
Combustion-Synthesized Porous CuO-CeO2-SiO2 Composites as Solid Catalysts for the Alkenylation of C(sp3)-H Bonds Adjacent to a Heteroatom via Cross-Dehydrogenative Coupling

 , and
, and
Abstract
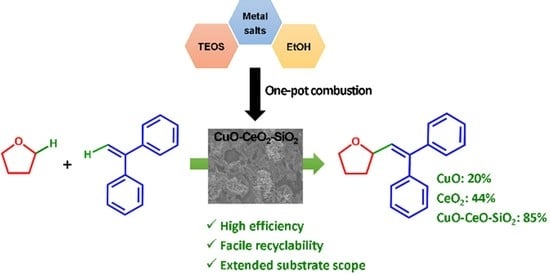
:
1. Introduction
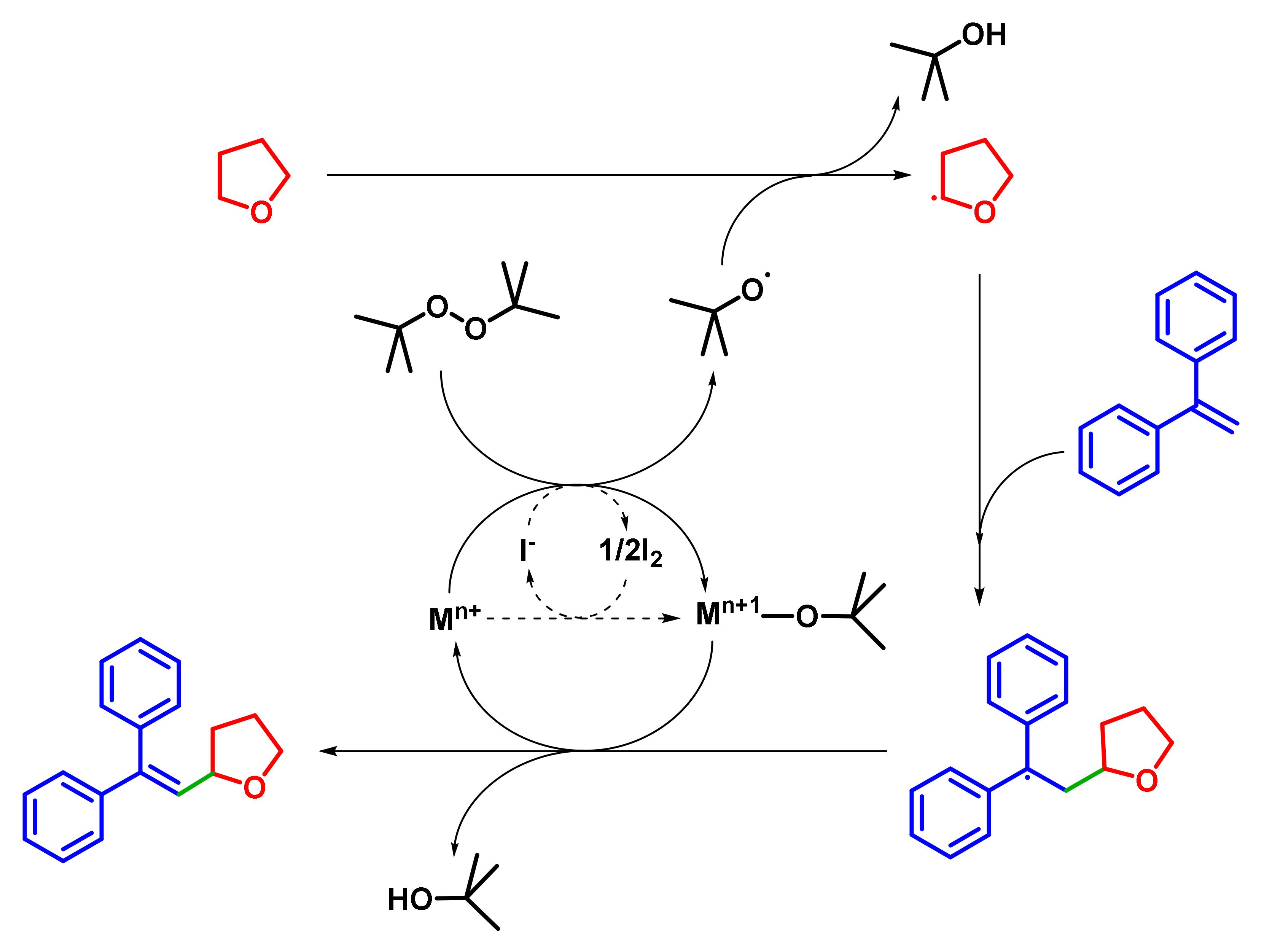
2. Results and Discussion
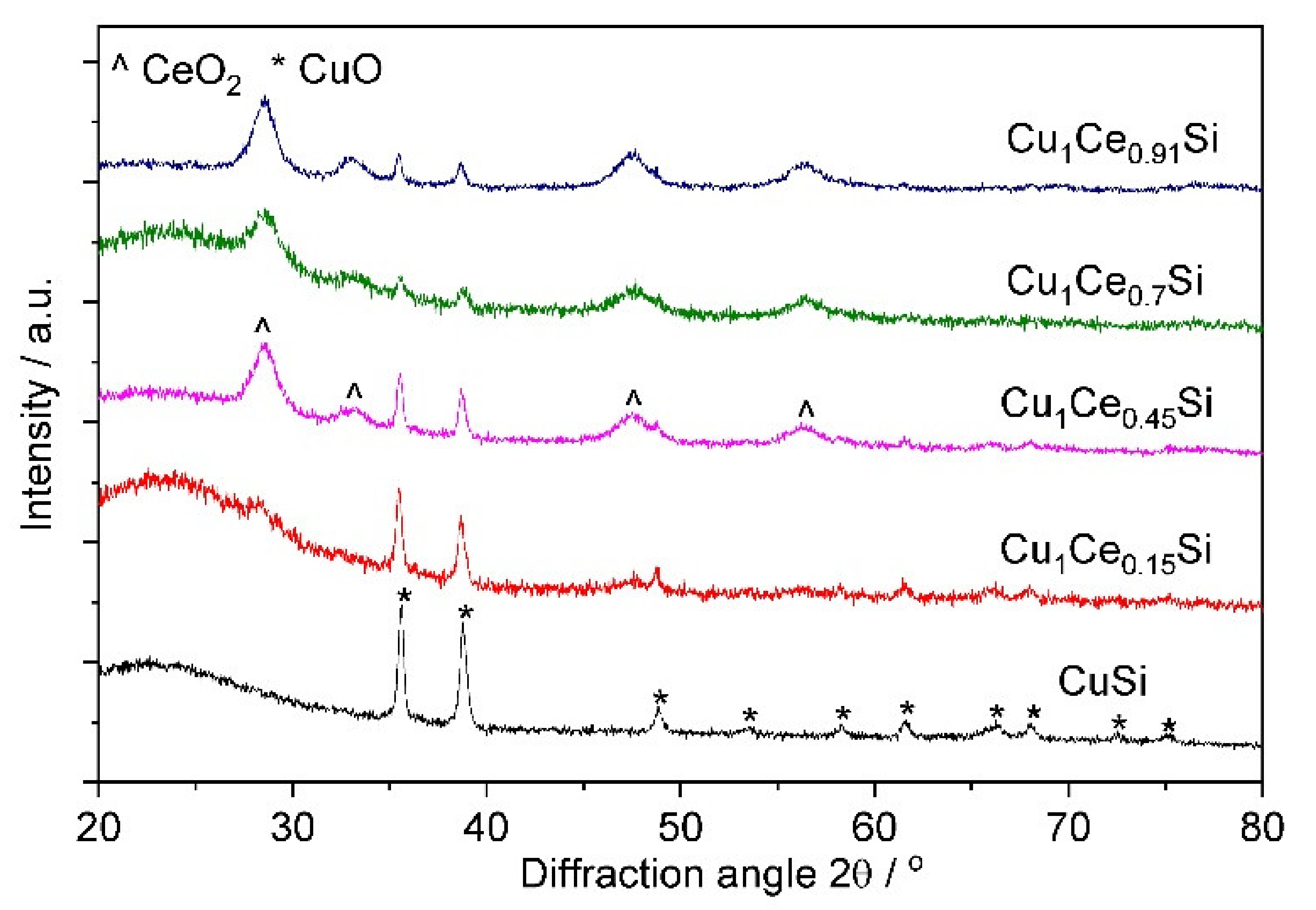
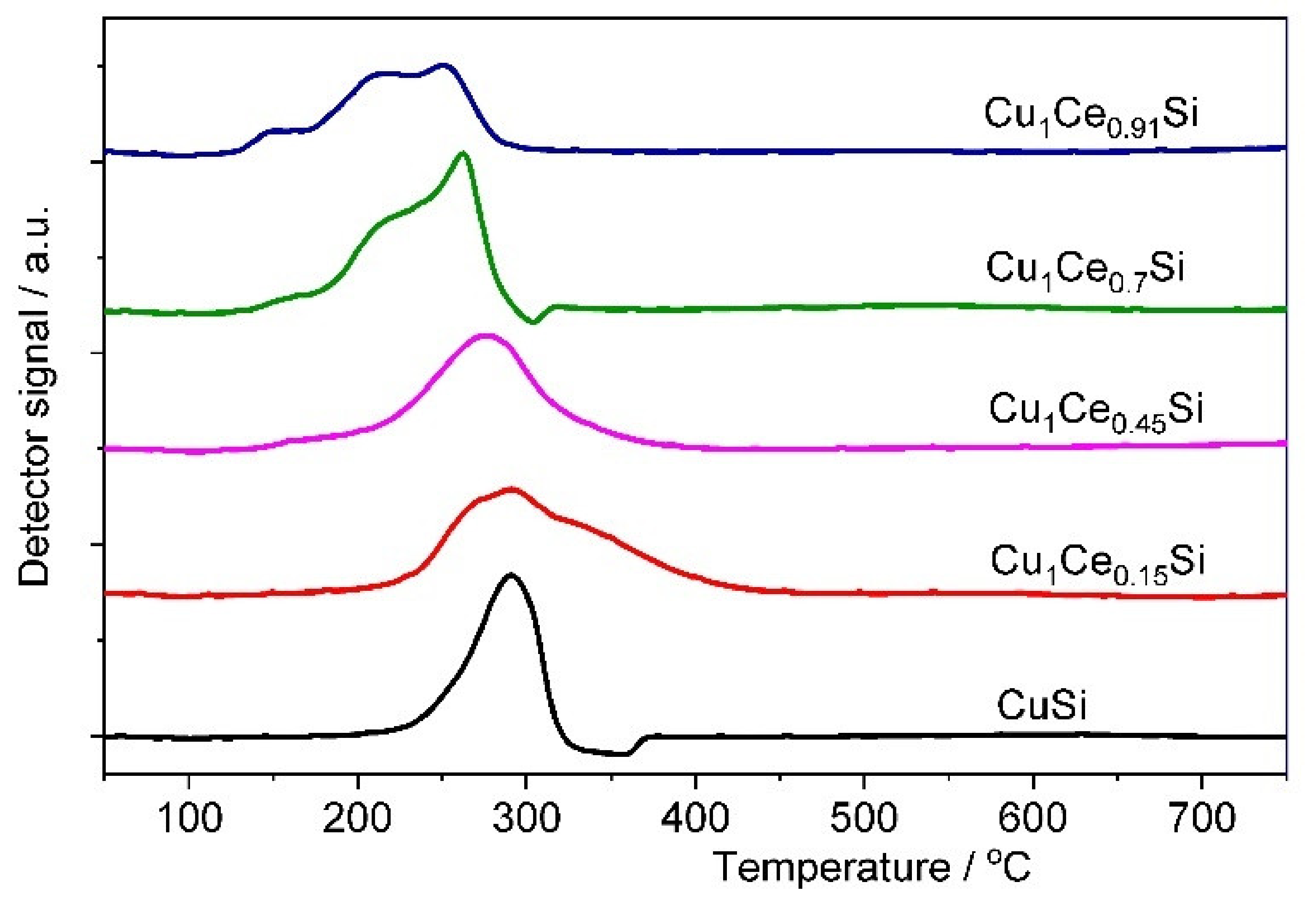
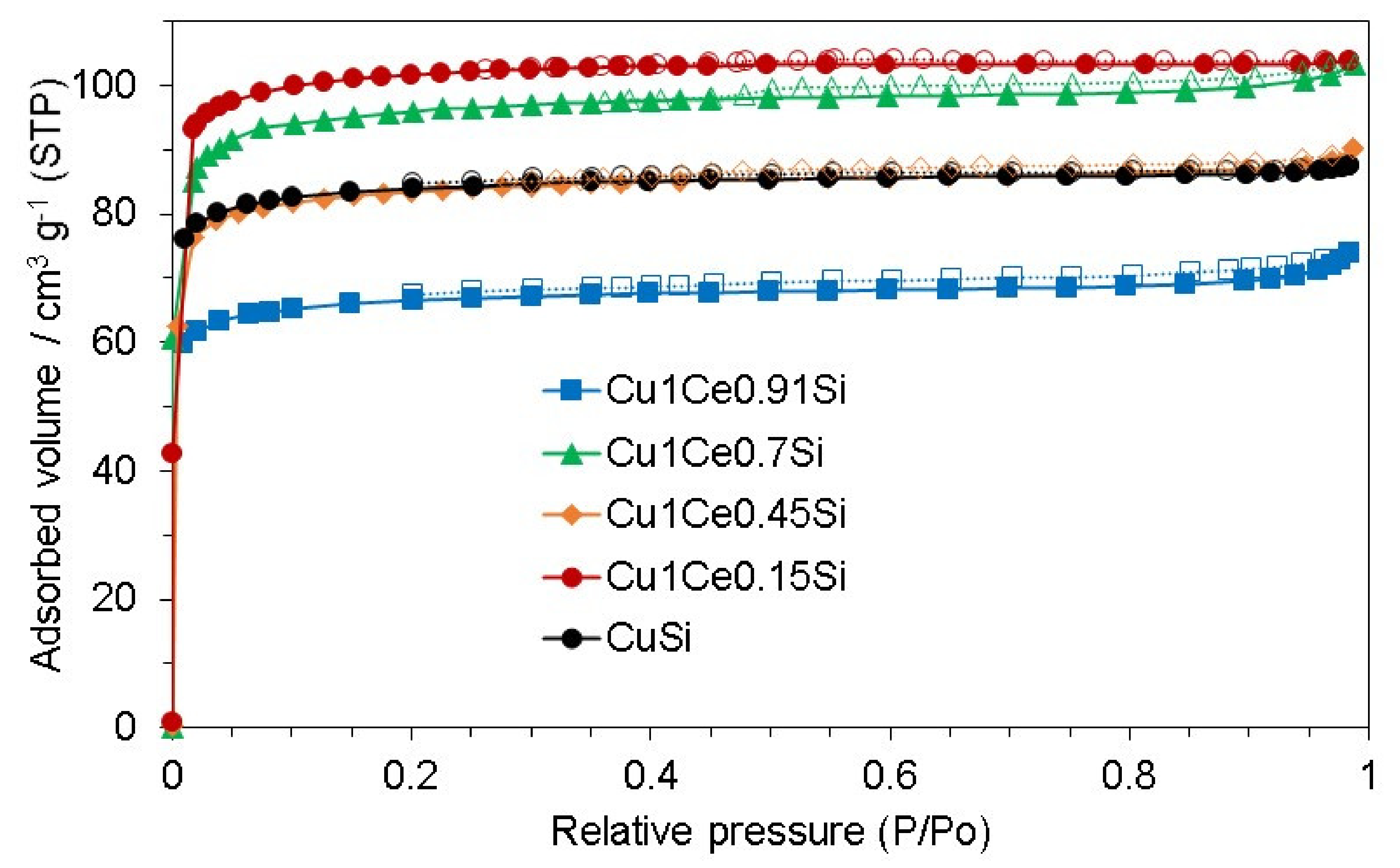
2.1. Catalyst Characterization
2.2. Catalytic Study
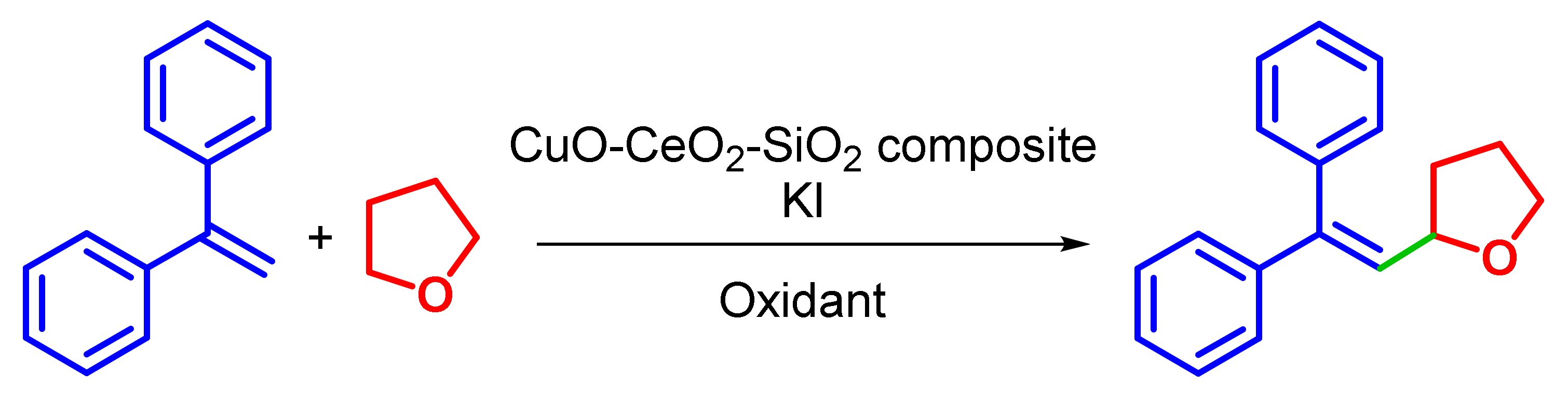
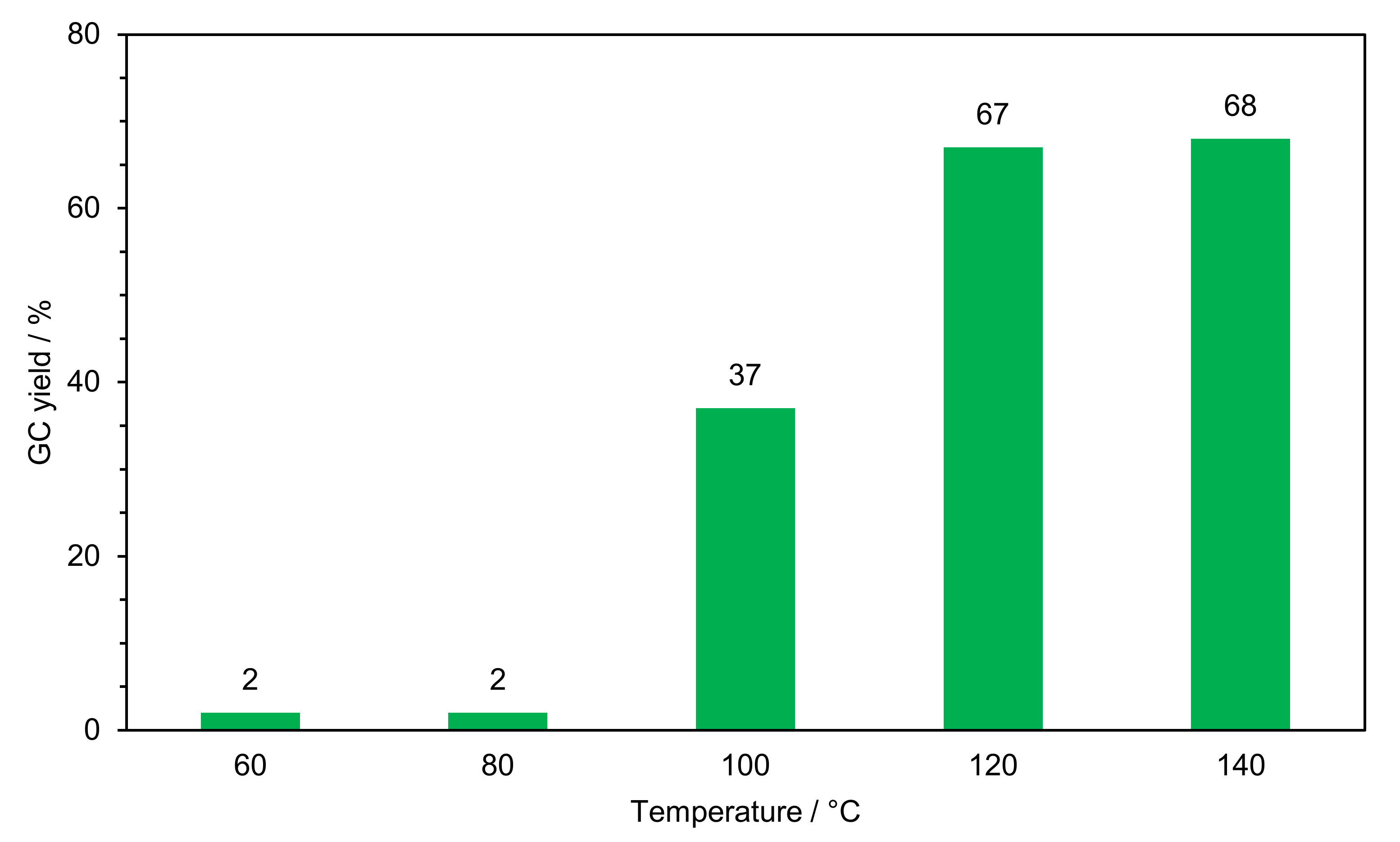
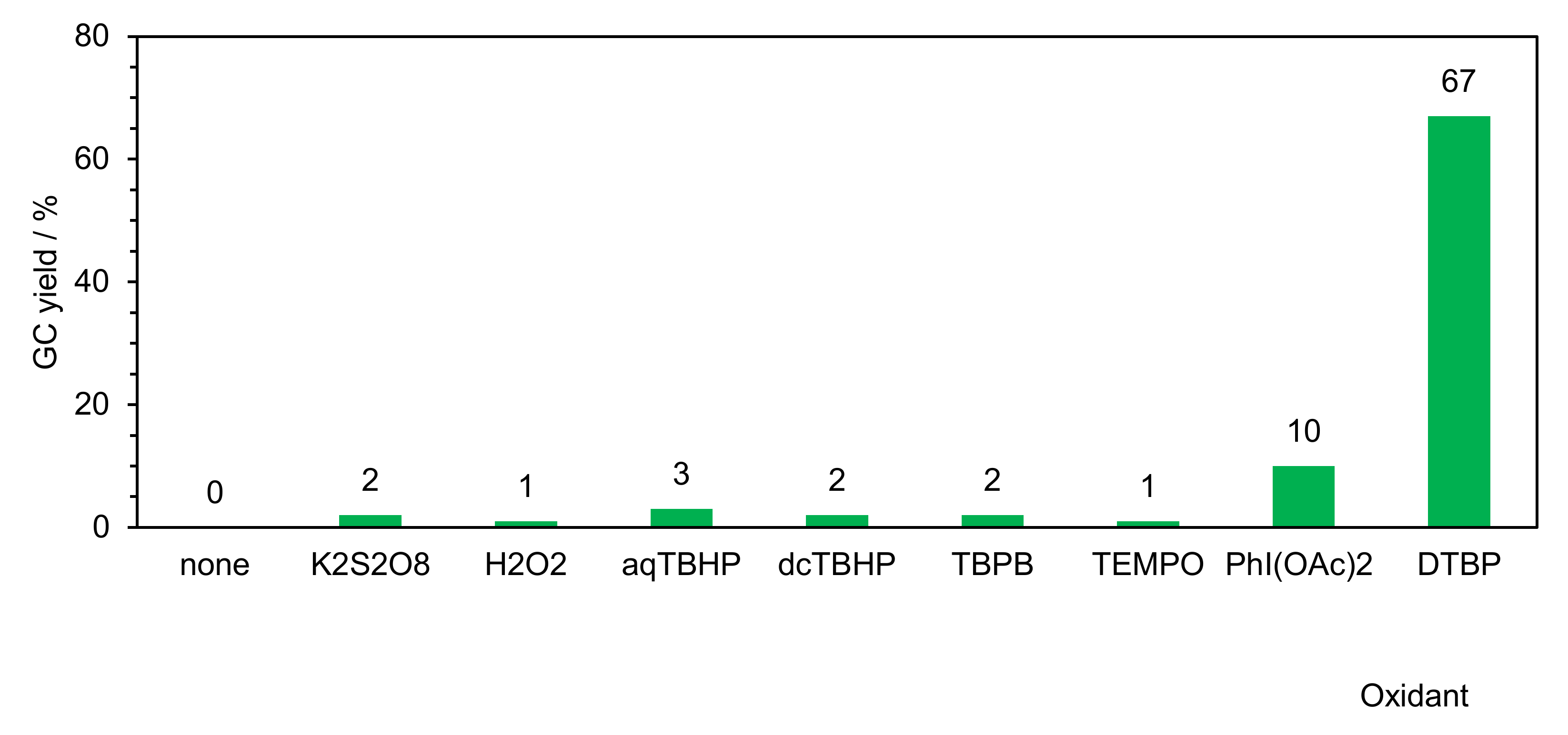
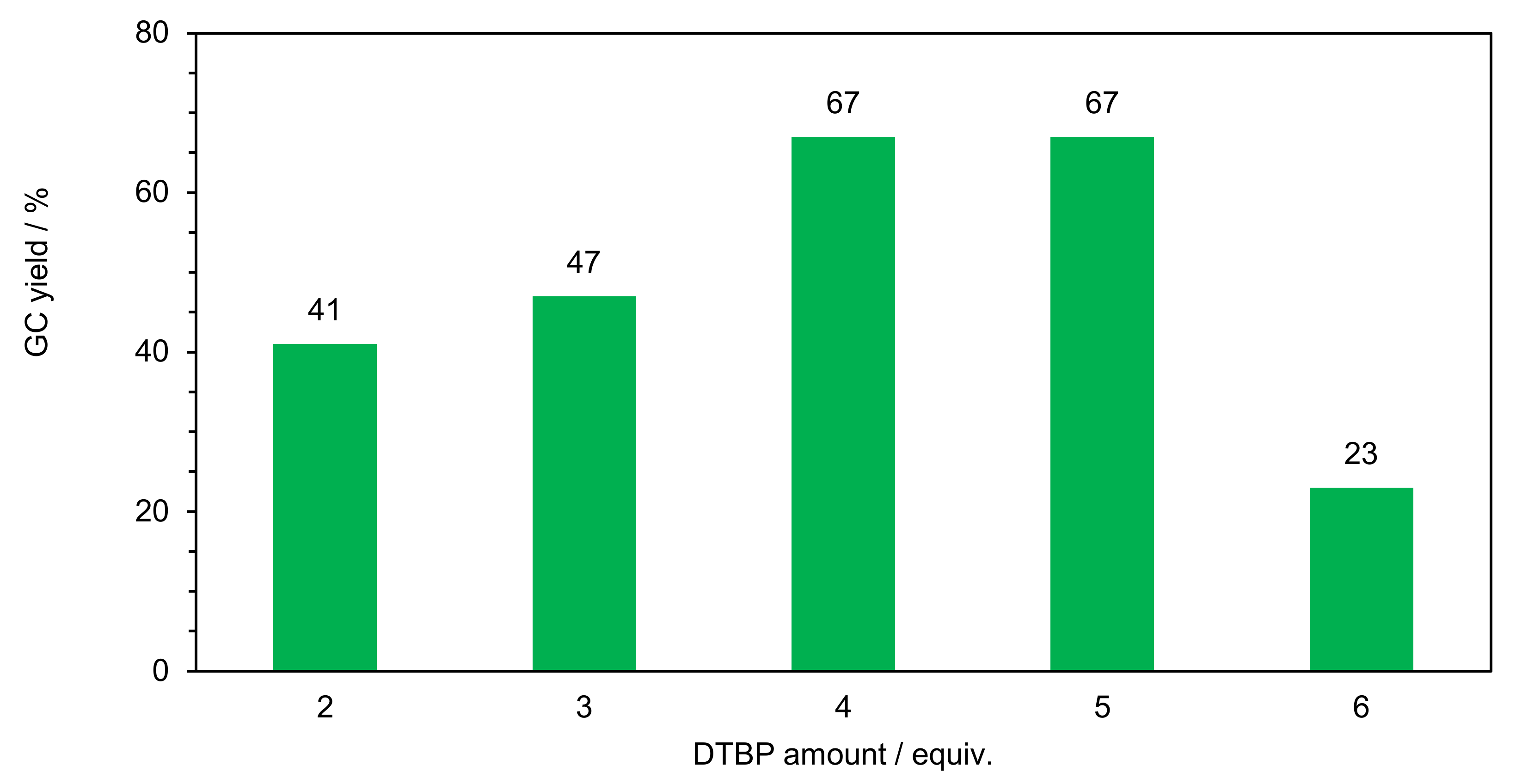
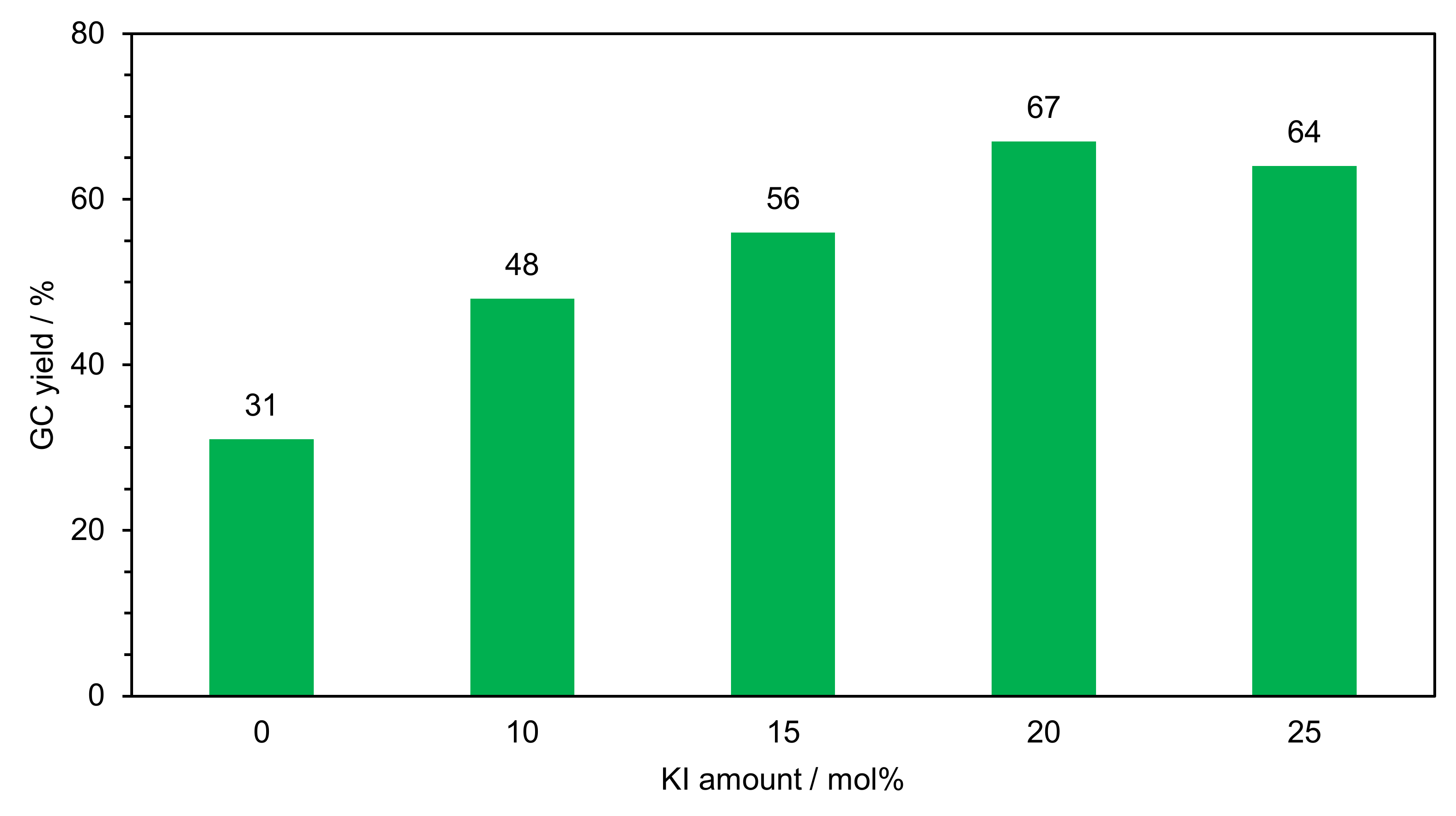
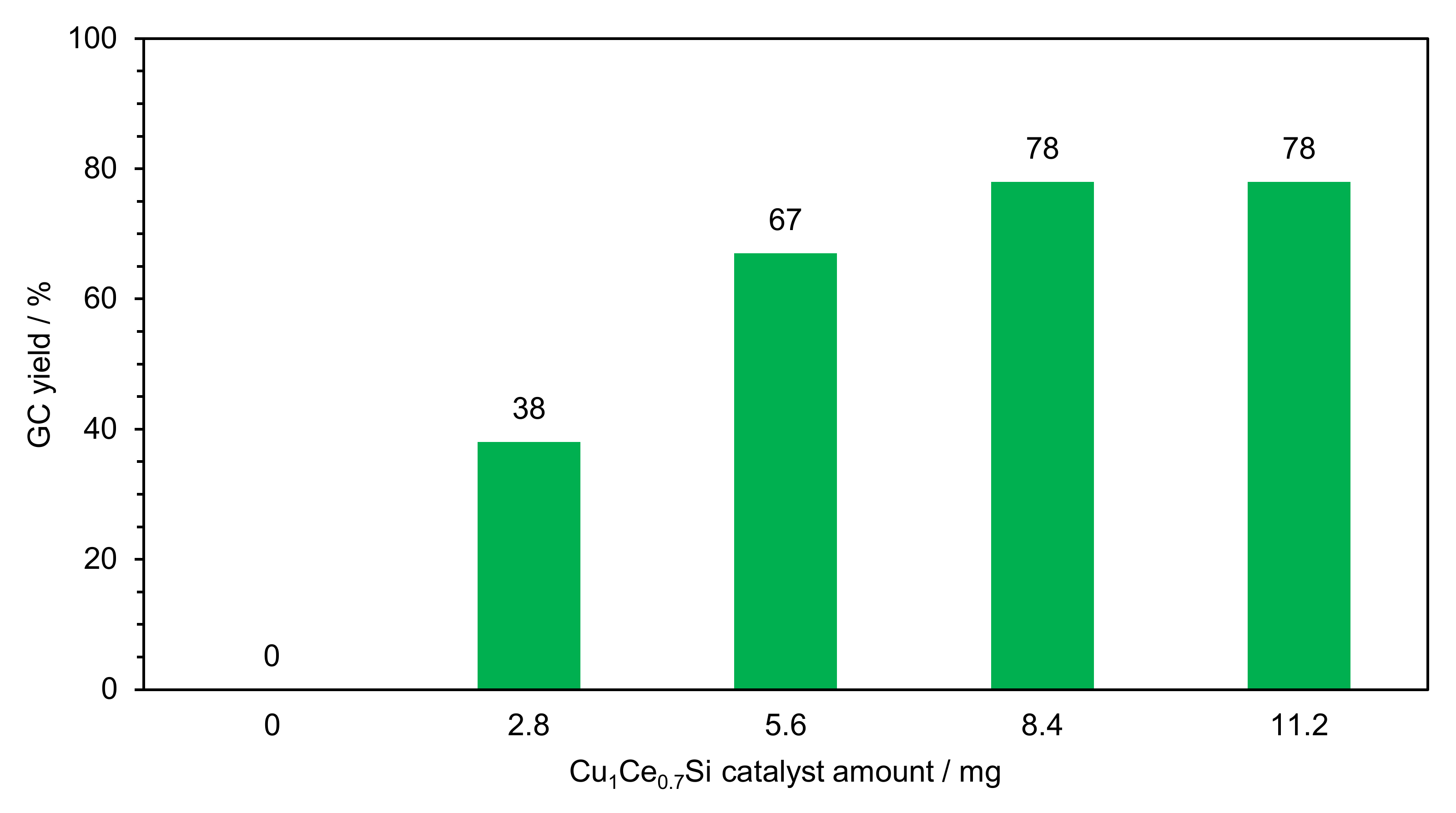
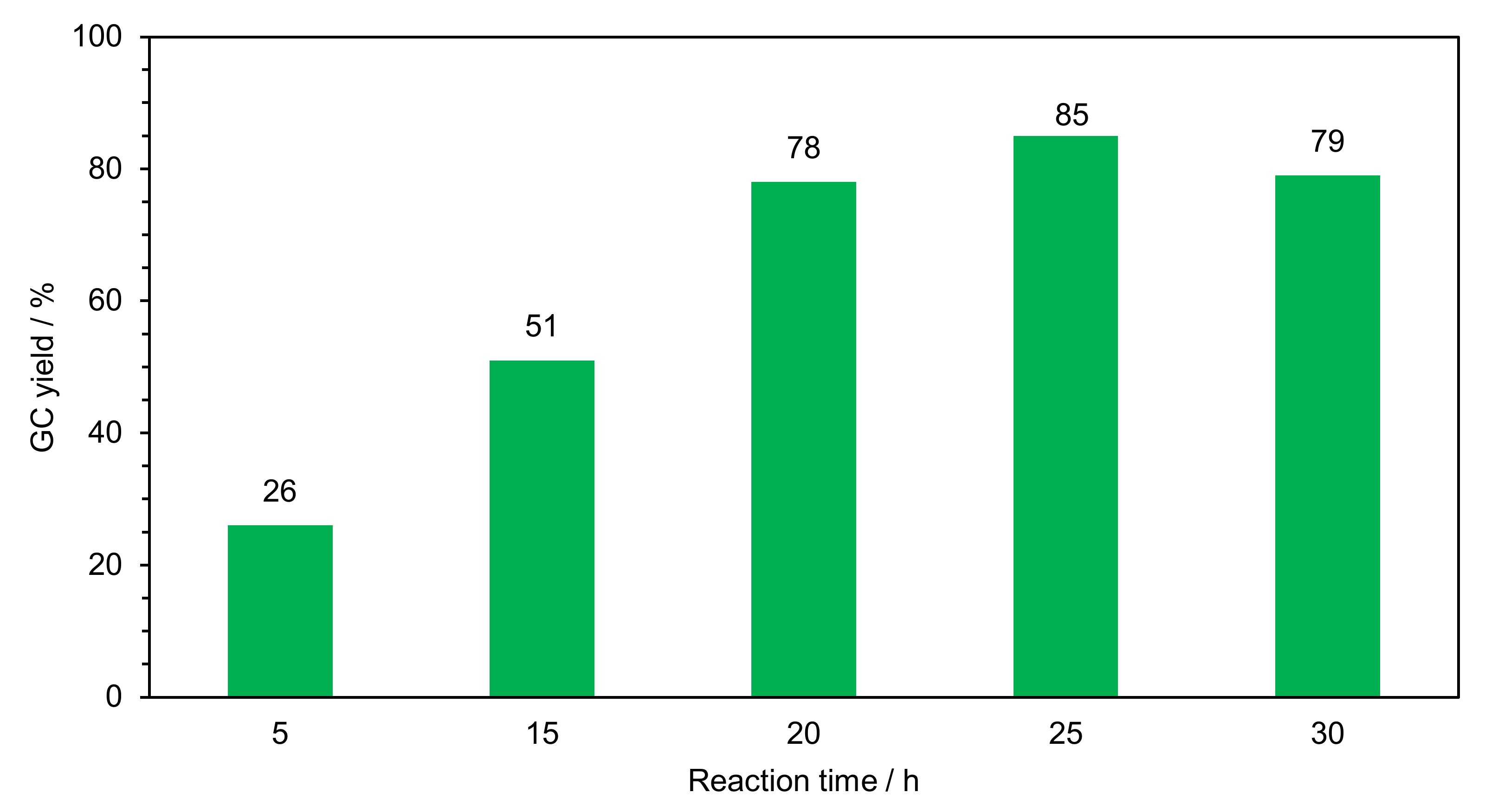
2.2.1. Optimization of Reaction Conditions
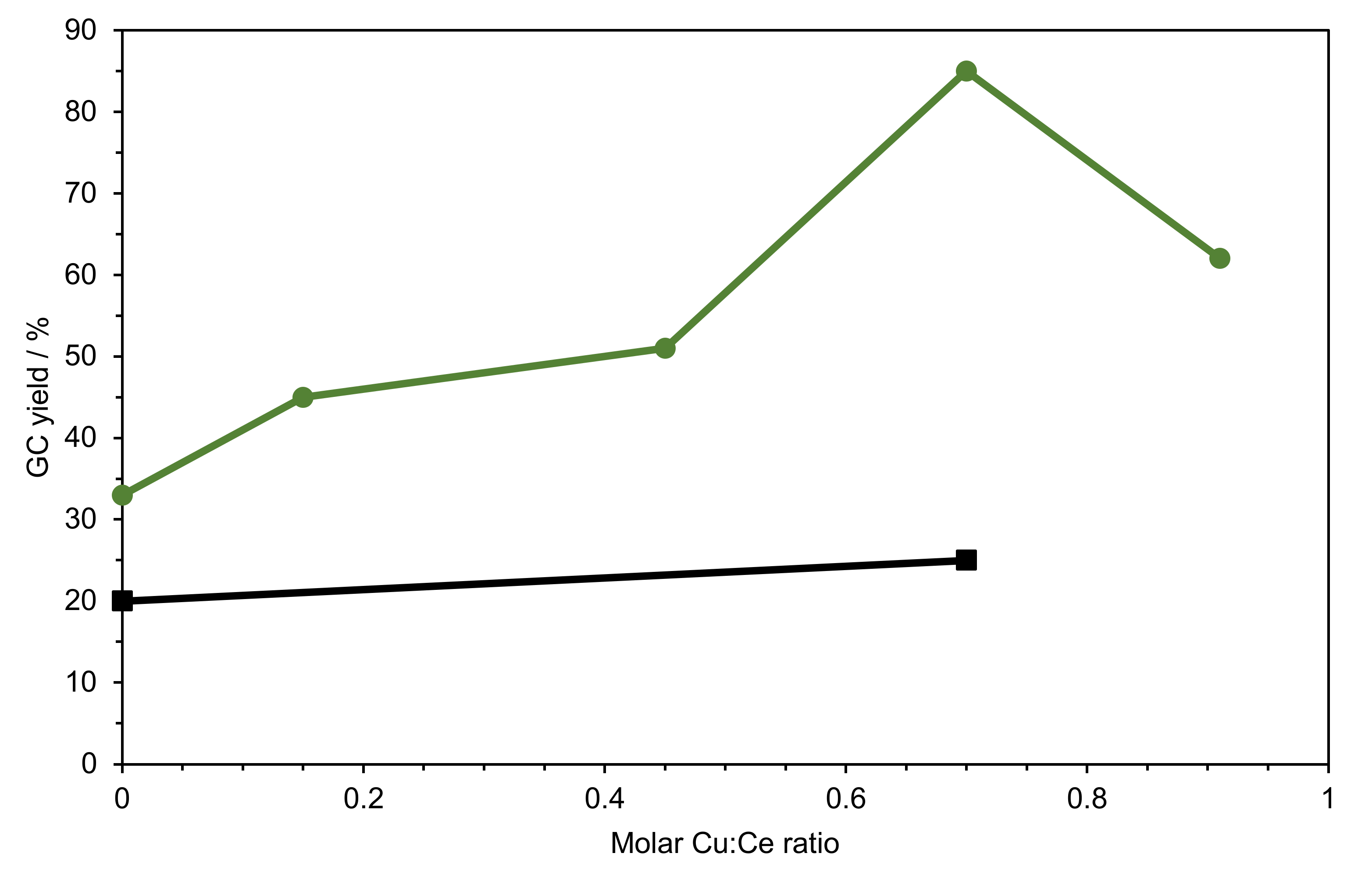
2.2.2. Comparison of Catalysts
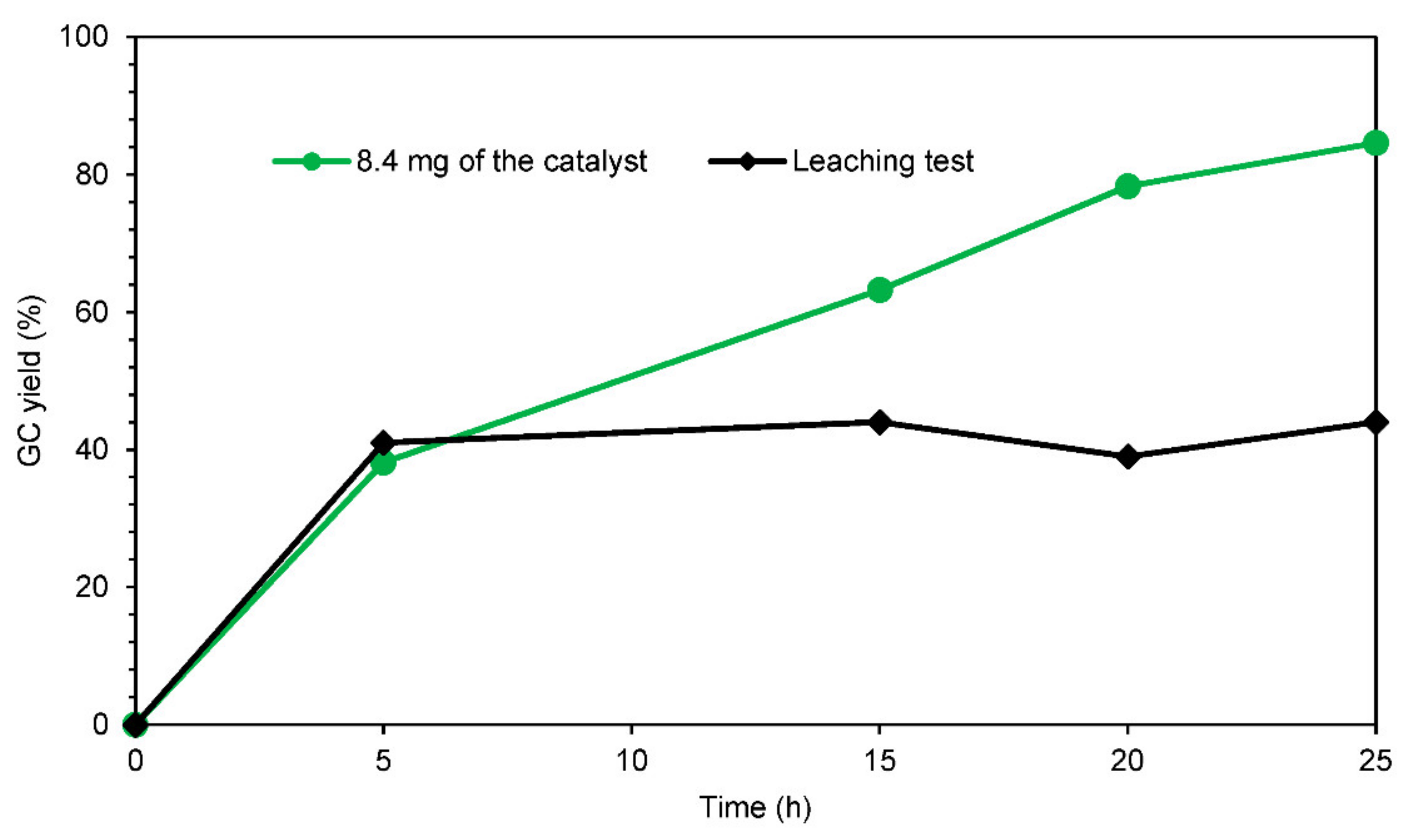
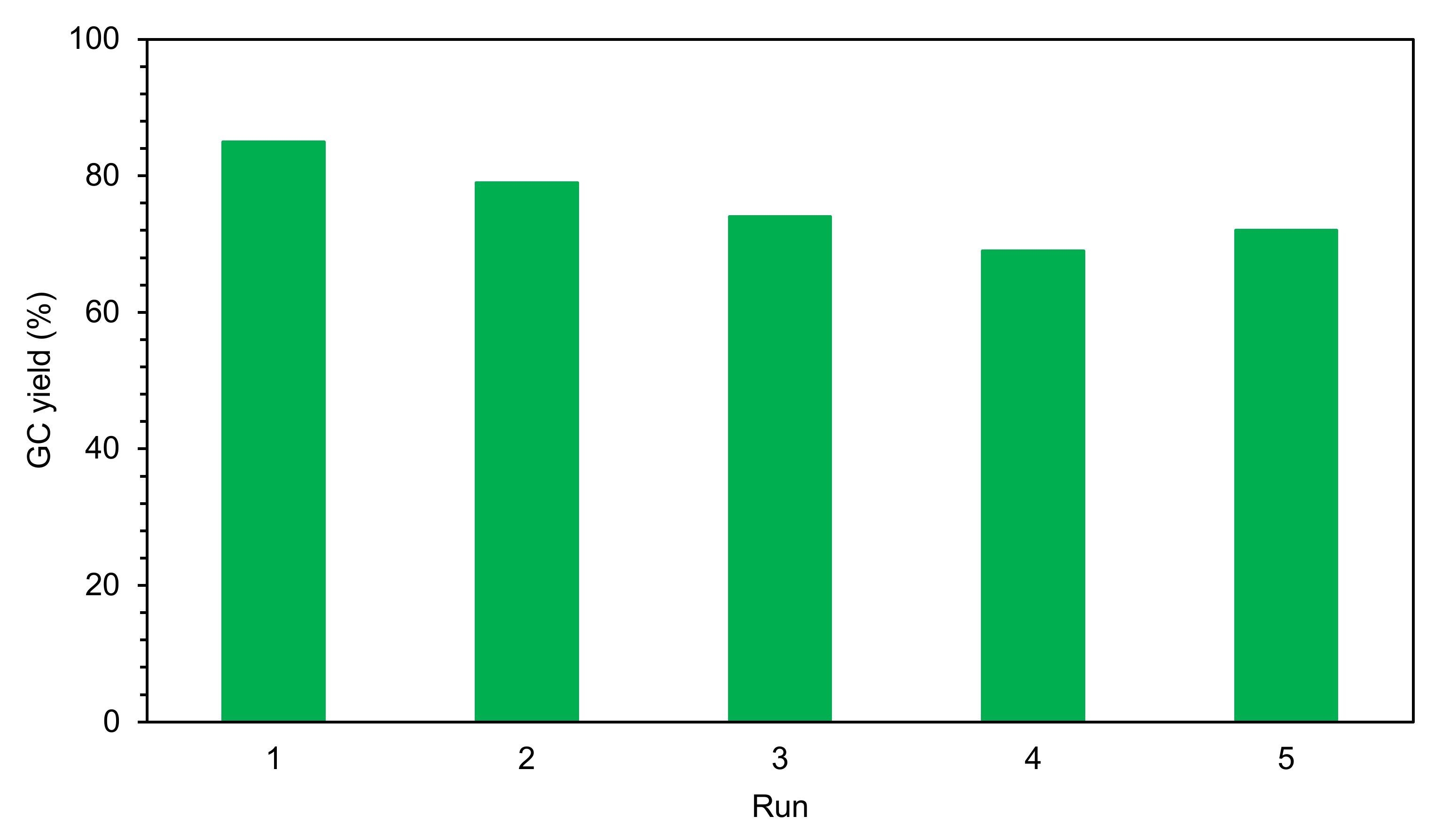
2.2.3. Leaching Test and Recycling of the Heterogeneous Catalyst
2.2.4. Dehydrogenative Coupling of Different Substrates
3. Materials and Methods
3.1. Synthesis of Composites
3.2. Characterization of As-Synthesized Composites
3.3. Catalytic Tests
3.4. Isolation and Identification of the Products
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yeung, C.S.; Dong, V.M. Catalytic dehydrogenative cross-coupling: Forming carbon-carbon bonds by oxidizing two carbon-hydrogen bonds. Chem. Rev. 2011, 111, 1215–1292. [Google Scholar] [CrossRef]
- Bras, J.L.; Muzart, J. Pd-Catalyzed Intermolecular Dehydrogenative Heck Reactions of Five-Membered Heteroarenes. Catalysts 2020, 10, 571. [Google Scholar] [CrossRef]
- Bosque, I.; Chinchilla, R.; Gonzalez-Gomez, J.C.; Guijarro, D.; Alonso, F. Cross-dehydrogenative coupling involving benzylic and allylic C–H bonds. Org. Chem. Front. 2020, 7, 1717–1742. [Google Scholar] [CrossRef]
- Huang, C.Y.; Kang, H.; Li, J.; Li, C.J. En Route to Intermolecular Cross-Dehydrogenative Coupling Reactions. J. Org. Chem. 2019, 84, 12705–12721. [Google Scholar] [CrossRef]
- Niu, B.; Zhao, W.; Ding, Y.; Bian, Z.; Pittman, C.U.; Zhou, A.; Ge, H. Regioselective Cross-Couplings of Coumarins and Flavones with Ethers via C(sp3)–H Functionalization. J. Org. Chem. 2015, 80, 7251–7257. [Google Scholar] [CrossRef]
- Zhang, M.; Yang, L.; Yang, H.; An, G.; Li, G. Visible Light Mediated C(sp3)-H Alkenylation of Cyclic Ethers Enabled by Aryl Ketone. ChemCatChem 2019, 11, 1606–1609. [Google Scholar] [CrossRef]
- Gandhi, S. Catalytic enantioselective cross dehydrogenative coupling of sp(3) C-H of heterocycles. Org. Biomol. Chem. 2019, 17, 9683–9692. [Google Scholar] [CrossRef]
- Guo, S.-R.; Kumar, P.S.; Yang, M. Recent Advances of Oxidative Radical Cross-Coupling Reactions: Direct α-C(sp3)-H Bond Functionalization of Ethers and Alcohols. Adv. Synth. Catal. 2017, 359, 2–25. [Google Scholar] [CrossRef]
- Faisca Phillips, A.M.; Pombeiro, A.J.L. Recent Developments in Transition Metal-Catalyzed Cross-Dehydrogenative Coupling Reactions of Ethers and Thioethers. ChemCatChem 2018, 10, 3354–3383. [Google Scholar] [CrossRef]
- He, C.; Whitehurst, W.G.; Gaunt, M.J. Palladium-Catalyzed C(sp3)–H Bond Functionalization of Aliphatic Amines. Chem 2019, 5, 1031–1058. [Google Scholar] [CrossRef]
- Varun, B.V.; Dhineshkumar, J.; Bettadapur, K.R.; Siddaraju, Y.; Alagiri, K.; Prabhu, K.R. Recent advancements in dehydrogenative cross coupling reactions for CC bond formation. Tetrahedron Lett. 2017, 58, 803–824. [Google Scholar] [CrossRef]
- Batra, A.; Singh, P.; Singh, K.N. Recent Advances in Functionalization of α-C(sp3)-H Centres in Inactivated Ethers through Cross Dehydrogenative Coupling. Eur. J. Org. Chem. 2017, 2017, 3739–3762. [Google Scholar] [CrossRef]
- Liu, D.; Liu, C.; Li, H.; Lei, A. Copper-catalysed oxidative C-H/C-H coupling between olefins and simple ethers. Chem. Commun. 2014, 50, 3623–3626. [Google Scholar] [CrossRef] [PubMed]
- Dian, L.; Zhao, H.; Zhang-Negrerie, D.; Du, Y. Cobalt-Catalyzed Twofold Direct C(sp2)−C(sp3) Bond Coupling: Regioselective C-3 Alkylation of Coumarins with (Cyclo)alkyl Ethers. Adv. Synth. Catal. 2016, 358, 2422–2426. [Google Scholar] [CrossRef]
- Trinh, K.H.; Tran, P.H.; Nguyen, T.T.; Doan, S.H.; Le, M.-V.; Nguyen, T.T.; Phan, N.T.S. Direct oxidative C(sp3)−H/C(sp2)−H coupling reaction using recyclable Sr-doped LaCoO3 perovskite catalyst. Appl. Organomet. Chem. 2020, 34, e5515. [Google Scholar] [CrossRef]
- Cam, T.S.; Vishnievskaia, T.A.; Popkov, V.I. Catalytic oxidation of CO over CuO/CeO2 nanocomposites synthesized via solution combustion method: Effect of fuels. Rev. Adv. Mater. Sci. 2020, 59, 131–143. [Google Scholar] [CrossRef]
- Liu, B.; Li, Y.; Cao, Y.; Wang, L.; Qing, S.; Wang, K.; Jia, D. Optimum Balance of Cu+ and Oxygen Vacancies of CuOx-CeO2 Composites for CO Oxidation Based on Thermal Treatment. Eur. J. Inorg. Chem. 2019, 2019, 1714–1723. [Google Scholar] [CrossRef]
- Chen, C.; Zhan, Y.; Zhou, J.; Li, D.; Zhang, Y.; Lin, X.; Jiang, L.; Zheng, Q. Cu/CeO2 Catalyst for Water-Gas Shift Reaction: Effect of CeO2 Pretreatment. ChemPhysChem 2018, 19, 1448–1455. [Google Scholar] [CrossRef]
- Zhu, C.; Ding, T.; Gao, W.; Ma, K.; Tian, Y.; Li, X. CuO/CeO2 catalysts synthesized from Ce-UiO-66 metal-organic framework for preferential CO oxidation. Int. J. Hydrog. Energy 2017, 42, 17457–17465. [Google Scholar] [CrossRef]
- Montini, T.; Melchionna, M.; Monai, M.; Fornasiero, P. Fundamentals and Catalytic Applications of CeO2-Based Materials. Chem. Rev. 2016, 116, 5987–6041. [Google Scholar] [CrossRef]
- Hu, C.; Zhu, Q.; Jiang, Z.; Zhang, Y.; Wang, Y. Preparation and formation mechanism of mesoporous CuO-CeO2 mixed oxides with excellent catalytic performance for removal of VOCs. Microporous Mesoporous Mater. 2008, 113, 427–434. [Google Scholar] [CrossRef]
- Águila, G.; Gracia, F.; Araya, P. CuO and CeO2 catalysts supported on Al2O3, ZrO2, and SiO2 in the oxidation of CO at low temperature. Appl. Catal. A Gen. 2008, 343, 16–24. [Google Scholar] [CrossRef]
- Delimaris, D.; Ioannides, T. VOC oxidation over CuO-CeO2 catalysts prepared by a combustion method. Appl. Catal. B Environ. 2009, 89, 295–302. [Google Scholar] [CrossRef]
- Baidya, T.; Mazumder, T.; Koltunov, K.Y.; Likhar, P.R.; Clark, A.H.; Tiwari, K.; Sobolev, V.I.; Payra, S.; Murayama, T.; Lin, M.; et al. Low-Temperature Propylene Epoxidation Activity of CuO-CeO2 Catalyst with CO + O2: Role of Metal–Support Interaction on the Reducibility and Catalytic Property of CuOx Species. J. Phys. Chem. C 2020, 124, 14131–14146. [Google Scholar] [CrossRef]
- Tiscornia, I.S.; Lacoste, A.M.; Gómez, L.E.; Boix, A.V. CuO-CeO2/SiO2 coating on ceramic monolith: Effect of the nature of the catalyst support on CO preferential oxidation in a H2-rich stream. Int. J. Hydrog. Energy 2020, 45, 6636–6650. [Google Scholar] [CrossRef]
- Akbar, M.; Tu, Z.; Jin, B.; Mushtaq, N.; He, Z.; Dong, W.; Wang, B.; Wang, X.; Xia, C. Demonstrating the dual functionalities of CeO2-CuO composites in solid oxide fuel cells. Int. J. Hydrog. Energy 2020, 46, 9938–9947. [Google Scholar] [CrossRef]
- The Luong, N.; Okumura, H.; Yamasue, E.; Ishihara, K.N. Structure and catalytic behaviour of CuO-CeO2 prepared by high-energy ball milling. R. Soc. Open Sci. 2019, 6, 181861. [Google Scholar] [CrossRef] [Green Version]
- Hossain, S.T.; Zell, E.T.; Balaz, S.; Wang, R. A γ to α type transition of CuO species over CeO2-SiO2 composites supported CuO catalysts. Appl. Surf. Sci. 2019, 491, 374–382. [Google Scholar] [CrossRef]
- Shang, H.; Zhang, X.; Xu, J.; Han, Y. Effects of preparation methods on the activity of CuO/CeO2 catalysts for CO oxidation. Front. Chem. Sci. Eng. 2017, 11, 603–612. [Google Scholar] [CrossRef]
- Sedmak, G.; Hočevar, S.; Levec, J. Transient kinetic model of CO oxidation over a nanostructured Cu0.1Ce0.9O2−y catalyst. J. Catal. 2004, 222, 87–99. [Google Scholar] [CrossRef]
- Zedan, A.F.; AlJaber, A.S. Combustion Synthesis of Non-Precious CuO-CeO(2) Nanocrystalline Catalysts with Enhanced Catalytic Activity for Methane Oxidation. Materials 2019, 12, 878. [Google Scholar] [CrossRef] [Green Version]
- Prasad, R.; Rattan, G. Preparation Methods and Applications of CuO-CeO2 Catalysts: A Short Review. Bull. Chem. React. Eng. Catal. 2010, 5, 7–30. [Google Scholar] [CrossRef]
- Piumetti, M.; Bensaid, S.; Andana, T.; Russo, N.; Pirone, R.; Fino, D. Cerium-copper oxides prepared by solution combustion synthesis for total oxidation reactions: From powder catalysts to structured reactors. Appl. Catal. B Environ. 2017, 205, 455–468. [Google Scholar] [CrossRef]
- Liu, G.; Chen, K.; Li, J. Combustion synthesis: An effective tool for preparing inorganic materials. Scr. Mater. 2018, 157, 167–173. [Google Scholar] [CrossRef]
- Thoda, O.; Xanthopoulou, G.; Vekinis, G.; Chroneos, A. Review of Recent Studies on Solution Combustion Synthesis of Nanostructured Catalysts. Adv. Eng. Mater. 2018, 20, 1800047. [Google Scholar] [CrossRef]
- Novitskaya, E.; Kelly, J.P.; Bhaduri, S.; Graeve, O.A. A review of solution combustion synthesis: An analysis of parameters controlling powder characteristics. Int. Mater. Rev. 2021, 66, 188–214. [Google Scholar] [CrossRef]
- Carlos, E.; Martins, R.; Fortunato, E.; Branquinho, R. Solution Combustion Synthesis: Towards a Sustainable Approach for Metal Oxides. Chemistry 2020, 26, 9099–9125. [Google Scholar] [CrossRef]
- Xanthopoulou, G.; Thoda, O.; Roslyakov, S.; Steinman, A.; Kovalev, D.; Levashov, E.; Vekinis, G.; Sytschev, A.; Chroneos, A. Solution combustion synthesis of nano-catalysts with a hierarchical structure. J. Catal. 2018, 364, 112–124. [Google Scholar] [CrossRef]
- Deganello, F.; Tyagi, A.K. Solution combustion synthesis, energy and environment: Best parameters for better materials. Prog. Cryst. Growth Charact. Mater. 2018, 64, 23–61. [Google Scholar] [CrossRef]
- Varma, A.; Mukasyan, A.S.; Rogachev, A.S.; Manukyan, K.V. Solution Combustion Synthesis of Nanoscale Materials. Chem. Rev. 2016, 116, 14493–14586. [Google Scholar] [CrossRef]
- Li, F.T.; Ran, J.; Jaroniec, M.; Qiao, S.Z. Solution combustion synthesis of metal oxide nanomaterials for energy storage and conversion. Nanoscale 2015, 7, 17590–17610. [Google Scholar] [CrossRef]
- Bera, P.; Aruna, S.T.; Patil, K.C.; Hegde, M.S. Studies on Cu/CeO2: A New NO Reduction Catalyst. J. Catal. 1999, 186, 36–44. [Google Scholar] [CrossRef]
- Purohit, R.D.; Sharma, B.P.; Pillai, K.T.; Tyagi, A.K. Ultrafine ceria powders via glycine-nitrate combustion. Mater. Res. Bull. 2001, 36, 2711–2721. [Google Scholar] [CrossRef]
- Hu, T.; Yang, J.; Zhao, J.; Wang, D.; Song, H.; Chou, L. Preparation of a Cu–Ce–O Catalyst by Urea Combustion for Removing CO from Hydrogen. Chin. J. Catal. 2007, 28, 844–846. [Google Scholar] [CrossRef]
- Marbán, G.; Fuertes, A.B. Highly active and selective CuOx/CeO2 catalyst prepared by a single-step citrate method for preferential oxidation of carbon monoxide. Appl. Catal. B Environ. 2005, 57, 43–53. [Google Scholar] [CrossRef]
- Avgouropoulos, G.; Ioannides, T. Selective CO oxidation over CuO-CeO2 catalysts prepared via the urea–nitrate combustion method. Appl. Catal. A Gen. 2003, 244, 155–167. [Google Scholar] [CrossRef]
- Luo, J.; Chu, W.; Xu, H.; Jiang, C.; Zhang, T. Low-temperature CO oxidation over CuO-CeO2/SiO2 catalysts: Effect of CeO2 content and carrier porosity. J. Nat. Gas Chem. 2010, 19, 355–361. [Google Scholar] [CrossRef]
- DeHoff, R.T.; Rummel, R.A.; LaBuff, H.P.; Rhines, F.N. The Relationship Between Surface Area and Density in the Second-Stage Sintering of Metals. In Modern Developments in Powder Metallurgy; Hausner, H.H., Ed.; Springer: Boston, MA, USA, 1966. [Google Scholar]
- Shi, L.; Yang, R.-Q.; Tao, K.; Yoneyama, Y.; Tan, Y.-S.; Tsubaki, N. Surface impregnation combustion method to prepare nanostructured metallic catalysts without further reduction: As-burnt Cu-ZnO/SiO2 catalyst for low-temperature methanol synthesis. Catal. Today 2012, 185, 54–60. [Google Scholar] [CrossRef]
- Reddy, B.M.; Reddy, G.K.; Ganesh, I.; Ferreira, J.M.F. Single step synthesis of nanosized CeO2-MxOy mixed oxides (MxOy = SiO2, TiO2, ZrO2, and Al2O3) by microwave induced solution combustion synthesis: Characterization and CO oxidation. J. Mater. Sci. 2009, 44, 2743–2751. [Google Scholar] [CrossRef]
- Voskanyan, A.A.; Chan, K.-Y.; Li, C.-Y.V. Colloidal Solution Combustion Synthesis: Toward Mass Production of a Crystalline Uniform Mesoporous CeO2 Catalyst with Tunable Porosity. Chem. Mater. 2016, 28, 2768–2775. [Google Scholar] [CrossRef] [Green Version]
- Papavasiliou, A.; Tsiourvas, D.; Deze, E.G.; Papageorgiou, S.K.; Katsaros, F.K.; Poulakis, E.; Philippopoulos, C.J.; Boukos, N.; Xin, Q.; Cool, P. Hyperbranched polyethyleneimine towards the development of homogeneous and highly porous CuO-CeO2-SiO2 catalytic materials. Chem. Eng. J. 2016, 300, 343–357. [Google Scholar] [CrossRef]
- Cannas, C.; Musinu, A.; Peddis, D.; Piccaluga, G. Synthesis and Characterization of CoFe2O4 Nanoparticles Dispersed in a Silica Matrix by a Sol−Gel Autocombustion Method. Chem. Mater. 2006, 18, 3835–3842. [Google Scholar] [CrossRef]
- Amaniampong, P.N.; Trinh, Q.T.; Li, K.; Mushrif, S.H.; Hao, Y.; Yang, Y. Porous structured CuO-CeO2 nanospheres for the direct oxidation of cellobiose and glucose to gluconic acid. Catal. Today 2018, 306, 172–182. [Google Scholar] [CrossRef]
- Liu, W.; Flytzanistephanopoulos, M. Total Oxidation of Carbon Monoxide and Methane over Transition Metal Fluorite Oxide Composite Catalysts: I. Catalyst Composition and Activity. J. Catal. 1995, 153, 304–316. [Google Scholar] [CrossRef]
- Wang, Z.-Q.; Xu, Z.-N.; Peng, S.-Y.; Zhang, M.-J.; Lu, G.; Chen, Q.-S.; Chen, Y.; Guo, G.-C. High-Performance and Long-Lived Cu/SiO2 Nanocatalyst for CO2 Hydrogenation. ACS Catal. 2015, 5, 4255–4259. [Google Scholar] [CrossRef]
- Le, H.V.; Parishan, S.; Sagaltchik, A.; Ahi, H.; Trunschke, A.; Schomäcker, R.; Thomas, A. Stepwise Methane-to-Methanol Conversion on CuO/SBA-15. Chem. A Eur. J. 2018, 24, 12592–12599. [Google Scholar] [CrossRef]
- Ho, P.H.; Ambrosetti, M.; Groppi, G.; Tronconi, E.; Fornasari, G.; Vaccari, A.; Benito, P. Electrodeposition of CeO2 and Pd-CeO2 on small pore size metallic foams: Selection of deposition parameters. Catal. Today 2019, 334, 37–47. [Google Scholar] [CrossRef]
- Astudillo, J.; Águila, G.; Díaz, F.; Guerrero, S.; Araya, P. Study of CuO-CeO2 catalysts supported on SiO2 on the low-temperature oxidation of CO. Appl. Catal. A Gen. 2010, 381, 169–176. [Google Scholar] [CrossRef]
- Sun, S.; Mao, D.; Yu, J.; Yang, Z.; Lu, G.; Ma, Z. Low-temperature CO oxidation on CuO/CeO2 catalysts: The significant effect of copper precursor and calcination temperature. Catal. Sci. Technol. 2015, 5, 3166–3181. [Google Scholar] [CrossRef]
- Qi, L.; Yu, Q.; Dai, Y.; Tang, C.; Liu, L.; Zhang, H.; Gao, F.; Dong, L.; Chen, Y. Influence of cerium precursors on the structure and reducibility of mesoporous CuO-CeO2 catalysts for CO oxidation. Appl. Catal. B Environ. 2012, 119–120, 308–320. [Google Scholar] [CrossRef]
- Ratnasamy, P.; Srinivas, D.; Satyanarayana, C.V.V.; Manikandan, P.; Senthil Kumaran, R.S.; Sachin, M.; Shetti, V.N. Influence of the support on the preferential oxidation of CO in hydrogen-rich steam reformates over the CuO-CeO2-ZrO2 system. J. Catal. 2004, 221, 455–465. [Google Scholar] [CrossRef]
- Le, H.V.; Parishan, S.; Sagaltchik, A.; Göbel, C.; Schlesiger, C.; Malzer, W.; Trunschke, A.; Schomäcker, R.; Thomas, A. Solid-State Ion-Exchanged Cu/Mordenite Catalysts for the Direct Conversion of Methane to Methanol. ACS Catal. 2017, 7, 1403–1412. [Google Scholar] [CrossRef]
- Cargnello, M.; Doan-Nguyen, V.V.T.; Gordon, T.R.; Diaz, R.E.; Stach, E.A.; Gorte, R.J.; Fornasiero, P.; Murray, C.B. Control of Metal Nanocrystal Size Reveals Metal-Support Interface Role for Ceria Catalysts. Science 2013, 341, 771–773. [Google Scholar] [CrossRef] [Green Version]
- Albonetti, S.; Lolli, A.; Morandi, V.; Migliori, A.; Lucarelli, C.; Cavani, F. Conversion of 5-hydroxymethylfurfural to 2,5-furandicarboxylicacid over Au-based catalysts: Optimization of active phase and metal–support interaction. Appl. Catal. B-Environ. 2015, 163, 520–530. [Google Scholar] [CrossRef]
- Gamarra, D.; Belver, C.; Fernández-García, M.; Martínez-Arias, A. Selective CO Oxidation in Excess H2 over Copper−Ceria Catalysts: Identification of Active Entities/Species. J. Am. Chem. Soc. 2007, 129, 12064–12065. [Google Scholar] [CrossRef]
- Gamarra, D.; Munuera, G.; Hungría, A.B.; Fernández-García, M.; Conesa, J.C.; Midgley, P.A.; Wang, X.Q.; Hanson, J.C.; Rodríguez, J.A.; Martínez-Arias, A. Structure−Activity Relationship in Nanostructured Copper−Ceria-Based Preferential CO Oxidation Catalysts. J. Phys. Chem. C 2007, 111, 11026–11038. [Google Scholar] [CrossRef]
- Aguila, G.; Guerrero, S.; Araya, P. Effect of the preparation method and calcination temperature on the oxidation activity of CO at low temperature on CuO-CeO2/SiO2 catalysts. Appl. Catal. A Gen. 2013, 462–463, 56–63. [Google Scholar] [CrossRef]
- Song, Y.-Y.; Du, L.-Y.; Wang, W.-W.; Jia, C.-J. CeO2@SiO2 Core–Shell Nanostructure-Supported CuO as High-Temperature-Tolerant Catalysts for CO Oxidation. Langmuir 2019, 35, 8658–8666. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.H.; Jabłońska, M.; Beltrami, G.; Martucci, A.; Cacciaguerra, T.; Paulus, W.; Di Renzo, F.; Fornasari, G.; Vaccari, A.; Benito, P.; et al. Promotion effect of rare earth elements (Ce, Nd, Pr) on physicochemical properties of M-Al mixed oxides (M = Cu, Ni, Co) and their catalytic activity in N2O decomposition. J. Mater. Sci. 2021, 56, 15012–15028. [Google Scholar] [CrossRef]
- Giordano, F.; Trovarelli, A.; de Leitenburg, C.; Giona, M. A Model for the Temperature-Programmed Reduction of Low and High Surface Area Ceria. J. Catal. 2000, 193, 273–282. [Google Scholar] [CrossRef]
- Zimmer, P.; Tschöpe, A.; Birringer, R. Temperature-Programmed Reaction Spectroscopy of Ceria- and Cu/Ceria-Supported Oxide Catalyst. J. Catal. 2002, 205, 339–345. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef] [Green Version]
- Meixner, D.L.; Dyer, P.N. Influence of Sol-Gel Synthesis Parameters on the Microstructure of Particulate Silica Xerogels. J. Sol-Gel Sci. Technol. 1999, 14, 223–232. [Google Scholar] [CrossRef]
- Newalkar, B.L.; Komarneni, S. Synthesis and Characterization of Microporous Silica Prepared with Sodium Silicate and Organosilane Compounds. J. Sol-Gel Sci. Technol. 2000, 18, 191–198. [Google Scholar] [CrossRef]
- Aerts, C.A.; Verraedt, E.; Mellaerts, R.; Depla, A.; Augustijns, P.; Van Humbeeck, J.; Van den Mooter, G.; Martens, J.A. Tunability of Pore Diameter and Particle Size of Amorphous Microporous Silica for Diffusive Controlled Release of Drug Compounds. J. Phys. Chem. C 2007, 111, 13404–13409. [Google Scholar] [CrossRef]
- Sebbar, N.; Bozzelli, J.W.; Bockhorn, H. Kinetic Study of Di-Tert-Butyl Peroxide: Thermal Decomposition and Product Reaction Pathways. Int. J. Chem. Kinet. 2015, 47, 133–161. [Google Scholar] [CrossRef]
- RajanBabu, T.V.; Simpkins, N.S.; RajanBabu, T.V. 1,1-Di-tert-butyl Peroxide. In Encyclopedia of Reagents for Organic Synthesis; John Wiley & Sons: Hoboken, NJ, USA, 2005. [Google Scholar]
- Chu, X.-Q.; Ge, D.; Shen, Z.-L.; Loh, T.-P. Recent Advances in Radical-Initiated C(sp3)–H Bond Oxidative Functionalization of Alkyl Nitriles. ACS Catal. 2017, 8, 258–271. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Liu, D.; Liu, C.; Hu, X.; Lei, A. Copper-catalyzed oxidative alkenylation of thioethers via Csp(3)-H functionalization. Org. Biomol. Chem. 2015, 13, 2264–2266. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Gong, M.; Huang, M.; Li, Y.; Kim, J.K.; Wu, Y. Copper-mediated alkylation of furan and thiophene derivatives with cyclic ethers. Tetrahedron 2016, 72, 7931–7936. [Google Scholar] [CrossRef]
- Trinh, K.H.; Doan, S.H.; Huynh, T.V.; Tran, P.H.; Pham, D.N.; Le, M.V.; Nguyen, T.T.; Phan, N.T.S. Alternative pathways to alpha,beta-unsaturated ketones via direct oxidative coupling transformation using Sr-doped LaCoO3 perovskite catalyst. R. Soc. Open Sci. 2019, 6, 191313. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Mi, X.; Li, Q.; Li, Y.; Huang, M.; Zhang, J.; Wu, Y.; Wu, Y. Copper-catalyzed cross-dehydrogenative-coulping (CDC) of coumarins with cyclic ethers and cycloalkane. Tetrahedron 2015, 71, 6689–6693. [Google Scholar] [CrossRef]
- Nam, W.; Han, H.J.; Oh, S.-Y.; Lee, Y.J.; Choi, M.-H.; Han, S.-Y.; Kim, C.; Woo, S.K.; Shin, W. New Insights into the Mechanisms of O−O Bond Cleavage of Hydrogen Peroxide and tert-Alkyl Hydroperoxides by Iron(III) Porphyrin Complexes. J. Am. Chem. Soc. 2000, 122, 8677–8684. [Google Scholar] [CrossRef]
- Gevorgyan, V.; Priede, E.; Liepiņš, E.; Gavars, M.; Lukevics, E. Radical addition of tetrahydrofuran and tetrahydro-2-furanone to alkenylsilanes in the presence of di(t-butyl)peroxide. J. Organomet. Chem. 1990, 393, 333–338. [Google Scholar] [CrossRef]
- Wu, X.; Wang, M.; Zhang, G.; Zhao, Y.; Wang, J.; Ge, H. Copper-catalyzed diastereoselective aerobic intramolecular dehydrogenative coupling of hydrazones via sp(3) C-H functionalization. Chem. Sci. 2015, 6, 5882–5890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imrich, H.-G.; Conrad, J.; Beifuss, U. Copper-Catalyzed Double Intramolecular Ullmann Coupling for the Synthesis of Diastereomerically and Enantiomerically Pure 4b,9b-Dihydrobenzofuro[3,2-b]benzofurans. Eur. J. Org. Chem. 2015, 2015, 7718–7734. [Google Scholar] [CrossRef]
- Maaliki, C.; Thiery, E.; Thibonnet, J. Emergence of Copper-Mediated Formation of C-C Bonds. Eur. J. Org. Chem. 2017, 2017, 209–228. [Google Scholar] [CrossRef]
- Li, J.; Han, Y.; Zhu, Y.; Zhou, R. Purification of hydrogen from carbon monoxide for fuel cell application over modified mesoporous CuO-CeO2 catalysts. Appl. Catal. B Environ. 2011, 108–109, 72–80. [Google Scholar] [CrossRef]
- Melchionna, M.; Fornasiero, P. The role of ceria-based nanostructured materials in energy applications. Mater. Today 2014, 17, 349–357. [Google Scholar] [CrossRef]
- Srikrishna, D.; Godugu, C.; Dubey, P.K. A Review on Pharmacological Properties of Coumarins. Mini Rev. Med. Chem. 2018, 18, 113–141. [Google Scholar] [CrossRef]
- Pereira, T.M.; Franco, D.P.; Vitorio, F.; Kummerle, A.E. Coumarin Compounds in Medicinal Chemistry: Some Important Examples from the Last Years. Curr. Top. Med. Chem. 2018, 18, 124–148. [Google Scholar] [CrossRef]
- Rouquerol, F.; Rouquerol, J.; Sing, K. CHAPTER 6—Assessment of Surface Area. In Adsorption by Powders and Porous Solids; Rouquerol, F., Rouquerol, J., Sing, K., Eds.; Academic Press: London, UK, 1999; pp. 165–189. [Google Scholar]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amount of Precursors (mmol) | Composite Sample [a] | ||
|---|---|---|---|
| Cu(NO3)2·3H2O | Ce(NO3)3·6H2O | TEOS | |
| 6.3 | 0 | 83.2 | CuSi |
| 6.3 | 1.0 | 83.2 | Cu1Ce0.15Si |
| 6.3 | 3.0 | 83.2 | Cu1Ce0.45Si |
| 6.3 | 4.4 | 83.2 | Cu1Ce0.7Si |
| 6.3 | 6.0 | 83.2 | Cu1Ce0.91Si |
| Entry | Composite Sample | Obtained Molar Composition of Cu:Ce [a] | Cu Content [a] (wt.%) | Ce Content [a] (wt.%) | SA [b] (m2 g−1) | Vmicro [c] (cm3 g−1) | Vtotal [d] (cm3 g−1) |
|---|---|---|---|---|---|---|---|
| 1 | CuSi | - | 7.44 | 0 | 340 | 0.129 | 0.136 |
| 2 | Cu1Ce0.15Si | 1Cu:0.15Ce | 7.17 | 2.37 | 411 | 0.156 | 0.160 |
| 3 | Cu1Ce0.45Si | 1Cu:0.45Ce | 6.69 | 6.64 | 337 | 0.127 | 0.140 |
| 4 | Cu1Ce0.7Si | 1Cu:0.7Ce | 6.48 | 10.00 | 389 | 0.145 | 0.155 |
| 5 | Cu1Ce0.91Si | 1Cu:0.91Ce | 6.22 | 12.35 | 262 | 0.102 | 0.115 |
| Entry | Catalyst(s) | Used Cu Amount (mol%) | Used Ce Amount (mol%) | Yield [a],[b] (%) |
|---|---|---|---|---|
| 1 | Cu1Ce0.91Si | 2.5 | 2.3 | 62 |
| 2 | Cu1Ce0.7Si | 2.8 | 2.0 | 85 |
| 3 | Cu1Ce0.45Si | 3.3 | 1.5 | 51 |
| 4 | Cu1Ce0.15Si | 4.2 | 0.6 | 45 |
| 5 | CuSi | 4.8 | - | 33 |
| 6 | CuSi + CeO2 | 2.8 | 2.0 | 64 |
| 7 | Cu(OAc)2 | 4.8 | - | 21 |
| 8 | Cu(OAc)2 + CeO2 | 2.8 | 2.0 | 43 |
| 9 | CuO | 4.8 | - | 20 |
| 10 | CuO + CeO2 | 2.8 | 2.0 | 25 |
| 11 | CeO2 | - | 4.8 | 30 |
| 12 | CuI | 4.8 | - | 74 |
| 13 | CuI + CeO2 | 2.8 | 2.0 | 64 |
| Entry | Reactant 1 | Reactant 2 | Product | Yield [a], [b] (%) |
|---|---|---|---|---|
| 1 |  |  |  | 75 |
| 2 |  |  |  | 70 |
| 3 |  |  |  | 65 |
| 4 |  |  |  | 30 |
| 5 |  |  |  | 28 |
| 6 |  |  |  | 58 |
| 7 |  |  |  | 57 |
| 8 |  |  |  | 32 |
| 9 |  |  |  | 41 |
| 10 |  |  |  | 38 |
| 11 |  |  |  | 37 |
| 12 |  |  |  | 40 |
| 13 |  |  |  | 30 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le, H.V.; Nguyen, V.B.; Pham, H.H.; Nguyen, K.D.; Ho, P.H.; Trens, P.; Di Renzo, F. Combustion-Synthesized Porous CuO-CeO2-SiO2 Composites as Solid Catalysts for the Alkenylation of C(sp3)-H Bonds Adjacent to a Heteroatom via Cross-Dehydrogenative Coupling. Catalysts 2021, 11, 1252. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11101252
Le HV, Nguyen VB, Pham HH, Nguyen KD, Ho PH, Trens P, Di Renzo F. Combustion-Synthesized Porous CuO-CeO2-SiO2 Composites as Solid Catalysts for the Alkenylation of C(sp3)-H Bonds Adjacent to a Heteroatom via Cross-Dehydrogenative Coupling. Catalysts. 2021; 11(10):1252. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11101252
Chicago/Turabian StyleLe, Ha V., Vy B. Nguyen, Hai H. Pham, Khoa D. Nguyen, Phuoc H. Ho, Philippe Trens, and Francesco Di Renzo. 2021. "Combustion-Synthesized Porous CuO-CeO2-SiO2 Composites as Solid Catalysts for the Alkenylation of C(sp3)-H Bonds Adjacent to a Heteroatom via Cross-Dehydrogenative Coupling" Catalysts 11, no. 10: 1252. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11101252