
Catalytic Wood Fractionation into Chemicals in Supercritical Ethanol and n-Heptane: Potential and Limitations

Institut de Recherches sur la Catalyse et l’Environnement de Lyon, Centre National de la Recherche Scientifique (CNRS), University Lyon 1, 2 Avenue Albert Einstein, 69626 Villeurbanne, France
*
Author to whom correspondence should be addressed.
Catalysts 2022, 12(11), 1333; https://0-doi-org.brum.beds.ac.uk/10.3390/catal12111333
Submission received: 20 September 2022
/
Revised: 20 October 2022
/
Accepted: 21 October 2022
/
Published: 1 November 2022
(This article belongs to the Special Issue Heterogeneous Catalysis for Valorization of Lignocellulosic Biomass II)
Abstract
:Direct selective wood fractionation into chemicals is an approach that has attracted recent attention. The application of sub- and supercritical (SC) alcohols to fractionate wood into solid cellulose and liquefy phenolic monomers is a process now widely known as “lignin first”. It is justified to study the potential of other SC organic solvents of variable polarities. Herein, we compare the abilities of SC ethanol and SC n-heptane to fractionate pine wood near their critical point. While near-critical ethanol has more affinity for lignin fraction, we show that near-critical n-heptane has preference for carbohydrate deconstruction. If SC ethanol favors biooil formation which contains important ethyl/ethoxy groups, the alkane greatly favors solid carbon products. The impact of addition of heterogeneous catalysts (acid, basic and Cu-based catalysts) on wood fractionation and light chemicals formation was investigated deeply in SC ethanol. In SC ethanol, catalysts favor light liquid products such as esters at the expense of biooil with a total oxygenates yield of 33 wt% relative to carbohydrates over β zeolite. However, we show that depending on the catalysts’ nature, wood components fractionation was completely changed, and this is particularly true with solid acid catalysts which promote cellulose deconstruction and the formation of solid carbon products. It is proposed that liquid products’ accumulation in the autoclave, in particular water, is at the origin of the wood fractionation changes which preclude its control by the choice of the SC organic solvent and conditions. Moreover, all the catalysts underwent severe leaching, which also contributed to the wood component fractionation changes.
1. Introduction
At the end of the 1970s, the application of sub- and supercritical fluids in the treatment of lignocellulosic biomass (LCB) appeared in the literature [1,2]. Supercritical fluids offer a lot of possibilities from total LCB liquefaction into bio-oils [3,4], chemicals upon catalysts’ addition [5,6,7,8] or selective wood component fractionation [9,10,11]. For instance, in 2011, we reported that in comparison to pure SC MeOH, the use of SC MeOH-10% water mixture allows complete cellulose liquefaction in shorter time [5]. A similar impact of water addition to SC ethanol was reported by other teams [4,8]. In catalyst-free conditions, only a few light chemicals were detected, while upon solid acid catalysts’ addition, up to 20% methyl-levulinate yield was obtained [5]. We also used SC but-2-ene for liquefication of cellulose and pine wood pre-impregnated with strong Br∅nsted acid solutions in a plug flow reactor forming directly sec-butyl levulinate formation [6]. If the catalysts drive the liquefaction towards more selective chemicals formation, a step further is the in situ control of the wood fractionation, in other words the selective liquefaction of one of the wood components which would ensure more selectivity of the light products. In principle, this approach is that of the “lignin first” process where sub-critical alcohols are reported to delignify LCB while metallic catalysts promote phenolic compounds formation, often in the presence of added hydrogen [12,13]. The lignin monomers or dimers are analyzed comprehensively by NMR technics and the remaining solid residue, enriched in cellulose, is often used for further enzymatic hydrolysis into glucose. However, the compositional analysis of the solid residue is scarcely provided in the published works related to the lignin first process. Thus, the hemicellulose become remains scarcely addressed and controversial, even though it is essential to understand and control the liquefaction process. For example, using sub-critical MeOH around 200 °C, some papers describe the preservation of hemicellulose and cellulose [13], whereas a few others mention that hemicellulose extraction is optional [12]. With regard to sub-critical ethanol, some papers report its inability to fractionate cellulose or hemicellulose [14], but other publications argue that sub-critical ethanol is efficient for the deconstruction of LCB to give a lignin-derived oil and a carbohydrate pulp [14] irrespective of whether hemicellulose is attacked or not. Other works describe high monomers yields from native wood lignin and hemicellulose in SC ethanol between 300–340 °C [15].
Only few articles have addressed the use of other SC organic solvents for selective wood deconstruction with the compositional analysis of the solid residue in order to quantify the conversion of each wood component. One can mention the pioneering work of Metzger et al. on a wide range of organic solvents. They investigated the abilities of alcohols to attack lignin while esters and alkanes would have more affinities for carbohydrates deconstruction [9]. In 2016, Li et al. explored how the nature of SC solvents affect the liquefaction of isolated wood component. They compared SC alcohols, n-heptane and cyclohexane at 320 °C for a short reaction time. They concluded that non-polar solvents were unable to break hydrogen bonds in cellulose. They also reported that methanol and ethanol have similar abilities towards lignin solubilization [16]. From the published literature, we can say that there is no agreement on how the nature of SC organic fluids impact wood fraction liquefaction, even with sub and SC alcohols. The lock in this area is probably the analysis of the solid residue which, in addition to the unconverted wood fraction, may contain solid carbon products. In 2015, to overcome this issue, we developed an analytical tool which combines acid hydrolysis of unconverted carbohydrate and direct FTIR quantification of unconverted lignin in order to solve the lock of the quantification of solid carbon products [17].
Thus, this article is a contribution to better understand the wood components deconstruction with one of the most used SC solvent, ethanol by comparing it to SC n-heptane, which is a non-polar solvent without functional group. Using deeper analysis of the solid residue, we show here that SC n-heptane has more ease to attack the carbohydrate fractions than ethanol but favors the formation of solid carbon products. On the other hand, ethanol, a reactive solvent, favors biooils with a significant contribution of ethyl or ethoxy groups in catalysts-free conditions. More light chemicals are formed upon addition of catalysts, in particular solid acid catalysts such as β zeolite that increases the total light chemicals’ yield to 33 wt% relative to carbohydrates. However, the enhanced formation of light chemicals is parallel to the enhanced formation of solid carbon products. In addition, a strong modification of the wood components fractionation is highlighted irrespective of the nature of the heterogeneous catalyst (basic, acid or a Cu-based one). This is tentatively explained by the accumulation of products in the autoclave, in particular water as well as leaching of the active catalytic species. This phenomenon prevents the control of wood components’ deconstruction when the transformation is implemented in batch reactor which makes even more difficult to control the catalytic conversion of the liquefied biopolymers into valuable chemicals.
2. Results and Discussion
2.1. Comparison of Pine Wood Fractionation in SC Ethanol and SC n-Heptane
2.1.1. Pine Wood Components’ Conversion and Analysis of the Solid Residue
The conversion of wood components near the critical temperatures of ethanol and n-heptane at 250 °C and 280 °C, respectively, is compared in Figure 1.
In near-critical ethanol conditions, at 250 °C, Figure 1 shows that only the two weakest components, lignin and hemicellulose, are converted: lignin is the most attacked biopolymer, with a conversion of 39%, then hemicellulose with a conversion of 28%. With 3% conversion, cellulose remains almost unchanged. In near-critical n-heptane at 280 °C, the three wood components are converted (probably due to the higher temperature), but in different %conversion: the hemicellulose becomes the most attacked one with a complete conversion, followed by lignin and cellulose which were almost half converted, at 46 and 47% conversions, respectively. To overcome the thermal effect, pine wood fractionation in SC ethanol was also performed at 280 °C (Figure 1). We see that lignin remains the most converted fraction at 280 °C, with 84% conversion while only 46% of lignin were converted in SC n-heptane at the same temperature. Hemicellulose conversion reached to 68% in SC ethanol at 280 °C. On the other hand, cellulose remains less attacked in SC ethanol at this temperature level, with 20% conversion against 47% in SC n-heptane. These results are in agreement with those reported by Metzger and Köll [9], i.e., SC hydrocarbons decompose carbohydrates preferentially, while SC alcohols decompose lignin preferentially.
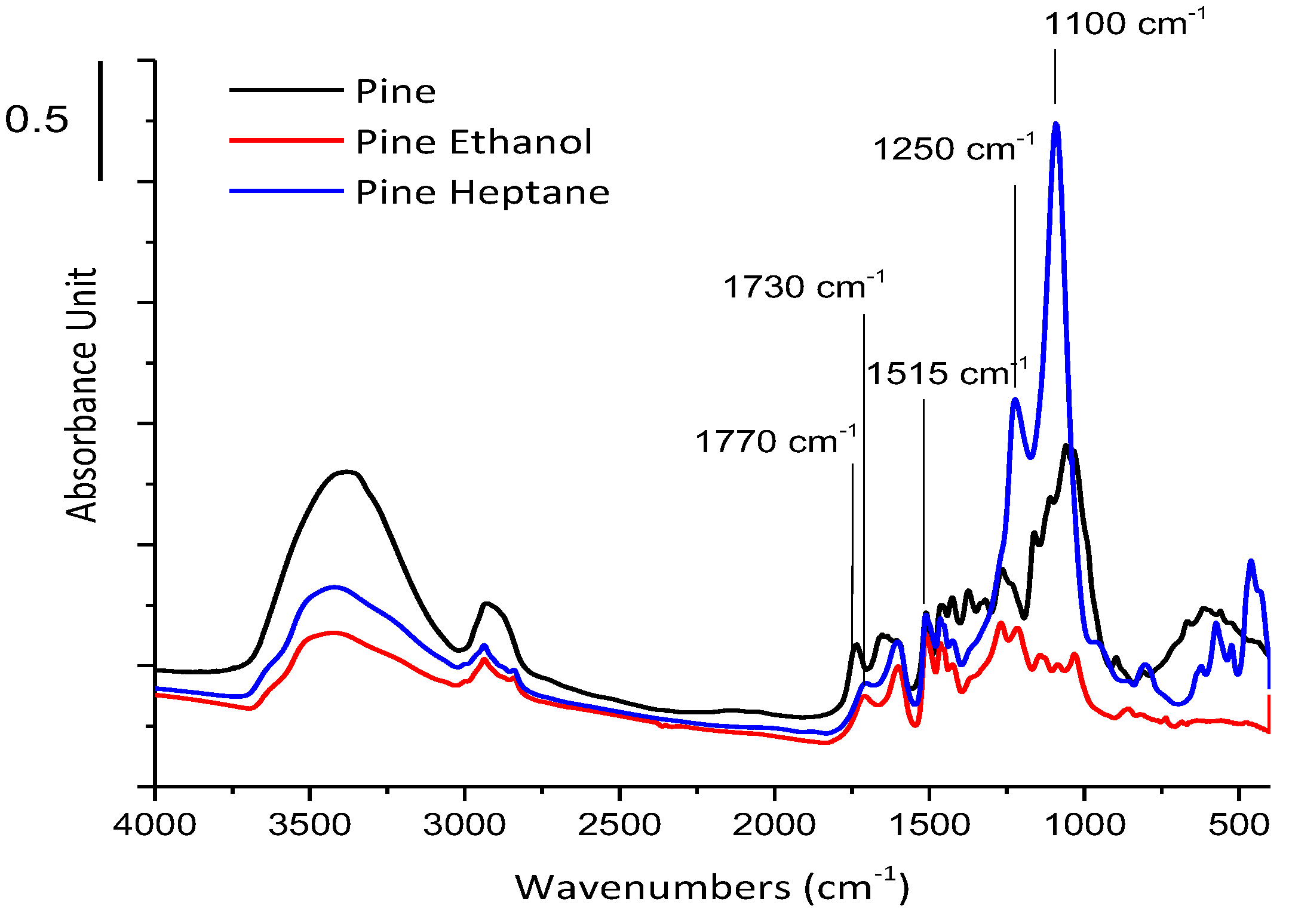
Figure 2 presents the infrared spectra of the solid residues obtained in near-critical ethanol at 250 °C and in near-critical n-heptane at 280 °C as compared to that of the initial pine. The νC=O vibration of the acetyl groups observed at 1770 cm−1 on the fresh wood is almost suppressed in the spectra of the residues. This vibration seems to be replaced by a vibration at lower frequency, 1730 cm−1, characteristic of the νC=O vibration of the carboxyl group COOH. It is deduced that the fractionation of pine in SC ethanol at 250 °C or SC n-heptane at 280 °C could break the ester bonds that bind lignin and hemicelluloses and could be replaced by carboxylic groups. Moreover, the characteristic bands from 800 to 1500 cm−1 are strongly modified in the two solvents: they are less intense after fractionnation in SC ethanol, while in SC n-heptane, the FTIR spectrum is dominated by two strong absorptions at 1250 cm−1 and 1100 cm−1, the vibration range δC-H of CH2 or CH3 groups. Note that, in the solid residues, the band characteristic of lignin, at 1515 cm−1 is clearly observed [17].
2.1.2. Products of Pine Fractionation in SC n-Heptane vs. SC Ethanol
The fractionation products were distributed into four families: gaseous ones, heavy liquid products recovered after solvent evaporation (bio-oil), light liquid products analyzed by GC-MS, and solid carbon products. Yields in each products’ family as a function of the SC solvent and temperature are presented in Figure 3.
The yields in the gas phase are low, 2–3% for SC ethanol at 250 °C and 280 °C, 5% for SC heptane at 280 °C. We can say that these values are much lower than the yields of the gas products obtained in SC water, which can easily reach 30% [18]. The yield of bio-oils is much higher using SC ethanol at 250 °C (33%) or 280 °C (34%), in comparison to 19% in SC n-heptane. Note that in SC ethanol, in the same range of temperature and in absence of hydrogen, bio-oil yields up to 60% were reported but only upon addition of a catalyst such as Zn/HZSM-5 [3]. We can observe a considerable difference as regards to solid carbon products. In SC ethanol, its yield is only of 2% at 250 °C, but rises to 15% at 280 °C, and in SC n-heptane at 280 °C it achieves 39%. Most likely, ethanol is a reactive solvent that can esterify, etherify or form acetal with the fragments from hemicellulose and lignin as soon as there are liquefied. However, it is also possible that the solid biomass functionalization occurs before its fractionation into liquids biooils or chemicals. This could explain the formation of bio-oil in high yield and the absence of solid products, contrary to what is observed in SC n-heptane. This is consolidated by the mass balance value of 245% in SC ethanol at 250 °C, which suggests the strong insertion of ethyl or ethoxy groups in the liquefied products. Note that at the higher temperature of 280 °C, the mass balance equals 128% in SC ethanol and 131% in SC n-heptane. In both cases, solid products are favored. Re-condensations between oligomers, monomers formed from the different components in addition to benzenic and furanic ring condensations must be at the origin of the solid products formed and may contribute to lower the mass balance values since less ethoxy/ethyl groups are introduced in the products. It is clear that the use of a saturated hydrocarbon, without any functional group, can hardly avoid the recombination of depolymerized oligomers or monomers. In SC ethanol, it seems that rise in the temperature may favour re-condensation of biopolymers fragments at the expense of their stabilization via reaction with the alcohol. Moreover, the nature of the oligomers and monomers, ex polysaccharides or ex lignin, could contribute to this result. Indeed, at iso temperature of 280 °C, we note the preferential attack of pine polysaccharide-type biopolymers in SC n-heptane while lignin is preferentially attacked in SC ethanol (Figure 1). This preferential carbohydrate fractionation could be at the origin of the formation of solid products in SC n-heptane. Indeed, carbohydrate deconstruction, in the absence of reactive solvent may undergo preferential monomolecular transformations such as dehydration into furanics known to polymerize easily into “humins”, reported to be a furan-structure based polymer [19].
As regards to the light liquid products that can be analysed in GC-MS, their formation remains low regardless of the solvent and the temperature. However, their yields are slightly higher in SC ethanol, 5% and 6% at 250 °C and 280 °C, respectively, than in SC heptane at 280 °C (2%).
The nature of the light products are displayed in the Figure 4.
In near critical ethanol, esters are the major products consisting mainly of ethyl formate and ethyl glycolate. They must be formed from the thermal transformation of hexoses and pentoses formed from hemicellulose. The two other most important products families, representing 14%, are acids and phenolic compounds. Acetic acids are the main acids formed by deacetylation, i.e., deconstruction of the covalent ester bonds between lignin and hemicellulose. One can also recall that lignin is the most converted component in near critical ethanol, with a conversion of 39%, followed by hemicellulose (28%). However, less lignin monomers are recovered as compared to the hemicellulose ones and isoeugenol is the main phenolic derivative.
The main difference between these two solvents lies in the absence of ester formation in SC n-heptane, which unlike alcohols is a non-reactive solvent. In SC n-heptane, the main light compound is acetic acid, followed by phenolic compounds (mainly isoeugenol), then furans (mainly furfural). Thus, acetic acid is recovered from the deconstructions of covalent bonds between hemicellulose and lignin, furanics come from hexoses/pentoses monomolecular transformation by dehydration, which seems to be the privileged way of carbohydrates depolymerization in absence of reactive solvent.
2.2. Coupling Pine Wood Fractionation in SC Ethanol and Heterogeneous Catalysts
With the aim to increase the selectivity in light chemicals, SC ethanol at 250 °C appeared as the most favorable medium, since only hemicellulose and lignin are converted into liquefied products without carbons lost in solid products as occurs prominently in SC n-heptane.
Different types of catalysts were tested: a basic one, La2O3/ZrO2, acid catalysts such as niobic acid (abbreviated NbOH) and β zeolite and a copper-based catalyst CuZnAlO. A basic catalyst could promote O-alkylation reactions of phenolic compounds by ethanol. Acid catalysts are expected to catalyse C-alkylation of aromatic groups, but also etherification, esterification or dehydration reactions [20,21,22,23]. As for Cu-based catalysts, they are very effective in alcohol dehydrogenation into aldehyde [24] supplying hydrogen to the reaction medium which promote hydrogen transfer reactions [25]. They could make it possible to limit re-condensations by formation of acetal between the OH functions of various monomers and acetaldehyde, formed in situ, but the in situ formed hydrogen could also allow hydrogen transfers and limit successive reactions from of un-saturations.
2.2.1. Evidence That Heterogeneous Catalysts Have an Unexpected Impact on Wood Components Fractionation
Surprisingly, we found that upon heterogeneous catalysts addition, wood conversion was enhanced in most cases: from 18% without catalyst to a maximum wood conversion of 75 % in the presence of β zeolite. The pine conversion remains largely in the same order of magnitude in the presence of the basic catalyst, La2O3/ZrO2 (Table 1).
However, even in the case of ZrO2/La2O3, there is a change in the level of conversion of the various components: the conversions of cellulose and hemicellulose are improved from 3% and 28% to 17% and 36 wt%, respectively, while that of lignin is greatly reduced, as compared to the reaction carried out in absence of a catalyst (Figure 5).
In the presence of the other catalysts, all the three fractions were converted in higher percentage. The acid catalysts, niobic acid and β zeolite, considerably promote the conversion of each fraction, especially β zeolite. This makes possible to obtain a total conversion of hemicellulose, almost a complete conversion of lignin and higher conversion of the cellulose (76 wt%). For niobic acid also, hemicellulose becomes the most converted fraction, 82%, then lignin with 72% and the cellulose conversion equals 48%. For these two catalysts, it is clear that the selectivity at the level of the wood components’ conversion, which, in SC ethanol, kept the cellulose unattacked, and converted more lignin than hemicellulose, is not more preserved at all. The CuZnAlO catalyst also deeply modifies the wood components’ conversion compared to the reference without any catalyst: a total conversion of hemicellulose is also found but the two other components are less converted than in the case of β zeolite, which shows 61% conversion of lignin and 37% conversion of cellulose. As in the case of niobic acid and β zeolite catalysts, CuZnAlO catalyst completely upsets the selectivity on the wood components characteristic of SC ethanol and makes hemicellulose the most converted fraction. Despite the interest of obtaining high wood conversions upon catalysts addition, it is found that heterogeneous catalysts do not allow to take benefit of the wood selective fractionation characteristic of near critical ethanol alone who attacks first of all, lignin. In the majority of cases, the addition of a heterogeneous catalyst in the medium considerably increases the conversions of all pine components, with the exception of the basic one, La2O3/ZrO2 (Figure 5).
Most likely, the liquid products of the catalytic wood fractionation, accumulated in the autoclave during the course of the reaction, must modify the original solvation ability of near-critical ethanol. This must be especially true for water formed by dehydration reactions in agreement with a previous studies showing that the addition of 10% water to SC MeOH makes cellulose liquefaction faster [4,5,8]. However, the possible catalysts leaching can also enhance wood deconstruction due to easier contact between solubilized active catalytic species and the solid wood components. Moreover, this phenomenon can be favored by water. To check the catalysts leaching, we performed calcination in a muffle furnace at 1000 °C under air the solid residues recovered after each test. The solid residues contain aged catalysts (the initial weight, 1 g, in absence of leaching), unconverted wood and solid carbon products. For each catalyst, we compared the solid residues’ remaining weight at 1000 °C, as compared to that determined on the fresh catalysts assuming that the remaining weight after calcination at 1000 °C correspond to the constitutive oxides of the mineral catalyst (see Supplementary Materials, Table S1). It was highlighted that all the catalysts suffer for severe leaching: 59 wt% for β zeolite, 75% for La2O3/ZrO2, 92% for NbOH and even 96% for CuZnAlO.
2.2.2. Impact of Addition of Heterogeneous Catalysts on the Products of Wood Fractionation in SC Ethanol at 250 °C
Globally, the presence of a catalyst has the expected influence on the increase in the light liquid products’ yields that can be analyzed by GC-MS (Figure 6). The catalytic effect is relatively low in the case of La2O3/ZrO2 and niobic acid. Over La2O3/ZrO2, the light liquid chemicals’ yield is 8 wt% and in the presence of niobic acid, it is of 9 wt% by comparison to 5 wt% without catalyst. In these two cases, the total yield of liquid products, light chemicals + bio-oil, changes little when compared to the solvolysis carried out in the absence of catalyst. The slight increase in light liquid products was made to the detriment of liquid heavy products (bio-oils). For La2O3/ZrO2 and niobic acid, the yields of bio-oils are reduced from 34% (without any catalyst) to 24 wt% and 22 wt%, respectively. As for that of the gaseous products with the two previous catalysts, it remains very low, of the order of 2–3 wt%, which is similar to reactions carried out in the absence of any catalyst.
The influence of catalysts’ addition on the light liquid products is the most important in the case of β zeolite with a yield increased to 21 wt%, not to the detriment of heavy liquid products whose yield remains as high as 31%, but this result comes directly from more wood components’ conversion. It is important to underline that the acid catalysts increase the yields in solid carbon products, 16% with NbOH and 19% with β zeolite. β zeolite also significantly increases the yield of gaseous products, 16%. With CuZnAlO, the gas phase yield is the highest, 34 %, probably overrated by the dehydrogenating properties of Cu. Moreover, the yield of light liquid products is 11%, in the same order of magnitude than that of bio-oil yield, 14%. This Cu-based catalyst leads to less formation of solid carbon products, 5% yield, as compared to the two most active catalysts, NbOH and β zeolite. We can conclude that β zeolite promotes the highest yield in the targeted products’ family, the light liquid chemicals. Note that the total yield of liquid products obtained with β zeolite, 52 wt%, is comparable to the bio-oil yield of 60% reported recently, in the same reaction medium, without hydrogen, in the presence of Zn/ZSM-5 [3]. However, for the most active catalysts, NbOH, β zeolite and Cu/ZnAlO, we also note a clear increase in the yields of solid carbon products which can be considered as a carbon loss whereas they are not formed in catalyst free near-critical ethanol.
We note that in the presence of the most active catalysts, NbOH, β Zeolite, CuZnAlO, the mass balances are between 91 and 128% (Table 2). How to interpret these changes in the mass balance following the addition of the most active catalysts? In the presence of La2O3/ZrO2, a catalyst which has little influence on wood conversion and which does not favor solid carbon products, the mass balance is as high as in the absence of catalyst. High values that we explain by the high participation of ethoxy and ethyl groups in the reaction liquid products. In the cases of NbOH, β zeolite, CuZnAlO, we note an important increase in solid carbon products’ yields, in particular for the acid catalysts NbOH and β zeolite. These data suggest that the solid carbon products would be formed by condensation of monomeric derivatives which did not react with ethanol first. For this family of solid carbon products, there would be no additional carbon contribution from ethoxy or ethyl groups from the solvent. Note that water produced by dehydration was not analyzed and is therefore not included in this mass balance. We can think that the acid catalysts are as efficient to promote the dehydration of the carbohydrates’ monomers into furanic and their further condensation into solid carbon products, than to promote bimolecular steps involving an ethanol molecule such as esterification, etherification or acetal formation. Moreover, it is known that the activity of acid catalysts in esterification is lowered in presence of water [26]. Thus, our results would suggest that monomolecular sugar dehydration into furanic compounds would be less sensitive to water. We can only conclude that adding solid acid for the purpose of improving the yield of light chemicals in near-critical ethanol conditions is a difficult balance that can easily fall towards the formation of solid carbon products.
One hundred percent distribution of light liquid products and the nature of main chemicals as a function of the catalysts are presented in Figure 7 and Table 3. Note that the products are gathered into different families as follows: we first gathered all the molecules with a furanic ring in the family name “furans” and the ones with a phenolic group in the family name “phenolic compounds”, then the other products were gathered with respect to their main functional groups: esters, aldehydes, acids, ketones, and alcohols.
In ethanol, at 250 °C without catalyst, esters are the main products’ family, with a proportion of 55%, then 14% of phenolic compounds and 14% of acids. Main representatives of these families are ethyl glycolate and isoeugenol. Acetic acid is the main detected carboxylic acid whatever be the conditions. Upon catalysts’ addition, esters remain the main formed products’ family except in the case of the Cu-catalyst where phenolic derivatives are equivalent to esters. In the case of La2O3/ZrO2, a basic catalyst, the proportion of esters is lower than that obtained in ethanol alone. In that case, the main ester is ethyl lactate and neither ethyl glycolate such as in catalyst free conditions. The proportion of furanic compounds is higher than that obtained in ethanol alone, at 250 °C. The major furan is furfuryl alcohol and among the phenolic compounds, isoeugenol is predominant. In the presence of niobic acid, esters are mainly formed, with the highest proportion of 58%, with low percentages for the other products’ families. The main esters are ethyl lactate and ethyl glycolate. Compared to niobic acid, β zeolite promotes larger distribution of products. The favored products are still esters, where ethyl levulinate obtained in a high proportion 26%, represents half of the esters, followed by ethyl glycolate. Among the phenols, isoeugenol is the most dominant. As regards to the furans, with mainly furfural, their proportion is low, this can be explained by their successive transformation into levulinates. In the presence of CuZnAlO, the following distribution of monomer derivatives is obtained: 14% for alcohols (of course, the products from ethanol transformation are not included), 14% for acids, mainly acetic acid, 27 % for esters with ethyl lactate as main ester, 6% for furan compounds, such as furfuryl alcohol which is still predominant, and 27% for phenolic compounds with mainly isoeugenol. This Cu-based catalyst is the most selective towards phenolic compounds.
Table 4 summarizes the yields in the main products’ families relative to the wood component from which they are derived. Trends lead to say that β zeolite allows the highest yields for the different families of compounds. The total yield of oxygenates reaches ~33% relative to wood carbohydrates accompanied by a 6% yield in phenolic derivatives relative to lignin. In particular, esters are mainly produced with ~16% yield relative to carbohydrates, and ethyl levulinate is the main product. Obtaining ethyl levulinate in the presence of β zeolite could account for distinct acidic properties compared to niobic acid which rather leads to the formation of glycolate and lactate. These last two products require C-C cleavages known to be promoted by basic or Lewis acid sites [27,28]. The formation of levulinates requires the intermediate formation of 5-hydroxymethylfurfural (or its acetal) which can be rehydrated in levulinic acid and then give levulinate by esterification (or levulinate directly from 5-HMF acetal). However, this pathway requires strong Br∅nsted acid sites [29]. This suggests that Br∅nsted acidity of β zeolite would retain sufficient acid strength in the reaction medium to promote 5-HMF (or its acetal) rehydration into levulinic acid or levulinates. In the case of niobic acid, its Lewis acidity seems to be the driving force since it favors products resulting from C-C cleavage of hexoses or pentoses and does not favor the sugars dehydration/rehydration pathway into levulinates. Moreover, Cu-based catalysts give the highest yield of phenolic derivatives relative to lignin, around 8%, with isoeugenol also as main product. Unlike β zeolite, this catalyst does not promote high yields of molecules from carbohydrates, ~14%.
Most of the studies on wood fractionation in sub or SC organic solvents target bio-oils production. In general, GC-MS analyses reflect the composition of the light fraction, but it is rare that the products’ yields are clearly expressed relative to the wood component from which they are derived. In a recent patent filing by Samec et al. in 2020, results are expressed in a similar way as ours. This patent describes the performance of β Zeolite to assist the direct fragmentation of birch wood into furfural and phenols in an ethanol/10% water medium at temperatures between 200 and 220 °C, in a batch reactor, in the absence of hydrogen. The maximum yield of phenolic compounds relative to lignin is 20 wt% at 220 °C. At best, they succeed in maximizing in parallel the yields of furfural and phenols, respectively, at 9 wt% relative to carbohydrates and 17 wt% relative to lignin at 220 °C [8]. In good agreement with these results, the β zeolite used in our study, with a Si/Al ratio of 10.5, allows high liquid monomers’ yields. We obtained 16 wt% ethyl levulinate and 6 wt% phenolic compounds, but the sum of the yields of oxygenates relative to carbohydrates reached 33%. The essential difference between our results and those of Samec et al. relies on the presence of 10 wt% water in the medium. Indeed, the presence of water is known to boost LCB deconstruction [4,5,8] but water is also known to lower the acid strength of catalysts, which may be unfavorable to the rehydration of furan derivatives into levulinic acid and which is also unfavorable to esterification reactions [26].
3. Materials and Methods
3.1. Materials
Feedstocks used in this work were sawdust of pine wood from Softwood. After sieving, only the particle sizes from 355 to 500 µm were used. A pretreatment by lyophilization (soft drying) was applied on fresh pine and on all the solid residues recovered after the fractionation tests to keep their moisture content constant. Ethanol and n-heptane (provided by Sigma Aldrich, France) have a purity > 99%. They were kept anhydrous by adding molecular sieves. A 72 wt% sulfuric acid (provided by Sigma Aldrich, France) was used for the acid hydrolysis of solid biomass for the compositional analysis.
3.2. Catalysts
The ammonium form of β zeolite, with Si/Al ratio of 10.5, was provided by IFPEN. Niobic acid, abbreviated NbOH, was provided by CBMM. Zirconia modified by Lanthanum (10% La2O3) was prepared in the team according to published exchange procedure [30] using zirconium hydroxide from MEL and calcined 6 h at 700 °C. XRD pattern shows only the diffraction peaks of tetragonal ZrO2 phase in line with the stabilization of this phase upon La dispersion within the zirconia framework (Figure S1). Before use, a calcination step of 2 h under air at 500 °C was applied in order to remove moisture and carbonates. CuZnAlO was also prepared in the team, using the Pecchini method to synthesize ZnAlO, which was amorphous upon calcination at 500 °C. Cu (20 wt%) was dispersed on calcined ZnAlO by incipient wetness impregnation using copper nitrate as a precursor, dried at 80 °C and calcined in a muffle furnace at 350 °C for 1 h [31]. The XRD pattern (Figure S2) shows the diffraction peaks of CuO only.
The main features of the catalysts are summarized in Table 5.
3.3. Protocol of Fractionation
We used a batch type reactor (Parr) with an internal volume of 74 mL which can be used at a pressure up to 600 bar and a temperature up to 350 °C. First, 2.5 g of lyophilized pine wood, 1 g of catalyst and 27 g of anhydrous ethanol or n-heptane were introduced in the autoclave. The reactor was closed, and leak tests were performed with pressurized helium. Then, the reactor was cooled down at −60 °C in a dry ice-ethanol mixture. Gases were rapidly evacuated from the autoclave to reach the final pressure of 100 mbar. The mixture of solvent and biomass generates autogenously the pressure without adding any gas. This allowed it to rigorously reach the critical coordinates since the volume was fixed by the apparatus and the initial mass of solvent. The reactor, once warmed up at ambient temperature, was heated up to the reaction temperature (250 °C or 280 °C) at 5 °C/min and kept at this temperature for 1 h. At the reaction end, the reactor was quenched in a cooled water batch (0 °C) to a rapid temperature decrease to ambient temperature. The gaseous products were collected in a vessel (95 mL) pre-evacuated at 100 mbar. This vessel was equipped with a manometer in order to measure the residual pressure. The gas yield was calculated by assuming that light gases contain only one carbon atom with the maximum molecular weight, i.e., CO2 based on previous analysis. The liquid and solid products were separated using a 0.45 µm porosity filter. Then, the unconverted biomass and solid products were washed three times with 30 mL of the solvent. The autoclave was also washed with 60 mL acetone to recover potential solids left on the autoclave walls. Solids recovered by filtration were lyophilized for further analysis and named “solid residue”. The liquid from reaction medium plus washing solvent were first analyzed by GC-MS and then evaporated under reduced pressure, in order to eliminate the solvents to obtain the heavy liquid products (bio-oil fraction).
The heavy liquid products (bio-oil) and the light liquid products yields were calculated as follows:
Yield heavy liquid products (wt%) = 100 × (weight of heavy liquid products)/weight of initial dried LCB)
Yield light liquid products (wt%) = 100 × (weight of light products/weight of initial dried LCB)
The reproducibility of this experimental protocol has been checked by carrying out three times the same experiment in absence of catalyst: a standard deviation equal to ±5% based on the yield of each fraction was determined.
3.4. Analytical Methods
The compositional analysis of the solid fresh LCB and of the solid residue applied in this work were adapted from a NREL method, combined with quantitative analysis of unconverted lignin by FTIR. The methodology is described in detail elsewhere [17].
Each component conversion was deduced from the compositional analysis of the fresh pine wood (%ini of cellulose, hemicellulose and lignin of the lyophilized fresh pine wood) and that of the solid residue (%final of cellulose, hemicellulose and lignin in the recovered solid residue). Knowing the weight of fresh lyophilized wood (mw) and that of the lyophilized recovered solid residue (msr), the conversion of one component is calculated as follows.
Ccomp (%) = 100 × [(mw × %ini/100) − (msr × %final/100)]/(mw × %ini/100)
Light liquid products (in ethanol/n-heptane and acetone solutions) were analyzed by a Shimadzu GC-MS 2010 apparatus equipped with a column NUKol (0.25 µm, 0.25 mm, and 30 m). The split ratio equals 50, He is the gas vector, the injector and mass source temperatures are 220 °C and 200 °C, respectively. The temperature program is the following one: successive plateaus of 5 min at increasing temperatures: 70 °C, 100 °C, 120 °C, 150 °C, and 200 °C using a temperature ramp of 20 °C.min−1 between each temperature. The highest molecule mass is ~200 g.mol−1.
Dodecane is used as internal standard. An example of GC chromatogram together with the corresponding table of products identified by MS were provided in the Supplementary Materials (Graph S1, Table S2) as well as relative coefficients with respect to dodecane determined for the mains products (Table S3).
The bio-oil was recovered by evaporation at a temperature of 50 °C and a pressure of 200 mBar.
The Fourier-transform infrared (FTIR) spectra were obtained from solid samples diluted in KBr pellet (1%) with a Bruker spectrometer with 2 cm−1 resolution, in absorbance mode.
4. Conclusions
If we were expecting, from catalysts’ addition, an increase in the yields of light monomer derivatives at the expense of bio-oils yields, we were not expecting such an impact of the catalyst on the transformation of the solid pine wood components. Indeed, we postulated that solely the nature of the solvent and the conditions should control the pine components transformation. Hence our choice of ethanol at 250 °C, which partially attacks hemicellulose and lignin keeping cellulose unchanged. We preferred near-critical ethanol to near-critical n-heptane because the latter converts the three components including cellulose, and moreover, favors solid carbon products, in contrast to near-critical ethanol at 250 °C.
Moreover, we chose heterogeneous catalysts based on zirconia, alumina or niobic acid on the basis of previous studies showing their relative good stability under hydrothermal conditions. Moreover, we could expect a relative better stability in SC organic medium at relative mild conditions. Our study shows the poor resistance of all the chosen catalysts in SC ethanol medium, at 250 °C, which is an unexpected result. Probably, water accumulation in the reaction medium is responsible for the leaching of the strong catalysts.
This study highlights the positive effect of catalysts, in particular β zeolite, to increase the direct chemical yields up to 33% relative to carbohydrates, as well as the parallel drawbacks such as changes in wood fractionation and leaching of catalysts which make the overall transformation hardly controllable. We believe that these phenomena contribute to hamper significant progress in that area.
Indeed, working on isolated biopolymers such as cellulose was already a new challenge in the field of heterogeneous catalyst since we faced the issue of solid–solid contact between the substrate and the catalyst, and that of in-situ solvolysis of the solid biopolymer for its further catalytic transformation. With a key point, the reliable measurement of the solid cellulose conversion, without making confusion between conversion and liquefaction [32]. Here, we face an even more difficult issue: the conversion of three solid substrates, the three wood components which present differentiate recalcitrance as regards to their solvation. Again, the issue of the individual component conversion survey is hardly solved. Moreover, the present study highlight that the presence of a catalyst influences the relative conversion of the wood solid components by phenomenon that can hardly be prevented such as modification of the solvation ability of the organic medium by the reaction products and the catalyst leaching. So many factors that make the catalytic wood fractionation difficult to control when implemented in batch reactor.
It is clear that the control of the pine wood fractionation independently but in close proximity to the catalytic selective conversion of the liquefied biopolymer at the early stage is the key, for achieving high selectivity in a family of products starting from raw wood. Accordingly, a plug flow reactor is certainly more appropriate
Supplementary Materials
The following supporting information can be downloaded at: https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/catal12111333/s1, Graph S1: Typical GC chromatogram of the Light liquid products; Figure S1: XRD Pattern of ZrO2/La2O3; Figure S2: XRD pattern of CuZnAlO; Table S1: Fresh catalysts and solid residues calcined atat 1000 °C under air in a muffle furnace: remaining weights on catalyst basis (wt%); Table S2: Table of identified products corresponding to the chromatogram shown in Graph S1; Table S3: Relative coefficients with respect to dodecane calibrated for the main products.
Author Contributions
M.E.: formal analysis, investigations. N.E.: writing—original draft preparation, supervision. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by CNRS.
Acknowledgments
The authors acknowledge the scientific services of IRCELYON.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Saka, S. Recent progress in supercritical fluid science for biofuel production from woody biomass. For. Stud. China 2006, 8, 9–13. [Google Scholar] [CrossRef]
- Bogolitsyn, K.G.; Krasikova, A.A.; Gusakova, M.A. Supercritical fluid technologies in the chemistry of wood and its components. Russ. J. Phys. Chem. B 2015, 9, 1065–1073. [Google Scholar] [CrossRef]
- Cheng, S.; Wei, L.; Julson, J.; Kharel, P.R.; Gao, Y.; Gu, Z. Catalytic liquefaction of pine sawdust for biodiesel development on bifunctional Zn/HZSM-5 catalyst in supercritical ethanol. J. Anal. Appl. Pyrolysis 2017, 126, 257–266. [Google Scholar] [CrossRef]
- Li, R.; Li, B.; Yang, T.; Kai, X.; Zhang, W. Hydrogenation of rice stalk in situ in supercritical ethanol-water co-solvent via catalytic ethanol steam reforming. J. Supercrit. Fluids 2018, 133, 309–317. [Google Scholar] [CrossRef]
- Rataboul, F.; Essayem, N. Cellulose reactivity in Supercritical methanol in the presence of solid acid catalysts: Direct synthesis of methyl-levulinate. Ind. Eng. Chem. Res. 2011, 50, 799–805. [Google Scholar] [CrossRef]
- Essayem, N.; Sapaly, G.; Eternot, M.; Rataboul, F. Method for Preparing Levulinic Acid Esters. WO 2014001486, 26 June 2013. [Google Scholar]
- Luo, B.; Zhou, L.; Tian, Z.; He, Y.; Shu, R. Hydrogenolysis of cornstalk lignin in supercritical ethanol over N-doped micro-mesoporous biochar supported Ru catalyst. Fuel Process. Technol. 2022, 231, 107218. [Google Scholar] [CrossRef]
- Subbotina, E.; Velty, A.; Corma, A.; Samec, J. Method of Forming Monomers and Furfural from Lignocellulose. WO2020/101563, 14 November 2019. [Google Scholar]
- Koll, P.; Bronstrup, B.; Metzger, J. Thermal-degradation of birch wood with supercritical gases (organic-solvents) in a high-pressure, high-temperature flow apparatus-liquefaction of wood and further evidence for an alternative cellulose pulp technology. Holzforschung 1979, 33, 112–116. [Google Scholar]
- Sunol, A.K.; Chen, S.L. Supercritical Delignification of Wood. WO 90/02836, 14 July 1989. [Google Scholar]
- Humphreys, L.; Rogers, P. Fractionation of Lignocellulosic Matter. WO 2010/034055, 23 September 2009. [Google Scholar]
- Renders, T.; V.den Bossche, G.; Vangeel, T.; Van Aelst, K.; Sels, B. Reductive catalytic fractionation: State of the art of the lignin first biorefinery. Curr. Opin. Biotechnol. 2019, 56, 193–201. [Google Scholar] [CrossRef]
- Anderson, E.M.; Stone, M.L.; Hulsey, M.; Beckham, G.T.; Roman-Leshkov, Y. Kinetic Studies of Lignin Solvolysis and Reduction by Reductive Catalytic Fractionation Decoupled in Flow-Through Reactors. ACS Sustain. Chem. Eng. 2018, 6, 7951–7959. [Google Scholar] [CrossRef]
- Kubilay Tekin, K.; Hao, N.; Karagoz, S.; Ragauskas, A.J. Ethanol: A Promising Green Solvent for the Deconstruction Lignocellulose. ChemSusChem 2018, 11, 3559–3575. [Google Scholar] [CrossRef]
- Huang, X.; Atay, C.; Zhu, J.; Palstra, S.W.; Korányi, T.I.; Boot, M.D.; Hensen, E.J. Catalytic Depolymerization of Lignin and Woody Biomass in Supercritical Ethanol: Influence of Reaction Temperature and Feedstock. ACS Sustain. Chem. Eng 2017, 5, 10864–10874. [Google Scholar] [CrossRef]
- Li, Q.; Liu, D.; Hou, X.; Wu, P.; Song, L.; Yan, Z. Hydro-liquefaction of microcrystalline cellulose, xylan and industrial lignin in different supercritical solvents. Bioressource Technol. 2016, 219, 281–288. [Google Scholar] [CrossRef]
- Bui, N.Q.; Fongarland, P.; Rataboul, F.; Dartiguelongue, C.; Charon, N.; Vallée, C.; Essayem, N. FTIR as a simple tool to quantify unconverted lignin from chars in biomass liquefaction process: Application to SC ethanol liquefaction of pinewood. Fuel Process. Technol. 2015, 134, 378–386. [Google Scholar] [CrossRef]
- Sharma, K.; Shah, A.A.; Toor, S.S.; Seehar, T.H.; Pedersen, T.H.; Rosendahl, L.A. Co-Hydrothermal Liquefaction of Lignocellulosic Biomass in Supercritical Water. Energies 2021, 14, 1708. [Google Scholar] [CrossRef]
- Zandvorrt, I.V.; Koers, E.J.; Weingarth, M.; Bruijnincx, A.; Baldus, M.; Weckhuysen, B.M. Structural characterization of 13C-enriched humins and alkali-treated 13C humins by 2D solid state NMR. Green Chem 2015, 17, 4383–4392. [Google Scholar] [CrossRef] [Green Version]
- Veiga, P.M.; Gomes, A.C.L.; Veloso, C.D.O.; Henriques, C.A. Etherification of different glycols with ethanol or 1-octanol catalyzed by acid zeolites. Mol. Catal. 2018, 458, 261–271. [Google Scholar] [CrossRef]
- Le, S.D.; Nishimura, S.; Ebitani, K. Direct esterification of succinic acid with phenol using zeolite beta catalyst. Catal. Commun. 2019, 122, 20–23. [Google Scholar] [CrossRef]
- Gallo, J.M.R.; Alonso, D.M.; Mellmer, M.A.; Yeap, J.H.; Wong, H.C.; Dumesic, J.A. Production of Furfural from Lignocellulosic Biomass Using Beta Zeolite and Biomass-Derived Solvent. Top. Catal. 2013, 56, 1775–1781. [Google Scholar] [CrossRef]
- Lopes de Souza, R.; Rataboul, F.; Essayem, N. 5-Hydroxymethylfurfural (5-HMF) production from hexoses: Limits of heterogeneous catalysis in hydrothermal conditions and potential of concentrated aqueous organic acids solutions as reactive solvent system. Challenges 2012, 3, 212–232. [Google Scholar] [CrossRef]
- Barrault, J.; Essayem, N. Hydrogenation and methylation of dodecyl nitrile in the presence of copper catalysts supported on alumina II-Catalytic properties. Appl. Catal. A Gen. 1993, 102, 137–150. [Google Scholar] [CrossRef]
- Liu, H.-M.; Li, H.-Y.; Li, M.-F. Cornstalk liquefaction in sub- and super-critical ethanol:Characterization of solid residue and the liquefaction mechanism. J. Energy Inst. 2016, 90, 734–742. [Google Scholar] [CrossRef]
- Nguyen, V.C.; Bui, N.Q.; Mascunan, P.; Vu, T.T.H.; Fongarland, P.; Essayem, N. Esterification of aqueous lactic acid solutions with ethanol using carbon solid acid catalysts: Amberlyst 15, sulfonated pyrolyzed wood and graphene oxide. Appl. Catal. A Gen. 2018, 552, 184–191. [Google Scholar] [CrossRef]
- Chambon, F.; Rataboul, F.; Pinel, C.; Cabiac, A.; Guillon, E.; Essayem, N. Cellulose hydrothermal conversion promoted by heterogeneous Bronsted and Lewis acids: Remarkable efficiency of solid Lewis acids to produce lactic acid. Appl. Catal. B Environ. 2011, 105, 171–181. [Google Scholar] [CrossRef]
- Abdouli, I.; Eternot, M.; Dappozze, F.; Guillard, C.; Essayem, N. Comparison of Hydrothermal and photocatalytic conversion of glucose with commercial TiO2: Superficial properties-Activities relationships. Catal. Today 2021, 367, 268–277. [Google Scholar] [CrossRef]
- Conti, F.; Begotti, S.; Ippolito, F. Process for Producing Levulinic Acid. WO2018/235012, 20 July 2018. [Google Scholar]
- Dandach, A.; Russbueldt, B.; Toufaily, J.; Karout, A.; Hamieh, T.; Essayem, N. Mesoporous zirconium oxide prepared by anchoring W, Mo, Nb, Ta using peroxo precursors: Influence of the oxoanions on the pores size and the hydrothermal catalysts stability for cellulose conversion. Catal. Lett. 2022. [Google Scholar] [CrossRef]
- Pinheiro Braga, T.; Essayem, N.; Valentini, A. Synthesis of Cu-MxOy/Al2O3 (M=Fe, Zn, W, Sb) catalysts for the conversion of glycerol to acetol: Effect of texture and acidity of the supports. RSC Adv. 2015, 5, 93394–93402. [Google Scholar] [CrossRef]
- Nguyen, V.C.; Bui, N.Q.; Eternot, M.; Vu, T.T.H.; Fongarland, P.; Essayem, N. Kinetic of ZrW catalyzed cellulose hydrothermal conversion: Deeper understanding of reaction pathway via analytic tools improvement. Mol. Catal. 2018, 458, 171–179. [Google Scholar] [CrossRef]
Figure 1.
Wood components’ conversion (wt%) in SC ethanol at 250 °C and 280 °C and in SC n-heptane at 280 °C. Conditions: Batch reactor. m pine wood = 2.5 g, m solvent = 27 g, 1 h at 250 °C or 280 °C in SC ethanol, 1 h at 280 °C in SC n-heptane.
Figure 1.
Wood components’ conversion (wt%) in SC ethanol at 250 °C and 280 °C and in SC n-heptane at 280 °C. Conditions: Batch reactor. m pine wood = 2.5 g, m solvent = 27 g, 1 h at 250 °C or 280 °C in SC ethanol, 1 h at 280 °C in SC n-heptane.

Figure 2.
FTIR spectra of solid residues recovered after pine wood fractionation in SC ethanol at 250 °C and in SC n-heptane at 280 °C for 1 h. Comparison to FTIR spectra of fresh pine wood.
Figure 2.
FTIR spectra of solid residues recovered after pine wood fractionation in SC ethanol at 250 °C and in SC n-heptane at 280 °C for 1 h. Comparison to FTIR spectra of fresh pine wood.

Figure 3.
Products’ yields of pine wood fractionation: influence of the nature of the SC fluid and of the temperature. Conditions: Batch reactor. m pine wood = 2.5 g, m solvent = 27 g, 1 h in SC ethanol or SC n-heptane at 250 °C or 280 °C.
Figure 3.
Products’ yields of pine wood fractionation: influence of the nature of the SC fluid and of the temperature. Conditions: Batch reactor. m pine wood = 2.5 g, m solvent = 27 g, 1 h in SC ethanol or SC n-heptane at 250 °C or 280 °C.

Figure 4.
Light products distribution in near SC ethanol and near SC n-heptane. Conditions: Batch reactor. m pine wood = 2.5 g, m solvent = 27 g, 1 h in near critical ethanol or near critical n-heptane at 250 °C or 280 °C.
Figure 4.
Light products distribution in near SC ethanol and near SC n-heptane. Conditions: Batch reactor. m pine wood = 2.5 g, m solvent = 27 g, 1 h in near critical ethanol or near critical n-heptane at 250 °C or 280 °C.

Figure 5.
Conversions (wt%) of cellulose, hemicelluloses and lignin in the presence of various catalysts, in SC ethanol at 250 °C. Conditions: batch reactor, 2.5 g pine, 1 g catalyst, 27 g ethanol, 250 °C, 1 h, 80 bar.
Figure 5.
Conversions (wt%) of cellulose, hemicelluloses and lignin in the presence of various catalysts, in SC ethanol at 250 °C. Conditions: batch reactor, 2.5 g pine, 1 g catalyst, 27 g ethanol, 250 °C, 1 h, 80 bar.

Figure 6.
Yields of gaseous, heavy liquid, light liquid and solid products resulting from the solvolysis of pine in SC ethanol at 250 °C in the presence of different types of catalysts. Conditions: 2.5 g of Pin, 1 g of catalyst, 27 g of ethanol, at 250 °C, 1 h.
Figure 6.
Yields of gaseous, heavy liquid, light liquid and solid products resulting from the solvolysis of pine in SC ethanol at 250 °C in the presence of different types of catalysts. Conditions: 2.5 g of Pin, 1 g of catalyst, 27 g of ethanol, at 250 °C, 1 h.

Figure 7.
100% distribution of light liquid products in the absence and presence of various catalysts. Fractionation of pine in SC ethanol at 250 °C. Conditions: Batch reactor, pine 2.5 g, catalyst 1 g, ethanol 27 g, 1 h at 250 °C.
Figure 7.
100% distribution of light liquid products in the absence and presence of various catalysts. Fractionation of pine in SC ethanol at 250 °C. Conditions: Batch reactor, pine 2.5 g, catalyst 1 g, ethanol 27 g, 1 h at 250 °C.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Pin conversions (wt%) obtained in SC ethanol in the presence of various catalysts.
| Catalysts | Without | La2O3/ZrO2 | NbOH | β Zeolite | CuZnAlO |
|---|---|---|---|---|---|
| Wood conversion (wt%) | 18 | 15 | 54 | 75 | 50 |
Conditions: 2.5 g of Pin, 1 g of catalyst, 27 g of ethanol, at 250 °C, 1 h.
Table 2.
Impact of heterogeneous catalysts’ addition on the mass balance of pine wood fractionation in SC ethanol at 250 °C.
Table 2.
Impact of heterogeneous catalysts’ addition on the mass balance of pine wood fractionation in SC ethanol at 250 °C.
| Catalyst Free | La2O3/ZrO2 | NbOH | β zeolite | CuZnAlO |
|---|---|---|---|---|
| 245 wt% | 233 wt% | 91 wt% | 116 wt% | 128 wt% |
Conditions: Batch reactor, 2.5 g of Pin, 1 g of catalyst, 27 g of ethanol, at 250 °C, 1 h.
Table 3.
Pine wood fractionation in SC ethanol: nature of the main light liquid products as a function of catalysts addition.
Table 3.
Pine wood fractionation in SC ethanol: nature of the main light liquid products as a function of catalysts addition.
| Products’ Families | Without Catalysts | La2O3/ZrO2 | NbOH | β Zeolite | Cu/ZnAlO |
|---|---|---|---|---|---|
| Phenolic derivatives | Isoeugenol | Isoeugenol | Acetyl eugenol | isoeugenol | Isoeugenol |
| Furans | Furfuryl alcohol | Furfuryl alcohol | / | Furfural | Furfuryl alcohol |
| Esters | Ethyl Glycolate | Ethyl Lactate d | Ethyl Glycolate  Ethyl Lactate  | Ethyl Levulinate Ethyl glycolate  | Ethyl Lactate |
Table 4.
Yields of chemicals relative to their original wood component.
| Catalysts | Without | La/ZrO2 | NbOH | β Zeolite | CuZnAlO |
|---|---|---|---|---|---|
| Esters/carbohydrates | 4.8 | 4 | 9.1 | 16.2 | 5.2 |
| Total oxygenates/carbohydrates | 7.2 | 12.5 | 15 | 32.7 | 13.9 |
| Phenolic derivatives/lignin | 1.8 | 2 | 0.9 | 5.9 | 7.6 |
Table 5.
Catalysts’ composition and BET surface areas.
| Catalysts | β Zeolite | NbOH | La2O3/ZrO2 | CuZnAlO |
|---|---|---|---|---|
| Composition | Si/Al = 10.5 Na = 83 ppm | - | 10% La2O3 | Cu:19.8 wt% Zn/Al = 0.15 |
| S BET (m2.g−1) | 609 | 144 | 112 | 88 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Eternot, M.; Essayem, N. Catalytic Wood Fractionation into Chemicals in Supercritical Ethanol and n-Heptane: Potential and Limitations. Catalysts 2022, 12, 1333. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12111333
AMA Style
Eternot M, Essayem N. Catalytic Wood Fractionation into Chemicals in Supercritical Ethanol and n-Heptane: Potential and Limitations. Catalysts. 2022; 12(11):1333. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12111333
Chicago/Turabian StyleEternot, Marion, and Nadine Essayem. 2022. "Catalytic Wood Fractionation into Chemicals in Supercritical Ethanol and n-Heptane: Potential and Limitations" Catalysts 12, no. 11: 1333. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12111333
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.