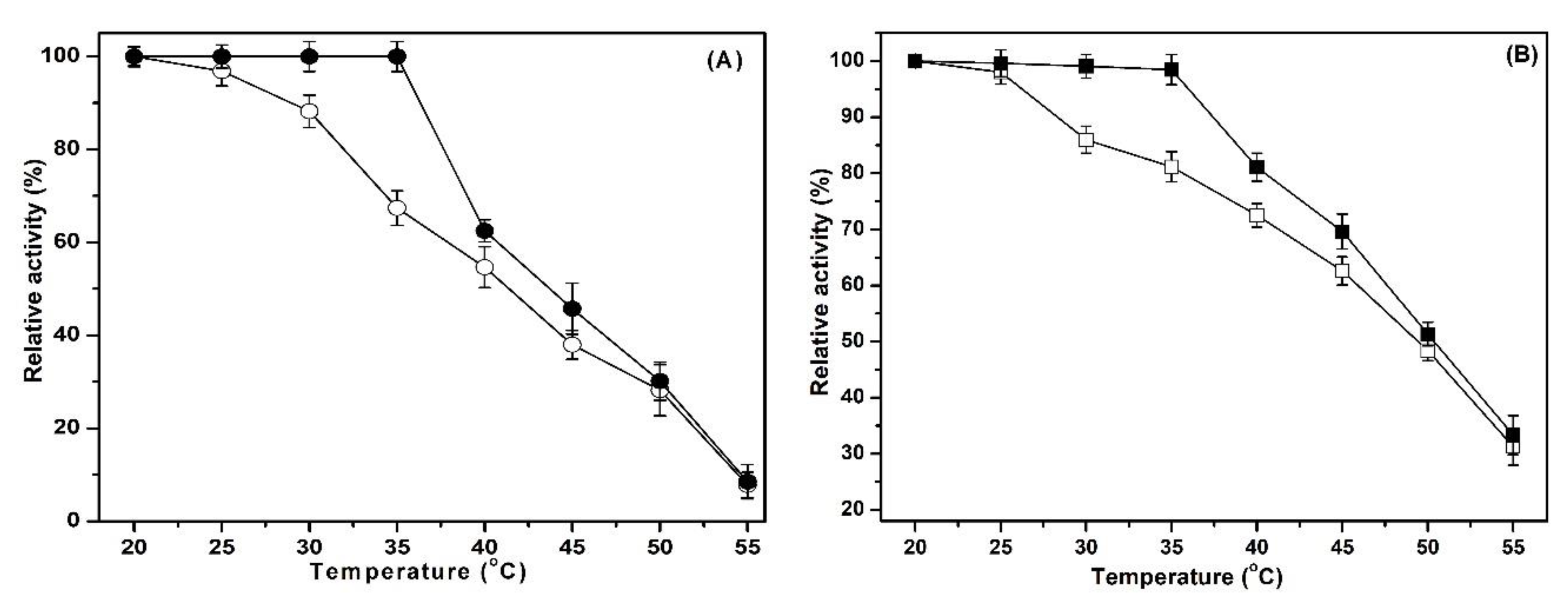
2.3. Effect of pH and Temperature on Activity and Stability of Enzymes
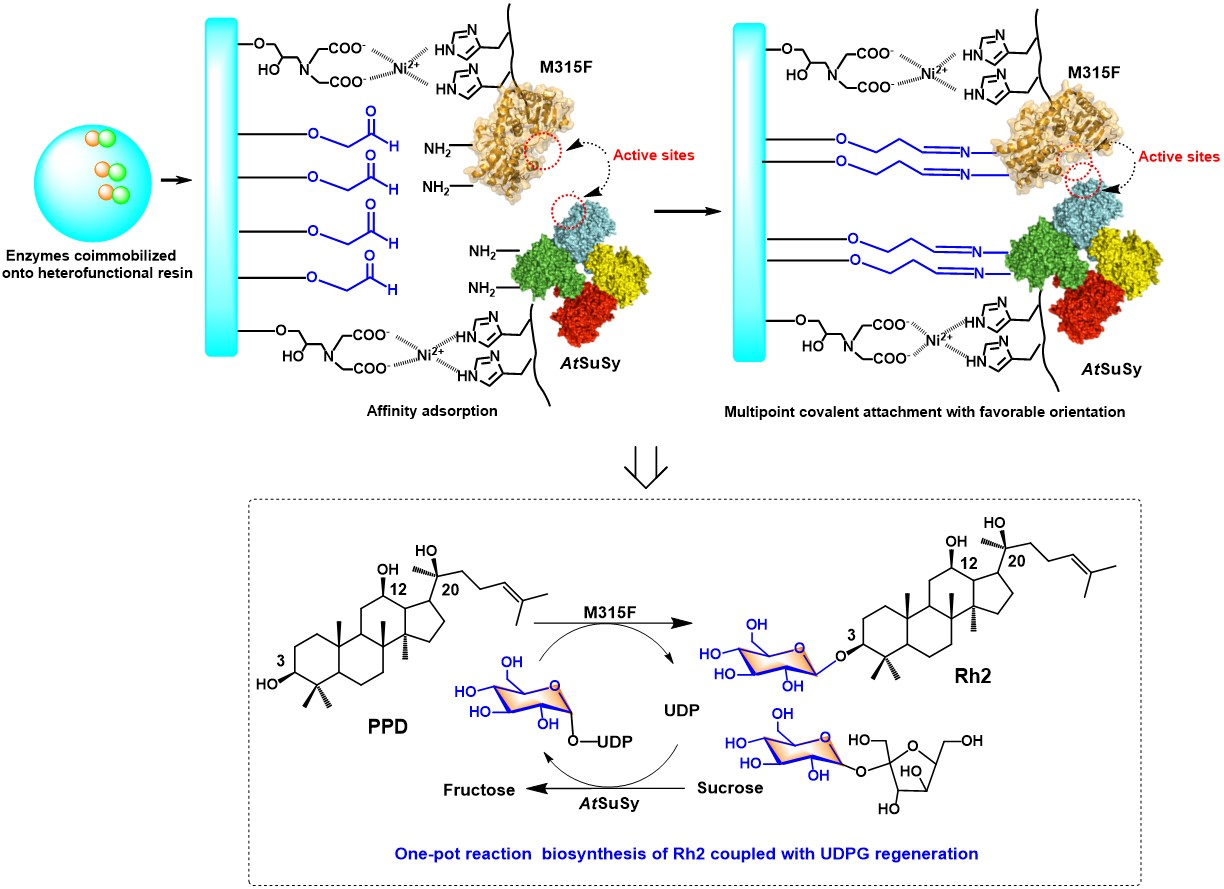
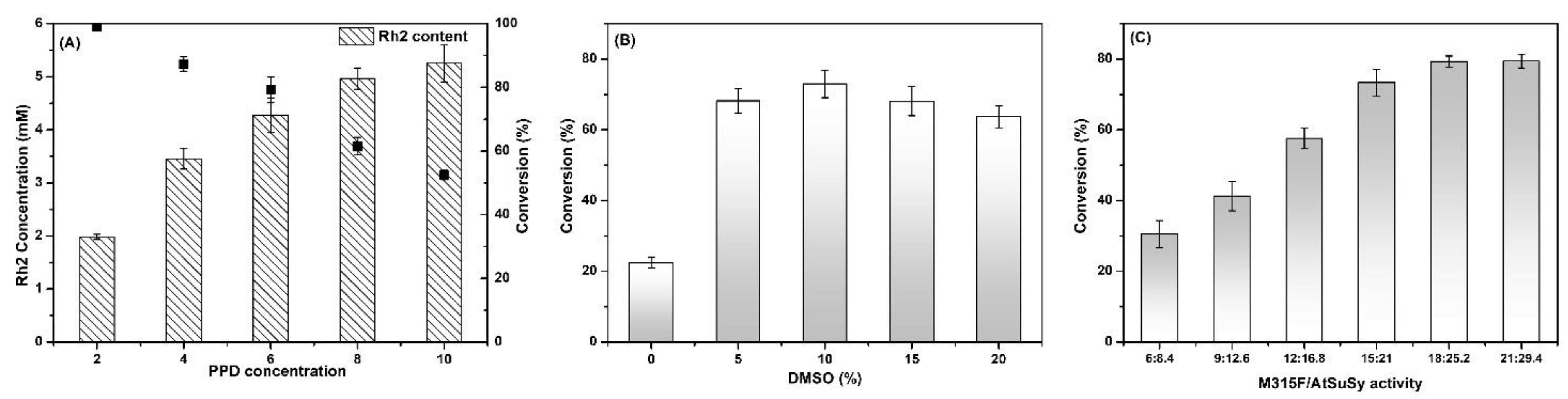
2.4. Conditions for the Co-Immobilized Enzymes Catalyzed Reactions
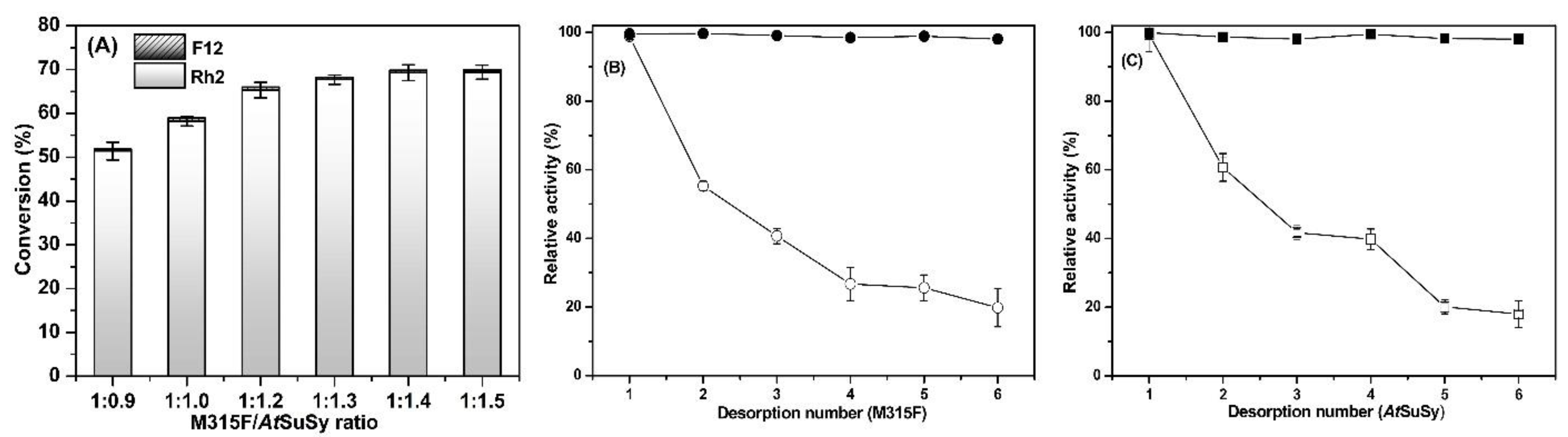
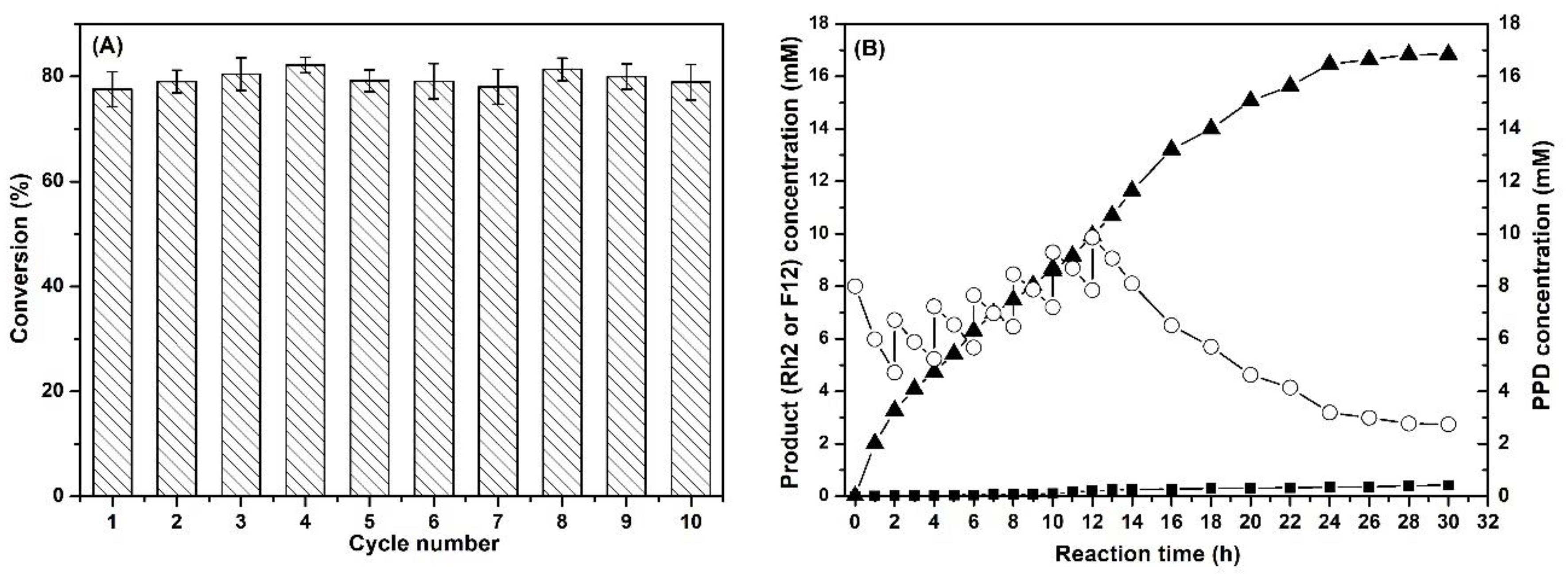
2.5. Reusability of Co-Immobilized Enzymes and Fed-Batch Strategy for Ginsenoside Rh2
3. Materials and Methods
3.1. Materials and Chemicals
3.2. Resin Modified with Bifunctional Groups and Carries Characterization
3.3. Dual Enzyme Co-Immobilization with Adsorption and Covalent Attachment
3.4. Enzyme Activity and Protein Assay
3.5. Biochemical Properties of Free and Co-Immobilized Enzymes
3.6. Optimization of the Co-Immobilized Enzymes Catalyzed Reaction Condition
3.7. Reusability of Co-Immobilized Enzymes and Fed-Batch Strategy
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, Z.Q. Chemical Insights into Ginseng as a Resource for Natural Antioxidants. Chem. Rev. 2012, 112, 3329–3355. [Google Scholar] [CrossRef] [PubMed]
- Christensen, L.P.; Jensen, M. Biomass and content of ginsenosides and polyacetylenes in American ginseng roots can be in-creased without affecting the profile of bioactive compounds. J. Nat. Med. 2009, 63, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.S.T.; Che, C.-M.; Leung, K.-W. Recent advances in ginseng as cancer therapeutics: A functional and mechanistic overview. Nat. Prod. Rep. 2015, 32, 256–272. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhao, J.; Wang, C.-Z.; Searle, J.S.; He, T.-C.; Yuan, C.-S.; Du, W. Ginsenoside Rh2 induces apoptosis and paraptosis-like cell death in colorectal cancer cells through activation of p53. Cancer Lett. 2011, 301, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.-S.; Lee, E.-H.; Ko, S.-R.; Choi, K.-J.; Park, J.-H.; Im, D.-S. Effects of ginsenosides Rg3 and Rh2 on the proliferation of prostate cancer cells. Arch. Pharmacal Res. 2004, 27, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Nakata, H.; Kikuchi, Y.; Tode, T.; Hirata, J.; Kita, T.; Ishii, K.; Kudoh, K.; Nagata, I.; Shinomiya, N. Inhibitory effects of ginsenoside Rh-2 on tumor growth in nude mice bearing human ovarian cancer cells. Jpn. J. Cancer Res. 1998, 89, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-E.; Oh, J.H.; Lee, S.K.; Oh, Y.J. Ginsenoside RH-2 induces apoptotic cell death in rat C6 glioma via a reactive oxygen- and caspase-dependent but Bcl-XL-independent pathway. Life Sci. 1999, 65, PL33–PL40. [Google Scholar] [CrossRef]
- Shibata, S. Chemistry and Cancer Preventing Activities of Ginseng Saponins and Some Related Triterpenoid Compounds. J. Korean Med. Sci. 2001, 16, S28–S37. [Google Scholar] [CrossRef] [Green Version]
- Kamra, P.; Gokhale, R.S.; Mohanty, D. SEARCHGTr: A program for analysis of glycosyltransferases involved in glycosylation of secondary metabolites. Nucleic Acids Res. 2005, 33, W220–W225. [Google Scholar] [CrossRef]
- Wang, P.P.; Wei, Y.J.; Fan, Y.; Liu, Q.F.; Wei, W.; Yang, C.S.; Zhang, L.; Zhao, G.P.; Yue, J.M. Production of bioactive gin-senosides Rh2 and Rg3 by metabolically engineered yeasts. Metab. Eng. 2015, 29, 97–105. [Google Scholar] [CrossRef]
- Hu, Z.-F.; Gu, A.-D.; Liang, L.; Li, Y.; Gong, T.; Chen, J.-J.; Chen, T.-J.; Yang, J.-L.; Zhu, P. Construction and optimization of microbial cell factories for sustainable production of bioactive dammarenediol-II glucosides. Green Chem. 2019, 21, 3286–3299. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.-C.; Kim, W.; Park, S.C.; Jeong, J.; Park, M.K.; Lim, S.; Lee, Y.; Im, W.-T.; Lee, J.H.; Choi, G.; et al. Two Ginseng UDP-Glycosyltransferases Synthesize Ginsenoside Rg3 and Rd. Plant Cell Physiol. 2014, 55, 2177–2188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Wei, W.; Ye, W.; Li, X.; Zhao, W.; Yang, C.; Li, C.; Yan, X.; Zhou, Z. Synthesizing ginsenoside Rh2 in Saccharomyces cerevisiae cell factory at high-efficiency. Cell Discov. 2019, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.M.; Xue, J.; Min, J.; Qin, L.J.; Zhang, J.K.; Dai, L.H. Biocatalytic synthesis of ginsenoside Rh2 using Arabidopsis thaliana glucosyltransferase-catalyzed coupled reactions. J. Biotechnol. 2020, 309, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Naoumkina, M.A.; Modolo, L.V.; Huhman, D.V.; Urbanczyk-Wochniak, E.; Tang, Y.; Sumner, L.W.; Dixon, R.A. Genomic and Coexpression Analyses Predict Multiple Genes Involved in Triterpene Saponin Biosynthesis in Medicago truncatula. Plant Cell 2010, 22, 850–866. [Google Scholar] [CrossRef] [Green Version]
- Augustin, J.M.; Drok, S.; Shinoda, T.; Sanmiya, K.; Nielsen, J.K.; Khakimov, B.; Olsen, C.E.; Hansen, E.H.; Kuzina, V.; Ekstrøm, C.T.; et al. UDP-Glycosyltransferases from the UGT73C Subfamily in Barbarea vulgaris Catalyze Sapogenin 3-O-Glucosylation in Saponin-Mediated Insect Resistance. Plant Physiol. 2012, 160, 1881–1895. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, Y.; Yang, G.-Y.; Chen, X.; Liu, Q.; Zhang, X.; Deng, Z.; Feng, Y. Biosynthesis of plant-derived ginsenoside Rh2 in yeast via repurposing a key promiscuous microbial enzyme. Metab. Eng. 2017, 42, 25–32. [Google Scholar] [CrossRef]
- Liang, H.C.; Hu, Z.F.; Zhang, T.T.; Gong, T.; Chen, J.J.; Zhu, P.; Li, Y.; Yang, J.L. Production of a bioactive unnatural gin-senoside by metabolically engineered yeasts based on a new UDP-glycosyltransferase from Bacillus subtilis. Metab. Eng. 2017, 44, 60–69. [Google Scholar] [CrossRef]
- Dai, L.; Liu, C.; Li, J.; Dong, C.; Yang, J.; Dai, Z.; Zhang, X.; Sun, Y. One-Pot Synthesis of Ginsenoside Rh2 and Bioactive Unnatural Ginsenoside by Coupling Promiscuous Glycosyltransferase from Bacillus subtilis 168 to Sucrose Synthase. J. Agric. Food Chem. 2018, 66, 2830–2837. [Google Scholar] [CrossRef]
- Dai, L.H.; Li, J.; Yang, J.G.; Zhu, Y.M.; Men, Y.; Zeng, Y.; Cai, Y.; Dong, C.X.; Dai, Z.B.; Zhang, X.L.; et al. Use of a Pro-miscuous Glycosyltransferase from Bacillus subtilis 168 for the Enzymatic Synthesis of Novel Protopanaxatriol-Type Ginseno-sides. J. Agric. Food Chem. 2018, 66, 943–949. [Google Scholar] [CrossRef]
- Ma, W.; Zhao, L.; Ma, Y.; Li, Y.; Qin, S.; He, B. Oriented efficient biosynthesis of rare ginsenoside Rh2 from PPD by compiling UGT-Yjic mutant with sucrose synthase. Int. J. Biol. Macromol. 2020, 146, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.Q. Immobilised enzymes: Science or art? Curr. Opin. Chem. Biol. 2005, 9, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A.; van Pelt, S. Enzyme immobilisation in biocatalysis: Why, what and how. Chem. Soc. Rev. 2013, 42, 6223–6235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pescador, P.; Katakis, I.; Toca-Herrera, J.L.; Donath, E. Efficiency of a Bienzyme Sequential Reaction System Immobilized on Polyelectrolyte Multilayer-Coated Colloids. Langmuir 2008, 24, 14108–14114. [Google Scholar] [CrossRef]
- Feng, J.; Balaji, N.; Surya, M. Materials-Based Strategies for Multi-Enzyme Immobilization and Co-Localization: A Review. Biotechnol. Bioeng. 2014, 111, 209–222. [Google Scholar] [CrossRef]
- Ley, C.; Holtmann, D.; Mangold, K.-M.; Schrader, J. Immobilization of histidine-tagged proteins on electrodes. Colloids Surf. B Biointerfaces 2011, 88, 539–551. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, J.; Chen, X.; Wang, P.G. Combined Biosynthetic Pathway for De Novo Production of UDP-Galactose: Catalysis with Multiple Enzymes Immobilized on Agarose Beads. ChemBioChem 2002, 3, 348–355. [Google Scholar] [CrossRef]
- Kim, H.; Kwon, H.-S.; Ahn, J.; Lee, C.-H.; Ahn, I.-S. Evaluation of a silica-coated magnetic nanoparticle for the immobilization of a His-tagged lipase. Biocatal. Biotransformation 2009, 27, 246–253. [Google Scholar] [CrossRef]
- de Melo, R.R.; Alnoch, R.C.; Vilela AF, L.; de Souza, E.M.; Krieger, N.; Ruller, R.; Sato, H.H.; Mateo, C. New Heterofunctional Supports Based on nanoparticle for the immobilization: A Tool for Enzyme Immobilization at Neutral pH. Molecules 2017, 22, 1088. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Huang, S.S.; Xu, L.; Yan, Y.J. Improving activity and enantioselectivity of lipase via immobilization on macroporous resin for resolution of racemic 1-phenylethanol in non-aqueous medium. BMC Biotechnol. 2013, 13, 92. [Google Scholar] [CrossRef] [Green Version]
- Blanco, R.M.; Terreros, P.; Muñoz, N.; Serra, E. Ethanol improves lipase immobilization on a hydrophobic support. J. Mol. Catal. B Enzym. 2007, 47, 13–20. [Google Scholar] [CrossRef]
- Mateo, C.; Bolívar, J.M.B.; Godoy, C.A.; Rocha-Martin, J.; Pessela, B.C.; Curiel, J.A.; Muñoz, R.; Guisán, J.M.; Fernández-Lorente, G. Improvement of Enzyme Properties with a Two-Step Immobilization Process on Novel Heterofunctional Supports. Biomacromolecules 2010, 11, 3112–3117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateo, C.; Abian, O.; Fernández-Lorente, G.; Pedroche, J.; Fernández-Lafuente, R.; Guisan, J.M.; Tam, A.; Daminati, M. Epoxy Sepabeads: A Novel Epoxy Support for Stabilization of Industrial Enzymes via Very Intense Multipoint Covalent Attachment. Biotechnol. Prog. 2002, 18, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Mokhtar, N.F.; Rahman, R.N.Z.R.A.; Noor, N.D.M.; Shariff, F.M.; Ali, M.S.M. The Immobilization of Lipases on Porous Support by Adsorption and Hydrophobic Interaction Method. Catalysts 2020, 10, 744. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Heterofunctional Support | Activity (mU/g Wet Support) | Enzyme Activity Recovery (%) | Protein Adsorption (mg/g Wet Support) | Specific Activity of Immobilized Enzyme (U/mg Protein) |
|---|---|---|---|---|---|
| M315F | Glyoxyl-Ni2+-chelate resin | 718.5 | 47.9% | 1.74 | 412.7 |
| Glyoxyl-Cu2+-chelate resin | 182.1 | 12.1% | 2.54 | 71.7 | |
| Glyoxyl-Co2+-chelate resin | 629.0 | 41.9% | 2.71 | 232.1 | |
| AtSuSy | Glyoxyl-Ni2+-chelate resin | 1959.5 | 39.1% | 1.18 | 1660.5 |
| Glyoxyl-Cu2+-chelate resin | 1862.5 | 37.2% | 2.46 | 350.6 | |
| Glyoxyl-Co2+-chelate resin | 1636.5 | 32.7% | 1.71 | 957.0 |
| Support | Specific Surface Area (m2/g) | Pore Volume (cm3/g) | Average Pore Diameter (nm) |
|---|---|---|---|
| Heterofunctional LX1000HG | 41.34 | 0.47 | 33.67 |
| Resin with co-immobilized enzymes | 58.93 | 0.37 | 27.64 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chu, J.; Yue, J.; Qin, S.; Li, Y.; Wu, B.; He, B. Biocatalysis for Rare Ginsenoside Rh2 Production in High Level with Co-Immobilized UDP-Glycosyltransferase Bs-YjiC Mutant and Sucrose Synthase AtSuSy. Catalysts 2021, 11, 132. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11010132
Chu J, Yue J, Qin S, Li Y, Wu B, He B. Biocatalysis for Rare Ginsenoside Rh2 Production in High Level with Co-Immobilized UDP-Glycosyltransferase Bs-YjiC Mutant and Sucrose Synthase AtSuSy. Catalysts. 2021; 11(1):132. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11010132
Chicago/Turabian StyleChu, Jianlin, Jiheng Yue, Song Qin, Yuqiang Li, Bin Wu, and Bingfang He. 2021. "Biocatalysis for Rare Ginsenoside Rh2 Production in High Level with Co-Immobilized UDP-Glycosyltransferase Bs-YjiC Mutant and Sucrose Synthase AtSuSy" Catalysts 11, no. 1: 132. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11010132