
Recent Progress in Two-Dimensional Materials for Electrocatalytic CO2 Reduction


1
Department of Energy and Petroleum Engineering, University of Stavanger, 4036 Stavanger, Norway
2
Beyonder AS, Kanalsletta 2, 4033 Stavanger, Norway
*
Author to whom correspondence should be addressed.
Catalysts 2022, 12(2), 228; https://0-doi-org.brum.beds.ac.uk/10.3390/catal12020228
Submission received: 17 January 2022
/
Revised: 8 February 2022
/
Accepted: 15 February 2022
/
Published: 17 February 2022
(This article belongs to the Special Issue Catalysis for CO2 Conversion)
Abstract
:Electrocatalytic CO2 reduction (ECR) is an attractive approach to convert atmospheric CO2 to value-added chemicals and fuels. However, this process is still hindered by sluggish CO2 reaction kinetics and the lack of efficient electrocatalysts. Therefore, new strategies for electrocatalyst design should be developed to solve these problems. Two-dimensional (2D) materials possess great potential in ECR because of their unique electronic and structural properties, excellent electrical conductivity, high atomic utilization and high specific surface area. In this review, we summarize the recent progress on 2D electrocatalysts applied in ECR. We first give a brief description of ECR fundamentals and then discuss in detail the development of different types of 2D electrocatalysts for ECR, including metal, graphene-based materials, transition metal dichalcogenides (TMDs), metal–organic frameworks (MOFs), metal oxide nanosheets and 2D materials incorporated with single atoms as single-atom catalysts (SACs). Metals, such as Ag, Cu, Au, Pt and Pd, graphene-based materials, metal-doped nitric carbide, TMDs and MOFs can mostly only produce CO with a Faradic efficiencies (FE) of 80~90%. Particularly, SACs can exhibit FEs of CO higher than 90%. Metal oxides and graphene-based materials can produce HCOOH, but the FEs are generally lower than that of CO. Only Cu-based materials can produce high carbon products such as C2H4 but they have low product selectivity. It was proposed that the design and synthesis of novel 2D materials for ECR should be based on thorough understanding of the reaction mechanism through combined theoretical prediction with experimental study, especially in situ characterization techniques. The gap between laboratory synthesis and large-scale production of 2D materials also needs to be closed for commercial applications.
1. Introduction
CO2 is the main component of greenhouse gases which lead to environmental concerns, and its concentration has increased from approximately 280 ppm in the early 1800s to 410 ppm today [1,2,3]. Without proper strategies to combat this problem, the steadily growing CO2 emissions will arouse the increase in the global average temperature, loss of glaciers, rise of sea level and other climate issues [4]. To reverse these negative courses, technologies such as carbon capture and storage (CCS) have been pursued [5,6,7]. Despite tremendous efforts on CCS, including separating CO2 from air or flue gas, storing the captured CO2 is still a daunting challenge due to the risk of leakage, massive energy consumption, high cost and social acceptance [8,9,10,11,12]. An alternative approach is to utilize CO2 as raw material and convert it into value-added products, which approach has received extensive attention in the past decades [13,14,15]. Therefore, the conversion of CO2 has been regarded as a promising route for closing the carbon cycle and producing value-added chemicals and fuels.
The conversion of CO2 can be realized by a variety of technologies such as thermochemical, electrochemical, photochemical, radiochemical and biochemical reactions [16,17,18,19,20,21,22,23,24]. Among these, the reduction of CO2 by electrochemical strategies is attractive and exhibits many advantages [25,26]. For instance, the energy required for electrochemical reduction reactions can be provided by renewable energy such as solar, geothermal or wind energy. In addition, the reaction can be performed under ambient pressure and temperature. Moreover, the external voltages as well as electrolyte solutions can be adjusted to produce specific products. However, the physicochemical properties of the CO2 molecule make electrochemical conversion processes challenging. To convert CO2 into other products, the dissociation of the C=O bond is essential and must overcome high-energy barriers of about 750 kJ/mol. In addition, electrolyte solutions are generally water-based, which makes the reaction inefficient due to the poor CO2 dissolution in water. In the past decades, both theoretical and experimental work have disclosed that the electrocatalysts play a vital role in catalytic processes [27,28,29,30,31,32]. For example, it has been proven that traditional metal electrocatalysts, such as copper, platinum and gold, can activate CO2 and reduce it into valuable chemical products [33,34,35]. However, these catalysts suffer drawbacks such as high overpotential, poor stability, low selectivity, complex separation and low efficiency for specific products. In addition, the prices of precious metals hamper the large-scale application of Pt catalysts. Therefore, developing efficient, cost-effective and durable alternatives to the traditional metal catalysts for electrocatalytic CO2 reduction (ECR) reaction is urgent.
In recent years, two-dimensional (2D) materials with novel geometric, electronic and optical properties greatly accelerated the development of catalysts. Two-dimensional materials, sometimes referred to as single-layer materials, are crystalline materials consisting of a single layer of atoms. These materials have promising applications in fields such as photovoltaics, semiconductors, electrodes and water purification. Two-dimensional materials can be categorized as either 2D allotropes of various elements or as compounds consisting of two or more covalently bonded elements. The elemental 2D materials generally carry the -ene suffix in their names, while the compounds have -ane or -ide suffixes. A wide range of 2D compounds, including metals, chalcogenides, group IV and V elements, oxides, carbides, nitrides, halides, hydroxides, hydrides, phosphates, phosphonates and covalent organic frameworks, have been investigated for various applications. Two-dimensional materials can exhibit semiconductive, insulative or even metallic behavior. Some of them with direct or indirect band gaps have great responses to ultraviolet and visible light. The electronic and optical properties of these 2D materials are tunable by adjusting the number of layers because of quantum confinement. Two-dimensional semiconductive materials display strong enhancements of Coulomb interactions among charge carriers and defects, contributing to longer-lived excitons compared to their bulk structures [36]. The specific surface area of 2D materials is usually quite high and more active atoms can be exposed. The physicochemical properties of 2D materials, especially, can be effectively modulated by different engineering strategies, including heterostructure, doping, chemical functionalization, etc. [37].
When employed as catalysts, 2D materials with nanosheets can show unique properties and excellent performance in catalytic reactions. Compared with conventional bulk materials, they have much higher specific surface areas and high percentages of bare surface atoms that can offer abundant active sites, boosting catalytic reactions. Note that the highly exposed surface atoms can potentially escape and form defect structures, leading to decreased coordination numbers of surface atoms, which are the preferential sites for the adsorption of reactants or intermediates. Similarly, the edge atoms of nanosheets with low coordination numbers can also exhibit novel catalytic properties. Therefore, 2D structures can promote the chemisorption of reactants and improve catalytic performance. A large class of 2D materials, such as noble metals, metal oxides, graphene, graphite carbon nitride (g–C3N4), transition metal dichalcogenides (TMDs), metal–organic frameworks (MOFs), etc., has been demonstrated to have great potential to catalyze CO2 conversion reactions [38,39,40,41,42,43]. Particularly, graphene, known as the first invented 2D material, has attracted extensive attention for many applications [44]. Its unique mechanical characteristics, electronic structures and optical properties make it applicable in fields such as solar cells, LED transistors, sensors and catalysts [45,46,47,48]. More importantly, the properties of 2D materials can be easily tuned via doping atoms, hybridizing with other nanostructures, surface engineering and defect generation, further improving their electronic properties and catalytic performance.
Numerous studies have been reported for ECR, and a few excellent reviews have been published. For example, Wang et al. reviewed the principles for electrocatalyst design and performance evaluation as well as the factors influencing ECR activity [49]. Nitopi et al. described the various experimental and theoretical approaches used to explore the mechanisms for the formation of ECR products on Cu catalysts. They concluded that nanostructuring and constructing bimetallic electrodes can be effective strategies to enhance activity and selectivity of Cu based catalysts [50]. Jin et al. highlighted the utilization of in situ spectroscopy, including X-ray absorption, X-ray photoelectron, Raman, infrared and mass spectroscopy, which greatly refined the understanding of ECR from intermediates, reaction pathways, active sites and the effects of a reaction environment on product distribution [51]. Zhu et al. summarized controlled synthesis methods for embedding a single atom on different carbon materials as an ECR catalyst. The effects of precursors and preparation environments on the structure of single-atom catalysts (SACs) were discussed. They also compared the intrinsic activity of different metal centers and methods for enhancing ECR efficiency [52]. Zhu et al. reviewed the latest progress on developing 2D materials for ECR from the perspective of theoretical simulation and discussed the reaction mechanisms of various 2D materials for ECR [53]. Zhang et al. reviewed recent advances on materials development for CO2 conversion by thermocatalysis, photocatalysis and photothermocatalysis, highlighting the reaction pathways and mechanism on C=O bond activation and intermediate formation [54]. Very recently, they also summarized CO2 conversion by electro- and photoelectrocatalysis, emphasizing the importance of operando characterization theoretical simulations [55]. Li et al. briefly reviewed bimetallic chalcogenides for ECR application in the past five years [56]. They disclosed that hybridization between metal atoms, such as that of intermetallic compounds, heterostructures and metal doping, shows positive effects on ECR selectivity and activity. Wang et al. reviewed recent advances on non-noble-metal-based ECR catalysts, focusing on the synthesis strategies for engineering the electrocatalysts and the ECR mechanism [57]. Therefore, a wide range of topics have been covered for ECR. In this work, we systematically review the recent progress on both theoretical and experimental work on 2D materials applied in ECR according to the materials type. We start with the introduction to 2D materials and CO2 conversion as well as the fundamentals of ECR. Thereafter, the emerging 2D materials for ECR, including metal, graphene-based materials, TMDs, MOFs, metal oxides and single-atom anchored 2D materials, are discussed in detail, and the effects of structure and modification strategies on catalytic performance for ECR are emphasized. We also summarize GDE and MEA electrolyzers which could solve the problem of limited CO2 solubility and energy loss of conventional H-type cells for potential industrial ECR applications. Based on these, future perspectives for ECR catalysts design and synthesis are proposed. We aim for this contribution to enlighten future research directions and strategies for exploiting novel 2D materials for ECR and energy conversion in general.
2. Fundamentals of Electrochemical CO2 Reduction
2.1. CO2 Properties and the Reduction Products
CO2 is one of the most stable molecules with a strong O=C=O double bond, and its bond energy is higher than that of C–H and C–C bonds. During electrocatalytic processes, the breaking of the O=C=O bond requires a high activation energy. In addition, since ECR consists of multiple elementary steps, these electrocatalytic processes are more demanding than, for example, water-splitting reactions. As ECR reactions are normally carried out in aqueous solutions, another critical issue is the low solubility of CO2 in water, which impedes the diffusion-controlled reactions. There are certain ways to improve CO2 dissolution, including using nonaqueous electrolytes and increasing CO2 partial pressure. For instance, some metal catalysts with low activity for ECR under atmospheric pressure, however, can reduce CO2 to CO and HCOOH efficiently at high pressures [58].
Electrocatalysts can effectively reduce activation energy, accelerate reactions or increase desired product selectivity in ECR. It should also dissociate water in solution to promote proton–electron transfer because the proton-assisted multiple-electron transfer can be beneficial for CO2 activation. Depending on the different pathways and the number of protons and electrons transferred, a range of products can be formed, including C1 products, such as carbon monoxide (CO), methanol (CH3OH), formic acid (HCOOH), methane (CH4), or C2 products, such as ethylene (C2H4), ethanol (C2H5OH) and others [59]. C2 products with higher energy densities are more valuable, but the synthesis of C2 products is more difficult than that of C1 products because of larger numbers of required protons during conversion. Meanwhile, high formation energy of the C–C bonds can decrease the efficiency of the reaction. To achieve specific product selectivity, the design and synthesis of high-performance ECR electrocatalysts is essential.
2.2. Single-Electron vs. Proton-Coupled Electron Reactions
Research on ECR has been conducted ever since the early 19th century. ECR contains two half reactions occurring in the anode and the cathode, where different numbers of proton-coupled electron transfers occur. The standard electrode potentials of different reactions with reference to standard hydrogen electrons (SHEs) for different products in aqueous solutions are summarized in Table 1. For a typical single-electron ECR, CO2 is reduced to the CO2 anion radical () at the cathode and water is oxidized to oxygen at the anode. Since the kinetics of the reactions are quite sluggish, the first step for converting CO2 into reduced carbon species is difficult. The thermodynamic potential for driving single-electron CO2 reduction to is about −1.90 V vs. SHE at a PH of 7, indicating that this reaction is highly energetic and unfavorable [60,61,62]. In addition, this first step is also the rate-limiting step, and the generation of the intermediate plays a significant role in forming 2e− reduction products. Interestingly, a more favorable route that bypasses the formation of by proton-coupled electron transfer processes has been identified. The transfer of proton-coupled electrons benefits ECR within the potential range of −0.20 to −0.60 V vs. SHE, and the final products are determined by the choices of electrocatalysts and electrolytes as well as the numbers of electrons and protons transferred [63,64]. For instance, two-proton-coupled electron transfer in a CO2 hydrogenation reaction mainly produces HCOOH and CO.
As shown in Figure 1, ECR starts by transferring a proton–electron pair to form carboxyl (*COOH) or formate (*OCHO) intermediate species [53]. These two intermediates can be further reduced by accepting different numbers of proton–electron pairs. Note that there is much competition between intermediate species. For example, the OHCO* species can be converted to HCOOH, and the biformate (H2COO*) can also be reduced to HCOOH. Meanwhile, the different products also show similar reduction potentials. Thus, further reactions of these intermediate species are important for the reaction rate and formation of the final products. Moreover, as the equilibrium potential of a hydrogen evolution reaction (HER) (0 V vs. reversible hydrogen electrode (RHE)) is more positive than that of CO2 reduction to HCOOH (−0.17 V vs. RHE), H2 will be the by-product, which seriously limits ECR efficiency [65].
2.3. Factors Determining Product Selectivity
Factors such as the type of electrocatalyst (morphology, composition, chemical state and crystallographic structure), type of electrolyte (composition, concentration and pH), temperature, pressure and applied potential all influence ECR. For instance, solid and aqueous electrolytes have been used in ECR, exhibiting remarkable differences in efficiency [66,67]. To date, NaHCO3 and KHCO3 solutions are frequently chosen as electrolytes because they can maintain the pH at the electrode surface. Meanwhile, the pH of the solution can affect the generation of by-products such as H2.
ECR, with a more positive potential vs. SHE, is thermodynamically more favourable. In this regard, the conversion of CO2 to alcohol or hydrocarbon products should be thermodynamically more favourable than to other products, including CO, HCOOH and HCHO (Table 1). Nevertheless, ECR not only needs to overcome the thermodynamic barrier but also the kinetic challenge determined by the concentration of available protons in reaction. Namely, the electrocatalysts prefer to transfer electrons from their catalytic sites to adjacent sites which can provide protons. Thus, the formation of hydrogenated C1 intermediates such as HCOOH can be kinetically more favourable than the formation of C–C bonds, which hinders the selectivity to C2 and higher hydrocarbon products. To date, only the Faradaic efficiency (FE) of C1 products CO and HCOOH can reach 100%, while the maximum FE of C2 product C2H4 is 80% and the maximum FE of C3 product C3H7OH is 30% [68,69].
Many theoretical studies proposed that for multielectron transfer reduction of CO2, the adsorption energies of intermediates follow linear scaling relationships, and breaking such linear scaling relationships can improve catalytic performance. Some strategies have been proposed, including reducing coordination numbers, doping with p-block elements, engineering oxyphilic sites and coating the catalyst surface with active ligands. Up to now, the majority of the ECR electrocatalysts have been metal- and carbon-based materials. It has been demonstrated that some heterogeneous electrocatalysts follow the two-electron-transfer mechanisms in CO2 reduction to HCOOH or CO. For example, Hori et al. investigated a series of metals for ECR reaction and indicated that metallic electrocatalysts, such as Cd, Sn, Hg and Pb, prefer HCOOH formation [70]. However, noble metals, including Pt, Ag and Au, can convert CO2 into CO. Copper-based electrocatalysts are the only electrocatalysts that have been reported for converting CO2 into alcohols and hydrocarbons with good activity and selectivity. However, as mentioned above, the interaction between intermediates and electrocatalysts plays a crucial role in forming final products, demonstrating the importance of electrocatalysts.
2.4. Key Parameters for Evaluating ECR Efficiency
In experimental ECR study, there are several key parameters for evaluating electrocatalyst performance, including overpotential, current density, Tafel slope, FE, turnover frequency (TOF) and stability of electrocatalysts. The overpotential can be regarded as the energy needed to drive the reaction. In other words, the higher the overpotential, the higher the energy required for the reaction and the more difficult the reaction to be realized. Current density reflects the rate of electrochemical reaction, and the larger the current density, the faster the electrocatalytic reaction. The Tafel slope can be obtained by fitting the linear region of the Tafel curve. With the increase in overpotential, if the current density increases dramatically, the slope will be small. FE is regarded as the fraction between charges transferred to specific products and the total charges in the electrocatalytic process, accounting for the selectivity and efficiency of the reaction. Larger FE suggests less energy loss, and smaller FE indicates more energy loss. TOF is the catalytic activity of each site under a certain overpotential, indicating the intrinsic activity of catalysts. The stability of electrocatalysts can be evaluated by long-term use under a specific voltage.
In summary, the electrocatalytic reaction is a complicated multistep reaction occurring at the multiphase interface. Small overpotential and high current density can boost ECR, but it is difficult to satisfy them simultaneously. Developing excellent electrocatalysts to overcome these problems is the key.
3. Two-Dimensional Materials as Efficient ECR Electrocatalysts
Compared to bulk materials, 2D materials have a disordered state in their normal direction and a long-range order in the plane, showing a sheet structure. In addition, 2D materials can be exfoliated to single- or multiple-atom thickness, exhibiting interatomic covalent bonds in the plane. The physicochemical properties of 2D materials are different from that of bulk materials. The high atomic exposure rate, large specific surface area and flexible structure make them promising ECR catalysts. Some reported 2D-materials-based electrocatalysts for ECR are summarized in Table 2 and are illustrated in detail in the following sections. Many metals (Ag, Cu, Au, Pt, Pd, etc.), graphene-based materials, Ni/Fe/Zn-doped nitric carbide, metal-doped TMDs and MOFs can produce CO. Particularly, single TM atoms embedded in 2D materials as SACs can exhibit great CO selectivity. Metal oxides and graphene-based materials can produce HCOOH. Interestingly, only Cu-based materials can produce CH4, C2H4 and other C2+ products.
3.1. Two-Dimensional Metallic Materials
Bulk metal catalysts in ECR have been investigated for decades [109,110,111]. Metal electrocatalysts can be divided into two groups: noble metal (Au, Ag, Pt, etc.) and other earth-abundant transition metals (Co, Ni, Cu, etc.). Theoretical work has well disclosed that noble metals are better electrocatalysts compared with earth-abundant metals. Nevertheless, some intermediates, such as CO, could poison the active sites, which affects their stability. Large-scale application of precious metals is also not possible due to their high price and scarcity.
It has been revealed that precious metals with 2D nanosheet structures can enhance their electrocatalytic performance effectively and improve the utilization of noble metals [72]. Huang et al. prepared a hexagonal Pd structure with 2.5 times higher electrocatalytic activity for the oxidation of formic acid compared to commercial Pd black [112]. The high performance could be attributed to nanosheet structure with more active sites, better electronic structure and larger surface area, as well as high atom utilization. Zhu et al. synthesized ultrathin Pd nanosheets for ECR, which exhibited an FE of 94% for CO production at a potential of −0.50 V vs. RHE [73]. Meanwhile, experimental and theoretical work revealed that ultrathin Pd nanosheets with high edge exposure ratios are beneficial for CO2 adsorption and CO desorption as well as suppressing HER. Nanostructured Au also shows great activity in ECR. Mistry et al. studied the electrocatalytic activity of Au nanoparticles (NPs) in the range of 1–8 nm for CO2 reduction to CO [113]. The results suggested that the current density increases with the decrease in particle size.
Moreover, multimetal composite nanosheets also exhibit excellent performance in electrocatalytic reactions because of the synergistic effect of different metals compared to single-metal electrocatalysts [114,115,116]. Wang et al. showed that a 2D Pd–Au bimetallic catalyst has lower activation energy than monometallic Au in ECR via density functional theory (DFT) calculation [74]. Very recently, Au–Cu bimetallic thin films with various ratios were synthesized for ECR [117]. The activity of the Au–Cu alloy was found to be correlated with the variation of electronic structures determined by alloy composition. As shown in Figure 2a, the surface valence bands of the Au–Cu catalysts present different patterns with different alloy compositions. The d-band center gradually moves away from the Fermi level with the increase in Au concentration (Figure 2b), which could influence the occupancy of antibonding states. Meanwhile, the interaction strength between substrate and intermediates decreases. The d-band center does not shift after ECR, implying great stability. Therefore, the catalytic performance for CO formation is enhanced as Au concentration increases, with the highest current density and FE at −0.80 V vs. RHE (Figure 2c,d).
Nonprecious metals, including Fe, Co, Ni, etc., have also attracted much attention in electrocatalysis due to low prices, abundant resources and promising electrochemical properties as well as tunable electronic structures [118,119]. Studies have shown that Fe, Ni and Ti produce H2 as the major product in ECR due to their low HER overpotential and strong CO adsorption capability. Sn, Pb and In have poor binding energy with intermediates and the final products are usually formate or formic acid. Moreover, Cu is the only metal catalyst that can produce C3 hydrocarbons by a C–C coupling mechanism [120]. Generally, modulating the electronic and surface structures of these metals could effectively enhance their electrocatalytic activity. Kuang et al. synthesized ultrathin Ni nanosheet arrays by an in situ topology reduction technique. In their study, the electrocatalyst showed excellent HER activity compared with Pt/C because ultrathin nanosheet structure could expose more active sites which boost electrocatalytic reactions [121]. In addition, 2D-structured bismuth (Bi) exhibited more active sites and promising selectivity to formate compared to bulk Bi. Zhang et al. prepared ultrathin Bi by a liquid phase stripping technique. The ultrathin Bi displayed excellent electrocatalytic performance for formate generation with an FE of 86% and a current density of 16.5 mA/cm2 at −1.1 V vs. RHE, which is obviously higher than bulk Bi and acetylene black decorated carbon paper (AB/CP) (Figure 3a,b). As presented in Figure 3c,d, CO2 conversion to HCOOH underwent lower Gibbs free energy change on edges (003) and (012) [76]. Special surface structure can also improve the catalytic efficiency and selectivity. It has been reported that ultrathin porous Cu nanosheets synthesized by a simple replacement method can achieve an FE of 74.1% and current density of 23.0 mA/cm2 at −1.0 V vs. RHE for CO2 conversion to CO, remarkably better than that of traditional bulk Cu [77]. DFT calculations clarified that the porous Cu nanosheet structure can accelerate the formation of CO intermediate, thus promoting ECR efficiency. Similarly, Wang et al. synthesized Cu nanocubes which demonstrated better electrocatalytic activity and selectivity in comparison with Cu nanospheres of the same size. The nanocubes could reach an FE of 60% and a partial current density of 144 mA/cm2 towards C2H4 production [78]. Zhang et al. prepared the pipet-like bismuth (Bi) nanorods semifilled in nitrogen-doped carbon nanotubes (BiNRs@NCNTs) for ECR [122]. The catalyst acted as a nanoscale conveyor which could facilitate mass transport and reactant adsorption on active sites. As a result, the FE for formate generation reached 90.9% at a moderate applied potential of −0.9 V (vs. RHE).
Therefore, metal catalysts are effective for electrocatalysis due to their intrinsic activity and good conductivity. When the metal catalysts are synthesized with 2D nanosheets or other special nanostructures, the atom utilization, activity and selectivity could be improved, thus increasing electrocatalytic efficiency. Precious metals, such as Pd or Au, or multimetals with different ratios show excellent selectivity for CO generation. Nonprecious metals, such as Fe, Co and Ni, show poor ECR performance, while Sn, Pb and In can have good activity for the production of formate or formic acid. Particularly, Cu-based catalysts with different morphologies can exbibit different activities and selectivities for different products. Accordingly, the choice of metal, small nanosheet size and different morphology will have a positive effect on ECR activity and selectivity.
3.2. Graphene and Graphene-Based Materials
Generally, graphene is a single layer of graphite with a zero-band gap. It is a promising electrocatalyst for CO2 reduction due to its high electron mobility, conductivity, unique electronic structure and large surface area. In addition, the high thermal conductivity can improve heat diffusion during the exothermic process, benefiting electrocatalysis. However, pristine graphene exhibits some drawbacks in ECR. For instance, the delocalized π bonding network of graphene negatively affects the adsorption of intermediates such as *COOH or *OCHO; therefore, graphene cannot effectively activate the CO2 molecule and presents high-energy barriers for intermediate formation, leading to low ECR activity [123]. Some studies have clarified that the undoped zigzag edge of graphene shows a metastable adsorbed CO2 state, and the energy barrier is quite high at about 1.3 eV [124]. As the modification of electronic structures can efficiently tackle these problems, many researchers have focused on tailing its electronic structures. For example, incorporating heteroatoms, such as B or N, can effectively modify graphene structures and decrease the CO2 adsorption barrier. Studies have disclosed that N-doped graphene can exhibit a low overpotential of around 0.19 V for converting CO2 into CO [75]. Duan et al. reported that doping boron can stabilize the negatively polarized O atoms of CO2 and improve CO2 chemisorption on carbon surfaces [125]. Similarly, Sreekanth et al. reported B-doped graphene as a metal-free electrocatalyst for CO2 reduction in 0.1 M KHCO3 solutions. They concluded that the presence of boron dopants can introduce spin density distribution and these atoms can be active sites for intermediate adsorption [79]. Some doped systems have been investigated in more detail by DFT calculations. Wu and coworkers systematically studied the effect of N-doped graphene for ECR. Three different N atom sites, including graphitic N, pyrrolic N and pyridinic N, were investigated as active sites for ECR. The authors suggested that the three different N sites can significantly decrease the free energy barrier for intermediate adsorption. The triple-pyridinic N can most effectively reduce the barrier for COOH adsorption, indicating that pyridinic N is a highly active site for converting CO2 to CO [75]. However, there are different views on the mechanism of N-doped graphene for ECR. Chai et al. reported that the graphitic N-doped edges of graphene sites have low CO2 activation barriers and are the most active sites for ECR among graphene-based materials [124]. Liu et al. demonstrated that a pyrrolic N site performs the best for CO2 reduction to HCOOH with a low overpotential [126]. It has also been reported that N-doped graphene quantum dots with predominant pyridinic N at the edges can electrochemically convert CO2 to C2 compounds with low overpotentials [59].
Compared with nonmetal dopants, introducing single or multiple metal dopants to construct special graphene nanostructures could also exploit materials with excellent catalytic performance. Au nanoparticles embedded in graphene nanoribbon exhibit low overpotential, high FE for CO generation and excellent stability compared to amorphous carbon-supported Au nanoparticles, attributed to the change in electronic properties and the increase in active sites [127]. Liu et al. synthesized Pd and Cu mono- and bimetallic nanoparticle-embedded graphene and suggested that 1 wt.% Pd–2 wt.% Cu/graphene had the highest ECR efficiency [80]. Su et al. prepared nickel–nitrogen comodified graphene (Ni–N–Gr) with more active centers by short-duration heat treatment and found that the Ni–N site was the active center for CO2 reduction [81]. As shown Figure 4a,c, the Ni–N–Gr and the Ni foil exhibit obvious enhancement of current density in the presence of CO2. However, there is a remarkable difference in FE of CO. The Ni–N–Gr showed an FE of 90% at −0.7 to −0.9 V vs. RHE for CO production, where Ni foil produced mostly H2 (Figure 4b,d), demonstrating that the synergistic effect of Ni and N is critical for improving CO selectivity.
Graphene-based composite materials also possess excellent conductivities and larger surface areas compared to pristine graphene. Huang et al. synthesized N-doped graphene monolayer-coated Sn foil, showing excellent flexibility with a high FE of 92% for formate at −1.0 V vs. RHE [82]. Lei et al. prepared Sn quantum sheets confined in graphene, offering more active sites for CO2 adsorption [83]. Li et al. successfully prepared SnS2 nanosheets supported on reduced graphene oxide for CO2 conversion to formate, displaying a low overpotential of 0.23 V and a maximum Faradaic efficiency of 84.5%. The spectroscopic and electrochemical characterizations suggested that the improvement of electrocatalytic performance can be attributed to the residual SnS2 [84].
For graphene-like materials, graphitic carbon nitride (g–C3N4) typically exhibits good chemical and thermal stability under ambient conditions. However, poor conductivity and less active sites limit its potential application in ECR. Strategies, including doping metal and constructing composite, have been developed to increase the activity of g–C3N4. Metal atoms can effectively modulate the electronic structure of g–C3N4, thus improving the catalytic activity. The g–C3N4 nanosheet and multiwalled carbon nanotubes composite have been shown to exhibit excellent stability and good electrocatalytic performance for CO2 reduction to CO [85]. The electrocatalytic activity of the composite arises from the C–N bonds, and the high conductivity allows numerous electrons to transfer rapidly to the C–N sites. Guo et al. synthesized Co3O4–CDots–g–C3N4 tricomponent electrocatalysts for syngas production [86]. They suggested that different catalytic components have different functionalities. For instance, g–C3N4 and Co3O4 provide active sites for ECR and HER, respectively, whereas CDots are the sites for proton generation.
It can be concluded that the electronic structure of graphene-based materials can be tuned to effectively improve ECR selectivity. Doping nonmetal or metal atoms is an efficient way for the design and synthesis of high-performance ECR catalysts. To date, nitrogen and metal atom codoping has been widely reported and shown excellent activity for CO production. Constructing highly active interfaces in graphene-based nanocomposite is also a good strategy in ECR catalyst design.
3.3. Two-Dimensional Transition Metal Dichalcogenides
Transition metal dichalcogenides (TMDs), such as MoS2, MoSe2 and WS2, have the general formula MX2, where M is a transition metal atom and X represents a chalcogen atom. The layers of X–M–X are stacked together by the van der Waals interaction. This is another class of 2D materials with exceptional physical and chemical properties to be applied as electrocatalysts [88]. The large surface area of 2D TMDs can provide more active sites for reactions such as CO2 reduction and water splitting [128,129]. DFT calculations suggested that the excellent catalytic properties for metal-terminated edges of 2D TMDs can be attributed to its metallic character and high d-electron density [87]. It has been confirmed that intermediates, such as COOH and CHO, prefer to adsorb on MoS2 and MoSe2 edges than the bridge S or Se atoms, and CO species adsorb selectively on the metal atoms during ECR [130,131]. Four different TMD materials, MoS2, MoSe2, WS2 and WSe2, have been investigated as ECR catalysts; of which, WSe2 nanoflakes exhibit the highest activity at an overpotential of 65 mV with an exceptional current density of 330 mA/cm2 and an FE of 85% for CO generation, surpassing other electrocatalysts [89]. The excellent performance can be attributed to WSe2 nanoflakes’ much lower charge transfer resistance and low work function. DFT calculations showed that the formation of CO is kinetically favorable.
Further efforts have been made to enhance intrinsic activity, suppress the competing HER reaction and improve catalyst stability. It has been reported that doping and alloying different metal atoms with TMDs could enhance electrocatalytic efficiency by tuning the binding strengths of reaction intermediates and the reaction energy barrier. For instance, in one study, 5% niobium (Nb)-doped vertically aligned MoS2 showed the largest CO2 reduction current density of ca. 237 mA/cm2 at the potential of −0.80 V, which was about two to three times higher than that of pristine and Ta-doped MoS2 (Figure 5a). Nb-doped MoS2 displayed an order of magnitude higher TOF for CO generation than pure MoS2 at an overpotential of 20–150 mV and two orders of magnitude higher TOF for CO formation than Ag nanoparticles at the potential range of 0–650 mV (Figure 5b). The Nb atom could facilitate rapid CO release from the TMD edge, which remarkably increased ECR efficiency [87]. Theoretical calculation elucidated that there is a linear scaling relationship between the adsorption energies of the crucial intermediates, including COOH*, *CHO and *CO. It means that the stabilities of these intermediates are closely related, which causes a persistent overpotential for ECR. Theoretically, to improve ECR activity, breaking the linear scaling relationship between the binding energies of intermediates would be a good solution. Norskøv et al. have revealed that if the intermediates bind to different active sites, the linear scaling relation can be broken at the edge site of MoS2 and MoSe2, which can increase ECR activity. They further showed that the doped S edge of MoS2 can meet this requirement with *CO bonding on the doped metal atoms and *COH, *CHO and *COOH binding on the S sites [130]. Therefore, the binding energies of these three intermediates are higher than that of CO*, suggesting the breaking of the linear scaling relationship.
Among four different TMD materials, MoS2, MoSe2, WS2 and WSe2, WSe2 exhibits good CO selectivity because of its lower work function and charge transfer resistance. Metal doping is also an effective way to improve ECR performance of TMD materials. More importantly, breaking the linear scaling relationship between the binding energies of intermediates will be a good strategy for designing excellent TMD catalysts.
3.4. MOF Materials
MOFs are a new type of porous material with metal ions or clusters coordinated with organic ligands. Due to their high porosity, large specific surface area and flexible structure, MOFs have been applied in electrocatalytic research recently [43,132]. However, some disadvantages, such as poor conductivity and less active centers, hamper their electrocatalytic activity. Studies have shown that low-dimensional MOFs can achieve high electrocatalytic performance because MOF nanosheets can expose more active atoms and exhibit excellent electron transfer. Moreover, the metal atoms, ligand and the connection of metal centers with ligands also play vital roles in the electrocatalytic activity of MOFs. Kornienko et al. synthesized a thin film cobalt–porphyrin MOF on a carbon substrate. The FE of this sample was 76% at −0.70 V vs. RHE for CO2 conversion to CO. In addition, it possessed a low Tafel slope of 165 mV/decade and the electrocatalytic stability was excellent [90]. A Zn MOF synthesized by ionic exchange of Zn nodes with adsorbed Ni salts achieved an FE of 71.9% for CO at 10.48 mA/cm2 under the potential of −0.89V vs. RHE [91]. Zhang et al. prepared a series of transition-metal-based MOF electrocatalysts by using phthalocyanine (Pc), which can convert CO2 to CO efficiently [92]. Under controlled potential electrolysis tests of different samples in CO2-saturated solution, the Co-based MOF exhibited the lowest onset potential and largest current density (Figure 6a). Remarkably, Co-based MOF also showed the largest CO current density in a wide potential range (Figure 6b). Moreover, Co-based MOF exhibited 99% FE of CO formation under the potential of −0.80 V, as shown in Figure 6c,d, demonstrating that Co-based MOF is the best catalyst for CO2 conversion to CO among others. DFT calculations suggested that the rate-limiting step for CO2 conversion to CO had the lowest Gibbs energy change (Figure 6e). As shown in the inset of Figure 6e, the adsorption energy of *COOH on N and C sites was 0.95 and 0.90 eV, which is much larger than that of −0.1 eV on the Co site, indicating that metal center is the most stable adsorption site. Moreover, two linear relationships disclosed a volcano curve (Figure 6f) in which the location of Co-based MOF is closest to the volcano peak. Therefore, it shows the best intrinsic activity for ECR into CO.
Recently, Perfecto-Irigaray et al. developed Cu-based MOF by doping metals (Zn, Pd and Ru) into HKUST-1 with different ratios [133]. They reported that the Ru-doped sample (10 wt.% Ru) showed an FE of 47.2% for methanol and ethanol generation, while other metal-doped samples performed worse compared to the pristine HKUST-1. Nevertheless, all doped samples exhibited increased ethanol selectivity. This is ascribed to the formation of C–C bonds on metal sites that boost the formation of C2 or C3 intermediates, which are finally converted into alcohol or other high-carbon products. Very recently, 2D copper tetrahydroxyquinone conductive MOF (Cu-THQ) was reported for ECR at a low overpotential. Cu-THQ nanoflakes with an average particle size of 140 nm showed the largest current density of about 173 mA/cm2 under −0.45 V vs. RHE, an average Faradaic efficiency FECO of about 91% and a remarkable TOF of 20.82 s−1. Spectroscopic characterization and DFT calculations uncovered the importance of reduced Cu+ during ECR, which was reversibly oxidized to Cu2+ after the reaction [134].
Overall, the metal atoms, ligand and the connection of metal center with ligand of MOF materials can determine electrocatalytic activity. Doping of a different metal to the original MOF materials, on the other hand, could either increase or decrease ECR activity.
3.5. Metal Oxide Nanosheets
Metal/metal oxide composites and oxide-derived metal have also been developed as ECR electrocatalysts with high performance [33,135,136]. Nevertheless, metal oxides still suffer problems such as instability and poor conductivity. It has been demonstrated that low-coordinated surface metal cations can be beneficial for CO2 adsorption during ECR. First principles calculation revealed that both the density of states (DOS) and the charge density around the conduction band edges can be greatly enhanced [137]. Therefore, faster carrier transport along the 2D ultrathin layer during ECR can be expected. One excellent example is the ultrathin Co3O4 layers, with thicknesses of 1.72 nm and 3.51 nm, synthesized by fast-heating treatment [93]. The ultrathin Co3O4 layer exhibited higher DOS at the conduction band edges than the bulk structure (Figure 7a,b). Moreover, the charge density of the ultrathin Co3O4 layer around the conduction band edge was larger and more delocalized than the bulk Co3O4 (Figure 7c,d). These changes can be beneficial for faster carrier transport, thus accelerating ECR. Co3O4 layers with 1.72 nm thickness displayed a maximum current density of 0.68 mA/cm2 and FE of 64.3% for formate production at −0.88 V vs. the saturated calomel electrode (SCE) (Figure 7e,f). Figure 7g indicates that the electrochemically active surface area (ECSA) increased with the decrease in thickness from bulk to 1.72 nm, which clearly evidences the close relationship between structure and activity. The intrinsic activity of Co3O4 with 1.72 nm thickness was over 1.5 times higher than the 3.51 nm thick Co3O4 layers and 20 times higher than the bulk structure (Figure 7h).
Moreover, the authors elucidated an atomic-level relationship between oxygen vacancies and ECR performance by constructing a model of oxygen vacancies confined in atomic Co3O4 single-unit cell layers [94]. DFT calculations uncovered that the main defect was oxygen (II), and X-ray absorption fine structure spectroscopy (XAFS) and X-ray photoelectron spectroscopy (XPS) indicated the distinct oxygen vacancy concentration. CO2 adsorption isotherms demonstrated that the presence of oxygen (II) vacancy can promote CO2 adsorption, while DFT calculations verified that oxygen (II) vacancy benefits spontaneous HCOO* desorption and prevents catalyst deactivation. More importantly, oxygen (II) vacancy facilitated the rate-limiting proton transfer step by stabilizing the HCOO* intermediate, which was confirmed by the drop of the activation energy barrier from 0.51 to 0.40 eV. This may also improve ECR kinetics because of the decreases in onset potential from 0.81 to 0.78 V vs. SCE and Tafel slope from 48 to 37mV dec−1. Thus, oxygen (II)-vacancy-rich Co3O4 nanosheets (0.84 nm) exhibited a current density of about 2.7 mA/cm2 with 85% formate selectivity for 40 h, which is promising for an ECR electrocatalyst. The researches further prepared a Co-based nanosheet with a thickness of only four atoms (about 0.84 nm) via solvothermal growth. The partial oxidation of the four-atom-thick Co layer can be formed on its surface by controlling the reaction time [28]. Interestingly, the four-atom-thick Co sheets exhibited great potential in ECR because the thinner 2D structures presented more surface-active sites and higher electrical conductivity. In LSV experiments, the partially oxidized four-atom-thick Co layers displayed a distinct CO2 reduction peak at −0.85 V vs. SCE with a current density of 10.59 mA/cm2. It was a performance enhancement by about 10, 40 and 260 times in comparison with Co four-atom-thick layer without oxidation, oxidized bulk Co and unoxidized bulk Co, respectively. More importantly, the partially oxidized four-atom-thick Co layers showed great stability with a current density of 10 mA/cm2 over 40 h and a high FE for formate generation of about 90% under a low overpotential of 0.24 V. Such a noticeable improvement can be partly attributed to the increase in ECSA. Another reason is that the partially oxidized four-atom-thick Co layer (atomic Co3O4) further increased their intrinsic activity and selectivity compared with pristine four-atom-thick Co layer.
Zhang’s group prepared mesoporous SnO2 nanosheets by a facile combination of hydrothermal reaction and calcination on carbon cloth [95]. As shown in Figure 8a,b, dense SnO2 nanosheets grew uniformly on carbon cloth, and these nanosheets also contained numerous pores. The study showed a high partial current density of about 50 mA/cm2 at −1.6 V vs. Ag/AgCl, with a high Faradaic efficiency of about 87% for formate generation, which outperformed most gas diffusion electrodes (Figure 8c,d). After electrolysis for 24 h, the current density and FE of formate were still stable (Figure 8e). Moreover, Figure 8f indicates that the current density also remained unchanged after folding or twisting the electrode 10 times. The excellent performance can be attributed to the following factors: Firstly, the SnO2 nanosheet could provide intrinsically active sites for ECR; secondly, the highly porous structure could increase the specific surface area, which facilitated charge and mass transfer during ECR; the third reason is that the robustness of the hierarchical structures ensured high stability of the electrocatalyst during long-term use.
Therefore, metal oxide nanosheets mainly produce formate during ECR. The low-coordinated surface metal cations can be active centers for ECR. Ultrathin metal oxides exhibit higher DOS at the conduction band compared to the bulk structure. The charge density is more delocalized, which benefits the carrier transport. In addition, building oxygen vacancy could also increase ECR performance by improving the reaction kinetics.
3.6. Two-Dimensional Materials Incorporated Single-Atom Catalysts
Single-atom catalysts (SACs) have enormous advantages in electrocatalysis in terms of 100% atom utilization and intriguing electronic structures [138,139]. The traditionally supported nanoparticles or clusters exhibit structural irregularities on the nanoparticle surface. The surface reactivity has a close relationship with surface free energy, and the ratio between fully coordinated surface atoms and the number of vacancies distinguishes the reactivity of atoms with identical chemical compositions but different positions [140,141,142]. Therefore, the conventional clusters with different surface reactivities can result in poor product selectivity. SACs are different from clusters because the surface free energy is homogeneous and the number of vacancies in the nearest neighbors is the same. Thus, SACs can show good stability and special intrinsic activity [143]. Single-atom catalysts exhibit uniform active sites as ideal catalysts; however, one major challenge for SACs is synthesis because SACs are unstable and can agglomerate quickly due to high surface energy. Furthermore, the supported structures in the neighboring environment increase the heterogeneity of SACs’ active sites, and thus uniform activity cannot be easily achieved. However, 2D materials can be utilized as supports to improve the uniformity and performance of SACs in electrocatalytic reactions [144,145]. Studies have evidenced that 2D materials, such as MoS2, graphene and MXene, could stabilize the SACs and maintain their single-atom state [96,146,147,148,149,150,151]. For instance, Back et al. investigated single transition metal atoms anchored on graphene with single or double vacancies as ECR catalysts by DFT calculations [152]. They suggested that many SACs exhibit high selectivity for ECR over the competitive HER because of favorable adsorption of *COOH or *OCHO over *H on the catalysts (Figure 9). SACs for CH3OH production, Ni and Pt, showed limiting potentials of −0.41 V and −0.27 V, respectively, while Os and Ru systems had the same limiting potential (−0.52 V) for CH4 production. The activity improvement of SACs can be partially attributed to the unique electronic structure of the SACs and orbital interaction.
Constructing a 3d transition metal-Nitrogen atom (M–Nx) system based on 2D materials for ECR is prevailing. Huan et al. investigated activity and selectivity of N-doped Fe–N4 and Fe nanoparticles’ moieties. They reported that samples with Fe–N4 active sites had an FE of 80% for CO generation, but Fe nanoparticles could mostly only produce H2 [97]. Zhang et al. synthesized Fe single atoms anchored on N-doped graphene and revealed that Fe–N4 with an Fe2+ oxidation state was the active site for ECR [98]. They also recognized that N dopants on the graphene substrate can be beneficial for ECR. However, there are some debates on the active sites and the corresponding ECR mechanisms on Fe SACs. For instance, Zhang et al. synthesized single Fe atoms anchored on N–doped graphene by prolonged thermal pyrolysis [99]. The SAC exhibited a high FE of about 97.0% for CO production under a low overpotential of 0.35 V. The excellent performance resulted from the presence of highly efficient dispersed Fe–N5 active sites. Theoretical calculations disclosed that a fifth N atom coordinated to Fe3+ to form Fe–N5 moieties, where the additional axial pyrrolic N ligand further depleted the electron density of Fe 3d orbitals and reduced the π back-donation of Fe–CO bonding, which resulted in rapid CO desorption and high selectivity for CO formation.
Zhang et al. reported on single Fe atoms confined in carbon foams forming Fe–N4 active sites [100]. However, they also detected an Fe3+ state in their samples because Fe2+ can be oxidized to Fe3+ when the samples are exposed to air, but the difference was not distinguishable in ECR. Gu et al. investigated single Fe atoms loaded on N-doped carbon with an overpotential as low as 0.08 V for CO production [101]. They confirmed the persistent presence of Fe3+ in the whole ECR process by in situ X-ray absorption near edge structure (XANES) measurements. Further studies indicated that Fe3+–N–C showed better performance for CO formation than that of Fe2+–N–C because of weaker CO absorption on the Fe3+ sites. Yang et al. prepared a N-anchored Zn single-atom catalyst on carbon [102]. From HAADF-STEM characterization, it can be clearly seen that an individual single Zn atom was supported on carbon (Figure 10a), and extended X-ray absorption fine-structure (EXAFS) spectra demonstrated the existence of a Zn-N4 moiety (Figure 10b). The catalyst could reduce CO2 to CO with high selectivity and a high FE of about 95% under the potential of −0.43 V (Figure 10c). It also exhibited remarkable durability over 75 h without any FECO decay (Figure 10d). Further experimental work and DFT calculations suggested that the remarkable activity can be attributed to the existence of Zn–N4, which was the main active site for ECR and had a low free energy barrier for the rate-limiting step of *COOH formation. Zhao et al. synthesized a single Ni atom loaded on graphene oxide. The catalyst showed an FE for CO production of 96.5% at a potential of −0.63V, with a TOF of 325.9/h [103].
Yang et al. prepared a single Ni atom catalyst supported on N-doped graphene. The TOF reached 14,800/h, and the FE of CO was 97% at a potential of −0.61 V [104]. The catalyst also displayed great stability with very stable electrocatalytic activity for 100 h. Liu et al. constructed a model SAC with precise structure using two-steps by linking Ni–TAPc to CNTs and by C–C coupling [105]. This model Ni SAC exhibited excellent activity, selectivity and durability in CO2 conversion to CO. Experimental study suggested that Ni+ in Ni–TAPc is highly active for CO2 activation and is the catalytically active site. In addition, the study suggested that * + H+ to *COOH is the rate-determining step. Gong et al. prepared a series of single-atom Ni catalysts (Ni–SA–Nx–C) with different N coordination numbers by controlling the pyrolysis temperature [106]. Figure 11a,b clearly indicates the existence of isolated Ni atoms from TEM characterizations. The homogeneous distribution of Ni, N and C is demonstrated by energy dispersive spectroscopy (EDS) mapping (Figure 11c). It was found that the sample with Ni–N2 moiety showed a higher current density than Ni–N3–C and Ni–N4 in CO2-saturated 0.5 M KHCO3, indicating the best activity of NiSA–N2–C among all three SACs (Figure 11d). In addition, it exhibited a maximum FECO of 98% at −0.8 V, which is also the highest (Figure 11e). The TOF for CO production on NiSA–N2–C reached 3467 h−1 at the potential of −1.0 V, which is much higher than those of NiSA–N4–C and NiSA-N3-C (Figure 11f), elucidating that the coordination environment plays a crucial role in ECR performance. The current density and FE for CO generation were almost unchanged on the NiSA–N2–C catalyst after electrolyzation for 10 h at the potential of −0.8 V vs. RHE (Figure 11g), demonstrating excellent stability. DFT calculations suggested that the reaction path is proton-coupled electron transfer (Figure 11h). The low N coordinated single-atom Ni sites in NiSA–N2–C are beneficial for the formation of *COOH intermediate, thereby enhancing ECR activity (Figure 11i).
Han et al. designed a free-standing ultrathin 2D SAC by self-assembly of a Co–porphyrin complex, which showed a CO production FE of 96% under an overpotential of 0.5 V [107]. They indicated that the improvement of activity was attributed to nitrogen coordination from the bottom layer, which increased the dz energy level of the Co atoms. Jiang et al. investigated a series of SACs supported on graphene nanosheets with different defect structures [153]. Ni SAC exhibited a CO formation FE of 95% under an overpotential of 0.55 V and excellent stability. Experimental study and theoretical calculations suggested that Ni sites with slightly larger vacancies benefit CO2 conversion to CO by decreasing CO desorption barrier. Recently, Zheng et al. investigated ECR performance of Cu SACs on a N-doped graphene matrix with a highly exposed and coordinatively unsaturated Cu–N2 center, as shown in Figure 12a [108]. Experimental work showed an onset potential of −0.33 V and FE for CO production of about 81% under a low potential of −0.50 V vs. RHE, obviously higher than the sample with Cu–N4 moiety (Figure 12b). DFT calculations manifested that Cu–N2 centers can facilitate CO2 activation and accelerate electron transfer from Cu–N2 sites to *CO2, which can greatly improve *COOH generation and the overall ECR activity. Jiao et al. investigated a single Cu atom catalyst anchored on g-C3N4 as an electrocatalyst for CO2 conversion to various hydrocarbons [154]. A slightly negative shift of photon energy demonstrated that N accepts extra charges from the Cu atom (Figure 12c). The authors reckoned that the d-orbital of Cu can be efficiently elevated by coordination with the g–C3N4 framework, enhancing the adsorption of carbonaceous intermediates. Therefore, Cu–g–C3N4 had better ECR activity with lower onset potential and exhibited a higher C2 product rate than conventional Cu supported on nitrogen-doped graphene (Figure 12d).
Single-metal-atom-loaded 2D materials are a class of very promising catalysts for ECR due to their high atom utilization and catalytic activity. Their activity is determined by the type of metal atom and the electronic structures of the metal atom. Furthermore, the coordination number of metal atoms can be tuned by the 2D materials’ support, which can further optimize the activity and selectivity of the SACs.
4. The Design of Electrolyzers
Numerous ECR studies are based on H-cells because they can be used conveniently to evaluate the activity of catalysts due to their easy operation and adaptability to different electrode materials and configurations [155,156]. A typical H-cell electrolyzer contains two chambers that are separated by an ion exchange membrane. The counter electrode is in the anodic chamber, whereas the working electrode and reference electrode are in the cathodic chamber. During the ECR process, CO2 is first dissolved in the liquid catholyte, such as KHCO3 and NaHCO3, and then transferred to the surface of the working electrode where ECR occurs. However, the thick diffusion layer and low solubility of CO2 in water limit CO2 mass transport to the catalyst surface, impeding its practical applications [157].
Commercial ECR application requires continuous operation to realize high production efficiency. A microfluidic reactor for ECR has been designed where the electrolyte can continuously flow through the electrolyzer [158,159]. The cathode and anode are separated by a very thin channel that is filled with flowing electrolytes. CO2 is supplied from the back of the cathodic gas diffusion electrode (GDE), and ECR occurs when CO2 reaches the surface of catalysts. The operation parameters, such as the composition and pH of the electrolyte, can be adjusted, which promotes ECR rates. GDEs are high-surface-area and porous electrodes. A typical GDE consists of a catalytic layer, a gas diffusion layer and a gas flow field. The structure and composition of GDE layers play a vital role in the transport of reactants and products because the transport processes influence the accumulation or depletion of intermediates on the catalysts, which determine ECR performance [160,161]. Lim et al. prepared Sn electrocatalysts with dense tips (SnDT) GDE, showing formate productivity of 65 mg h−1. It was stable for 72 h without changes in FE or current density [162]. A 2D nanosheet Cu-loaded GDE achieved the highest partial current density of 131 mA cm−2 for acetate production [163]. Although high current density was realized, the microfluidic reactor still suffered product crossover, where the reduction of oxygen diffused to the cathode and the reoxidation of cathodic products diffused to the anode, decreasing the overall energy efficiency and productivity.
The membrane electrode assembly (MEA) electrolyzers have been exploited to solve problems of the microfluidic reactors. The catalytic layer of cathodic GDEs is in direct contact with the polymer electrolyte membranes. MEAs can decrease ohmic losses and prevent the poison of cathode catalysts by impurities through removing catholyte between the membrane and GDE. As a result, MEAs can exhibit excellent stability and high energy efficiency [164]. Gas and liquid products flow out of the cathode flow plate with little diffusion into catholyte through the membrane, resulting in high concentration liquid product. Cobalt phthalocyanine (Co-Pc) has been widely studied for ECR to CO in both immobilized and homogeneous forms and was also used in an MEA system with FE of CO >90% over 8 h at 50 mA cm−2 [165]. Gabardo et al. designed an MEA electrolyzer that achieved an FE of ~80% for higher carbon products such as ethylene, ethanol, acetate and 1-propanol with current densities >100 mA cm−2 for 100 h [164].
5. Conclusions and Perspectives
Electrochemical reduction reactions are a promising strategy to convert CO2 into valuable fuels and chemical products, which can offer potential technological solutions for energy problems and greenhouse effects. An effective electrocatalyst is the key to the potential deployment of commercial ECR processes. In this contribution, we review a series of 2D materials, including metal, graphene-based materials, TMDs, MOFs materials, metal oxide nanosheets and 2D materials incorporated with single atoms as ECR catalysts. Compared to the conventional metal-based electrocatalysts, 2D materials show unique physicochemical properties and electronic structures benefiting ECR activity.
Nevertheless, there are still challenges in the design and synthesis of 2D electrocatalysts for ECR. To date, most ECR catalysts can only produce C1 products, especially CO, while producing more valuable products, such as C2 and C3 products, is desired. In general, the intrinsic activity can be modified in various ways such as doping, constructing special structures and morphologies, etc. Meanwhile, the choices of different classes of 2D materials are important for producing specific products. Strategies, such as constructing composite electrocatalyst materials and surface engineering of electrocatalysts, could be employed. Composite electrocatalyst materials could have different properties and catalytic performance in comparison with individual components due to synergistic effects. They could possess excellent properties for ECR such as better conductivity, high specific surface area, more active sites, better transport property and high durability. Another strategy is surface engineering of electrocatalysts by synthesizing nanotubes, nanosheets, nanospheres, isolated single atoms and defects. These properties are beneficial for effective contact for electrolyte ions and adsorption and desorption of intermediates. It is worth noting that single metal atoms anchored on the surface of 2D materials as SACs have rather high efficiency in ECR. However, the synthesis of SAC loaded on 2D materials is still a challenge, and key aspects, including suitable mononuclear metal complex precursors and stabilization of single metal atoms on the 2D materials, should be considered. Future research opportunities could be to develop new 2D materials such as MXenes and COFs.
Combining theoretical calculation and experimental work is necessary to obtain a fundamental understanding on the design and synthesis of 2D electrocatalysts. Although theoretical calculations are conducted with simplified models under idealized conditions, they could always be beneficial for revealing the reaction mechanism and providing guidance for design principles. For instance, breaking the scaling relationships between adsorption energies of different intermediates is of great significance to improve the activity of catalysts, which can be revealed from theoretical calculations. Similarly, to mitigate catalyst deactivation in ECR, it is necessary to understand the deactivation mechanisms. Theoretical models could be constructed to predict the intermediates which affect the stability of electrocatalysts. Finally, rapid development of characterization techniques, especially in situ spectroscopy, can greatly promote the development of catalysts of ECR. In situ spectroscopy can investigate catalytic reactions under reaction conditions and provide insights into the true active sites and reaction mechanisms. Through in situ techniques, such as infrared spectroscopy, Raman spectroscopy, X-ray absorption spectroscopy, X-ray photoelectron spectroscopy, mass spectrometry, etc., crucial issues in ECR, including identifying reaction intermediates, active sites and reaction pathways, can be resolved.
H-type cells are widely used in ECR. The current density of an H-type cell is limited to tens of milliamps, which is far from industrial application due to limited CO2 solubility and energy loss. The design of microfluidic flow cells has made great progress recently. For example, the GDE and MEA electrolyzers can effectively tune reaction conditions and local environments in ECR. GDEs utilize a porous catalyst layer along with diffusion media, which can promote reactant transport. Thus, high current density can be realized, and transport losses can be effectively avoided. In the flow cell, higher carbon products are favored at high pH values, which can be hardly realized in an H-type cell. Flow cells and MEA make the separation of products convenient. These advantages could effectively accelerate the large-scale application of ECR at the industrial level.
It has been demonstrated that 2D nanomaterials possess great potential for ECR. Nevertheless, most of the 2D materials discussed in this review are still far from practical application. It is still demanding to synthesize 2D electrocatalysts with excellent performance at a large scale. Therefore, it is important to narrow the gap between laboratory- and commercial-scale production. Currently, some techniques, such as strip and salt–temperature, have been applied in the large-scale synthesis of 2D materials, but scalable routes to prepare ultrathin nanosheets remain to be developed.
Author Contributions
Conceptualization, S.L., F.L. and Z.Y.; investigation, S.L. and Z.Y.; writing—original draft preparation, S.L. and Z.Y.; writing—review and editing, S.L., F.L. and Z.Y.; funding acquisition, Z.Y.; supervision, F.L. and Z.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the Norwegian Ministry of Education and Research.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Caldeira, K.; Wickett, M.E. Anthropogenic carbon and ocean pH. Nature 2003, 425, 365. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Sun, D.; Chen, Y.; Huang, R.; Ding, Z.; Fu, X.; Li, Z. An amine-functionalized titanium metal-organic framework photocatalyst with visible-light-induced activity for CO2 reduction. Angew. Chem. Int. Ed. 2012, 51, 3364–3367. [Google Scholar] [CrossRef]
- Li, J.R.; Ma, Y.; McCarthy, M.C.; Sculley, J.; Yu, J.; Jeong, H.K.; Balbuena, P.B.; Zhou, H.C. Carbon dioxide capture-related gas adsorption and separation in metal-organic frameworks. Coord. Chem. Rev. 2011, 255, 1791–1823. [Google Scholar] [CrossRef]
- Li, X.; Yu, J.; Jaroniec, M.; Chen, X. Cocatalysts for Selective Photoreduction of CO2 into Solar Fuels. Chem. Rev. 2019, 119, 3962–4179. [Google Scholar] [CrossRef] [PubMed]
- Rubin, E.S.; Davison, J.E.; Herzog, H.J. The cost of CO2 capture and storage. Int. J. Greenh. Gas Control 2015, 40, 378–400. [Google Scholar] [CrossRef]
- Keith, D.W.; Holmes, G.; St Angelo, D.; Heidel, K. A Process for Capturing CO2 from the Atmosphere. Joule 2018, 2, 1573–1594. [Google Scholar] [CrossRef] [Green Version]
- Drage, T.C.; Snape, C.E.; Stevens, L.A.; Wood, J.; Wang, J.; Cooper, A.I.; Dawson, R.; Guo, X.; Satterley, C.; Irons, R. Materials challenges for the development of solid sorbents for post-combustion carbon capture. J. Mater. Chem. 2012, 22, 2815–2823. [Google Scholar] [CrossRef] [Green Version]
- Orr, J.F.M. CO2 capture and storage: Are we ready? Energy Environ. Sci. 2009, 2, 449–458. [Google Scholar] [CrossRef]
- Maginn, E.J. What to Do with CO2. J. Phys. Chem. Lett. 2010, 1, 3478–3479. [Google Scholar] [CrossRef]
- Ramdin, M.; de Loos, T.W.; Vlugt, T.J.H. State-of-the-Art of CO2 Capture with Ionic Liquids. Ind. Eng. Chem. Res. 2012, 51, 8149–8177. [Google Scholar] [CrossRef]
- Wang, J.Y.; Huang, L.; Yang, R.Y.; Wu, J.W.; Gao, Y.S.; Wang, Q.; O’Hare, D.; Zhong, Z.Y. Recent advances in solid sorbents for CO2 capture and new development trends. Energy Environ. Sci. 2014, 7, 3478–3518. [Google Scholar] [CrossRef]
- Yu, C.H.; Huang, C.H.; Tan, C.S. A Review of CO2 Capture by Absorption and Adsorption. Aerosol Air Qual. Res. 2012, 12, 745–769. [Google Scholar] [CrossRef] [Green Version]
- Inoue, T.; Fujishima, A.; Konishi, S.; Honda, K. Photoelectrocatalytic reduction of carbon dioxide in aqueous suspensions of semiconductor powders. Nature 1979, 277, 637–638. [Google Scholar] [CrossRef]
- Halmann, M. Photoelectrochemical reduction of aqueous carbon dioxide on p-type gallium phosphide in liquid junction solar cells. Nature 1978, 275, 115–116. [Google Scholar] [CrossRef]
- Li, W. Electrocatalytic Reduction of CO2 to Small Organic Molecule Fuels on Metal Catalysts. In Advances in CO2 Conversion and Utilization; ACS Symposium Series; ACS Publisher: Washington, DC, USA, 2010; Volume 1056, pp. 55–76. [Google Scholar] [CrossRef]
- Kortlever, R.; Peters, I.; Koper, S.; Koper, M.T.M. Electrochemical CO2 Reduction to Formic Acid at Low Overpotential and with High Faradaic Efficiency on Carbon-Supported Bimetallic Pd–Pt Nanoparticles. ACS Catal. 2015, 5, 3916–3923. [Google Scholar] [CrossRef]
- Zafar, Q.; Mattisson, T.; Gevert, B. Integrated Hydrogen and Power Production with CO2 Capture Using Chemical-Looping ReformingsRedox Reactivity of Particles of CuO, Mn2O3, NiO, and Fe2O3 Using SiO2 as a Support. Ind. Eng. Chem. Res. 2005, 44, 3485–3496. [Google Scholar] [CrossRef]
- Chen, X.; Wang, J.; Huang, C.; Zhang, S.; Zhang, H.; Li, Z.; Zou, Z. Barium zirconate: A new photocatalyst for converting CO2 into hydrocarbons under UV irradiation. Catal. Sci. Technol. 2015, 5, 1758–1763. [Google Scholar] [CrossRef]
- Ding, J.; Bu, Y.; Ou, M.; Yu, Y.; Zhong, Q.; Fan, M. Facile decoration of carbon fibers with Ag nanoparticles for adsorption and photocatalytic reduction of CO2. Appl. Catal. B Environ. 2017, 202, 314–325. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S.; Ma, X.; Gong, J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 3703–3727. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.F.; Jiang, Y.J.; Jiang, Z.Y.; Wang, X.Y.; Wang, X.L.; Zhang, S.H.; Han, P.P.; Yang, C. Enzymatic conversion of carbon dioxide. Chem. Soc. Rev. 2015, 44, 5981–6000. [Google Scholar] [CrossRef]
- Gong, Q.; Ding, P.; Xu, M.; Zhu, X.; Wang, M.; Deng, J.; Ma, Q.; Han, N.; Zhu, Y.; Lu, J.; et al. Structural defects on converted bismuth oxide nanotubes enable highly active electrocatalysis of carbon dioxide reduction. Nat. Commun. 2019, 10, 2807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.T.; Shan, B.; Xu, W.; Meyer, T.J. Electrocatalytic CO2 Reduction with a Ruthenium Catalyst in Solution and on Nanocrystalline TiO2. ChemSusChem 2019, 12, 2402–2408. [Google Scholar] [CrossRef] [PubMed]
- Grodkowski, J.; Neta, P. Copper-Catalyzed Radiolytic Reduction of CO2 to CO in Aqueous Solutions. J. Phys. Chem. B 2001, 105, 4967–4972. [Google Scholar] [CrossRef]
- Agarwal, A.S.; Zhai, Y.; Hill, D.; Sridhar, N. The electrochemical reduction of carbon dioxide to formate/formic acid: Engineering and economic feasibility. ChemSusChem 2011, 4, 1301–1310. [Google Scholar] [CrossRef]
- Pritchard, J.; Filonenko, G.A.; van Putten, R.; Hensen, E.J.; Pidko, E.A. Heterogeneous and homogeneous catalysis for the hydrogenation of carboxylic acid derivatives: History, advances and future directions. Chem. Soc. Rev. 2015, 44, 3808–3833. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Sun, X.; Yang, D.; Ma, J.; Kang, X.; Zheng, L.; Zhang, J.; Wu, Z.; Han, B. Carbon dioxide electroreduction to C2 products over copper-cuprous oxide derived from electrosynthesized copper complex. Nat. Commun. 2019, 10, 3851. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; Lin, Y.; Jiao, X.; Sun, Y.; Luo, Q.; Zhang, W.; Li, D.; Yang, J.; Xie, Y. Partially oxidized atomic cobalt layers for carbon dioxide electroreduction to liquid fuel. Nature 2016, 529, 68–71. [Google Scholar] [CrossRef]
- Kaneco, S.; Katsumata, H.; Suzuki, T.; Ohta, K. Electrochemical reduction of carbon dioxide to ethylene at a copper electrode in methanol using potassium hydroxide and rubidium hydroxide supporting electrolytes. Electrochim. Acta 2006, 51, 3316–3321. [Google Scholar] [CrossRef]
- Lu, Q.; Rosen, J.; Zhou, Y.; Hutchings, G.S.; Kimmel, Y.C.; Chen, J.G.; Jiao, F. A selective and efficient electrocatalyst for carbon dioxide reduction. Nat. Commun. 2014, 5, 3242. [Google Scholar] [CrossRef]
- Durand, W.J.; Peterson, A.A.; Studt, F.; Abild-Pedersen, F.; Nørskov, J.K. Structure effects on the energetics of the electrochemical reduction of CO2 by copper surfaces. Surf. Sci. 2011, 605, 1354–1359. [Google Scholar] [CrossRef] [Green Version]
- Siahrostami, S.; Jiang, K.; Karamad, M.; Chan, K.; Wang, H.; Nørskov, J. Theoretical Investigations into Defected Graphene for Electrochemical Reduction of CO2. ACS Sustain. Chem. Eng. 2017, 5, 11080–11085. [Google Scholar] [CrossRef]
- Chen, Y.; Li, C.W.; Kanan, M.W. Aqueous CO2 reduction at very low overpotential on oxide-derived Au nanoparticles. J. Am. Chem. Soc. 2012, 134, 19969–19972. [Google Scholar] [CrossRef]
- Sui, S.; Wang, X.; Zhou, X.; Su, Y.; Riffat, S.; Liu, C.j. A comprehensive review of Pt electrocatalysts for the oxygen reduction reaction: Nanostructure, activity, mechanism and carbon support in PEM fuel cells. J. Mater. Chem. A 2017, 5, 1808–1825. [Google Scholar] [CrossRef]
- Gattrell, M.; Gupta, N.; Co, A. A review of the aqueous electrochemical reduction of CO2 to hydrocarbons at copper. J. Electroanal. Chem. 2006, 594, 1–19. [Google Scholar] [CrossRef]
- Chernikov, A.; van der Zande, A.M.; Hill, H.M.; Rigosi, A.F.; Velauthapillai, A.; Hone, J.; Heinz, T.F. Electrical Tuning of Exciton Binding Energies in Monolayer WS2. Phys. Rev. Lett. 2015, 115, 126802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Yuan, H.; Sae Hong, S.; Li, Y.; Cui, Y. Physical and chemical tuning of two-dimensional transition metal dichalcogenides. Chem. Soc. Rev. 2015, 44, 2664–2680. [Google Scholar] [CrossRef]
- Wang, H.; Chen, Y.; Hou, X.; Ma, C.; Tan, T. Nitrogen-doped graphenes as efficient electrocatalysts for the selective reduction of carbon dioxide to formate in aqueous solution. Green Chem. 2016, 18, 3250–3256. [Google Scholar] [CrossRef]
- Ao, C.; Feng, B.; Qian, S.; Wang, L.; Zhao, W.; Zhai, Y.; Zhang, L. Theoretical study of transition metals supported on g-C3N4 as electrochemical catalysts for CO2 reduction to CH3OH and CH4. J. CO2 Util. 2020, 36, 116–123. [Google Scholar] [CrossRef]
- Zhi, X.; Jiao, Y.; Zheng, Y.; Qiao, S.Z. Impact of Interfacial Electron Transfer on Electrochemical CO2 Reduction on Graphitic Carbon Nitride/Doped Graphene. Small 2019, 15, e1804224. [Google Scholar] [CrossRef]
- Kong, D.; Cha, J.J.; Wang, H.; Lee, H.R.; Cui, Y. First-row transition metal dichalcogenide catalysts for hydrogen evolution reaction. Energy Environ. Sci. 2013, 6, 3553–3558. [Google Scholar] [CrossRef]
- Chhowalla, M.; Shin, H.S.; Eda, G.; Li, L.J.; Loh, K.P.; Zhang, H. The chemistry of two-dimensional layered transition metal dichalcogenide nanosheets. Nat. Chem. 2013, 5, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Lin, H.; Zhu, J. Recent Advances in Metal-Organic Frameworks for Photo-/Electrocatalytic CO2 Reduction. Chem. Eur. J. 2019, 25, 14026–14035. [Google Scholar] [CrossRef] [PubMed]
- Olabi, A.G.; Abdelkareem, M.A.; Wilberforce, T.; Sayed, E.T. Application of Graphene in Energy Storage Device—A Review. Renew. Sustain. Energy Rev. 2021, 135, 110026. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, T.; Chen, X.; Zhang, L.; Wang, Y.; Niu, Y.; Yu, J.; Ma, H.; Li, X.; Liu, F.; et al. Demonstration of epitaxial growth of strain-relaxed GaN films on graphene/SiC substrates for long wavelength light-emitting diodes. Light Sci. Appl. 2021, 117. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, R.; Yamazaki, S.; Watanabe, S.; Tsuboi, N. Layer dependency of graphene layers in perovskite/graphene solar cells. Carbon 2021, 172, 597–601. [Google Scholar] [CrossRef]
- Nag, A.; Mitra, A.; Mukhopadhyay, S.C. Graphene and its sensor-based applications: A review. Sens. Actuators A Phys. 2018, 270, 177–194. [Google Scholar] [CrossRef]
- Nemiwal, M.; Zhang, T.C.; Kumar, D. Graphene-based electrocatalysts: Hydrogen evolution reactions and overall water splitting. Int. J. Hydrogen Energy 2021, 46, 21401–21418. [Google Scholar] [CrossRef]
- Wang, G.; Chen, J.; Ding, Y.; Cai, P.; Yi, L.; Li, Y.; Tu, C.; Hou, Y.; Wen, Z.; Dai, L. Electrocatalysis for CO2 conversion: From fundamentals to value-added products. Chem. Soc. Rev. 2021, 50, 4993–5061. [Google Scholar] [CrossRef]
- Nitopi, S.; Bertheussen, E.; Scott, S.B.; Liu, X.; Engstfeld, A.K.; Horch, S.; Seger, B.; Stephens, I.E.L.; Chan, K.; Hahn, C.; et al. Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte. Chem. Rev. 2019, 119, 7610–7672. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Seifitokaldani, A. In Situ Spectroscopic Methods for Electrocatalytic CO2 Reduction. Catalysts 2020, 10, 481. [Google Scholar] [CrossRef]
- Zhu, Y.; Yang, X.; Peng, C.; Priest, C.; Mei, Y.; Wu, G. Carbon-Supported Single Metal Site Catalysts for Electrochemical CO2 Reduction to CO and Beyond. Small 2021, 17, e2005148. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Li, Y. Review of two-dimensional materials for electrochemical CO2 reduction from a theoretical perspective. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2019, 9, e1416. [Google Scholar] [CrossRef]
- Zhang, W.; Ma, D.; Pérez-Ramírez, J.; Chen, Z. Recent Progress in Materials Materials Exploration for Thermocatalytic, Photocatalytic, and Integrated Photothermocatalytic CO2 to Fuel Conversion. Adv. Sustain. Syst. 2021, 3, 2100169. [Google Scholar] [CrossRef]
- Zhang, W.; Jin, Z.; Chen, Z. Rational-Designed Principles for Electrochemical and Photoelectrochemical Upgrading of CO2 to Value-Added Chemicals. Adv. Sci. 2022, e2105204. [Google Scholar] [CrossRef]
- Li, Q.; Wang, Y.C.; Zeng, J.; Zhao, X.; Chen, C.; Wu, Q.M.; Chen, L.M.; Chen, Z.Y.; Lei, Y.P. Bimetallic chalcogenides for electrocatalytic CO2 reduction. Rare Met. 2021, 40, 3442–3453. [Google Scholar] [CrossRef]
- Wang, J.J.; Li, X.P.; Cui, B.F.; Zhang, Z.; Hu, X.F.; Ding, J.; Deng, Y.D.; Han, X.P.; Hu, W.B. A review of non-noble metal-based electrocatalysts for CO2 electroreduction. Rare Met. 2021, 40, 3019–3037. [Google Scholar] [CrossRef]
- Sun, Z.; Ma, T.; Tao, H.; Fan, Q.; Han, B. Fundamentals and Challenges of Electrochemical CO2 Reduction Using Two-Dimensional Materials. Chem 2017, 3, 560–587. [Google Scholar] [CrossRef] [Green Version]
- Zou, X.; Liu, M.; Wu, J.; Ajayan, P.M.; Li, J.; Liu, B.; Yakobson, B.I. How Nitrogen-Doped Graphene Quantum Dots Catalyze Electroreduction of CO2 to Hydrocarbons and Oxygenates. ACS Catal. 2017, 7, 6245–6250. [Google Scholar] [CrossRef]
- García Rey, N.; Dlott, D.D. Structural Transition in an Ionic Liquid Controls CO2 Electrochemical Reduction. J. Phys. Chem. C 2015, 119, 20892–20899. [Google Scholar] [CrossRef]
- Ma, T.; Fan, Q.; Li, X.; Qiu, J.; Wu, T.; Sun, Z. Graphene-based materials for electrochemical CO2 reduction. J. CO2 Util. 2019, 30, 168–182. [Google Scholar] [CrossRef]
- Benson, E.E.; Kubiak, C.P.; Sathrum, A.J.; Smieja, J.M. Electrocatalytic and homogeneous approaches to conversion of CO2 to liquid fuels. Chem. Soc. Rev. 2009, 38, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Liu, Y.; Hong, F.; Zhang, J. A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem. Soc. Rev. 2014, 43, 631–675. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Talreja, N.; Tao, H.; Texter, J.; Muhler, M.; Strunk, J.; Chen, J. Catalysis of Carbon Dioxide Photoreduction on Nanosheets: Fundamentals and Challenges. Angew. Chem. Int. Ed. 2018, 57, 7610–7627. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, Z.J.; Gong, J. Nanostructured Materials for Heterogeneous Electrocatalytic CO2 Reduction and their Related Reaction Mechanisms. Angew. Chem. Int. Ed. 2017, 56, 11326–11353. [Google Scholar] [CrossRef] [PubMed]
- DeCiccio, D.; Ahn, S.T.; Sen, S.; Schunk, F.; Palmore, G.T.R.; Rose-Petruck, C. Electrochemical reduction of CO2 with clathrate hydrate electrolytes and copper foam electrodes. Electrochem. Commun. 2015, 52, 13–16. [Google Scholar] [CrossRef]
- Appel, A.M.; Bercaw, J.E.; Bocarsly, A.B.; Dobbek, H.; DuBois, D.L.; Dupuis, M.; Ferry, J.G.; Fujita, E.; Hille, R.; Kenis, P.J.; et al. Frontiers, opportunities, and challenges in biochemical and chemical catalysis of CO2 fixation. Chem. Rev. 2013, 113, 6621–6658. [Google Scholar] [CrossRef] [Green Version]
- Zhong, M.; Tran, K.; Min, Y.; Wang, C.; Wang, Z.; Dinh, C.T.; De Luna, P.; Yu, Z.; Rasouli, A.S.; Brodersen, P.; et al. Accelerated discovery of CO2 electrocatalysts using active machine learning. Nature 2020, 581, 178–183. [Google Scholar] [CrossRef]
- Geioushy, R.A.; Khaled, M.M.; Alhooshani, K.; Hakeem, A.S.; Rinaldi, A. Graphene/ZnO/Cu2O electrocatalyst for selective conversion of CO2 into n-propanol. Electrochim. Acta 2017, 245, 456–462. [Google Scholar] [CrossRef]
- Hori, Y. Electrochemical CO2 Reduction on Metal Electrodes. Mod. Aspect. Electroc. 2008, 42, 89–189. [Google Scholar] [CrossRef]
- Li, F.; Zhao, S.F.; Chen, L.; Khan, A.; MacFarlane, D.R.; Zhang, J. Polyethylenimine promoted electrocatalytic reduction of CO2 to CO in aqueous medium by graphene-supported amorphous molybdenum sulphide. Energy Environ. Sci. 2016, 9, 216–223. [Google Scholar] [CrossRef]
- Zhu, W.; Zhang, Y.J.; Zhang, H.; Lv, H.; Li, Q.; Michalsky, R.; Peterson, A.A.; Sun, S. Active and selective conversion of CO2 to CO on ultrathin Au nanowires. J. Am. Chem. Soc. 2014, 136, 16132–16135. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Zhang, L.; Yang, P.; Hu, C.; Luo, Z.; Chang, X.; Zhao, Z.J.; Gong, J. Low-Coordinated Edge Sites on Ultrathin Palladium Nanosheets Boost Carbon Dioxide Electroreduction Performance. Angew. Chem. Int. Ed. 2018, 57, 11544–11548. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cao, L.; Libretto, N.J.; Li, X.; Li, C.; Wan, Y.; He, C.; Lee, J.; Gregg, J.; Zong, H.; et al. Ensemble Effect in Bimetallic Electrocatalysts for CO2 Reduction. J. Am. Chem. Soc. 2019, 141, 16635–16642. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, M.; Sharma, P.P.; Yadav, R.M.; Ma, L.; Yang, Y.; Zou, X.; Zhou, X.D.; Vajtai, R.; Yakobson, B.I.; et al. Incorporation of Nitrogen Defects for Efficient Reduction of CO2 via Two-Electron Pathway on Three-Dimensional Graphene Foam. Nano Lett. 2016, 16, 466–470. [Google Scholar] [CrossRef]
- Zhang, W.; Hu, Y.; Ma, L.; Zhu, G.; Zhao, P.; Xue, X.; Chen, R.; Yang, S.; Ma, J.; Liu, J.; et al. Liquid-phase exfoliated ultrathin Bi nanosheets: Uncovering the origins of enhanced electrocatalytic CO2 reduction on two-dimensional metal nanostructure. Nano Energy 2018, 53, 808–816. [Google Scholar] [CrossRef]
- Pan, J.; Sun, Y.; Deng, P.; Yang, F.; Chen, S.; Zhou, Q.; Park, H.S.; Liu, H.; Yu Xia, B. Hierarchical and ultrathin copper nanosheets synthesized via galvanic replacement for selective electrocatalytic carbon dioxide conversion to carbon monoxide. Appl. Catal. B Environ. 2019, 255, 117736. [Google Scholar] [CrossRef]
- Wang, Y.; Shen, H.; Livi, K.J.T.; Raciti, D.; Zong, H.; Gregg, J.; Onadeko, M.; Wan, Y.; Watson, A.; Wang, C. Copper Nanocubes for CO2 Reduction in Gas Diffusion Electrodes. Nano Lett. 2019, 19, 8461–8468. [Google Scholar] [CrossRef]
- Sreekanth, N.; Nazrulla, M.A.; Vineesh, T.V.; Sailaja, K.; Phani, K.L. Metal-free boron-doped graphene for selective electroreduction of carbon dioxide to formic acid/formate. Chem. Commun. 2015, 51, 16061. [Google Scholar] [CrossRef]
- Liu, X.; Zhu, L.; Wang, H.; He, G.; Bian, Z. Catalysis performance comparison for electrochemical reduction of CO2 on Pd–Cu/graphene catalyst. RSC Adv. 2016, 6, 38380–38387. [Google Scholar] [CrossRef]
- Su, P.; Iwase, K.; Nakanishi, S.; Hashimoto, K.; Kamiya, K. Nickel-Nitrogen-Modified Graphene: An Efficient Electrocatalyst for the Reduction of Carbon Dioxide to Carbon Monoxide. Small 2016, 12, 6083–6089. [Google Scholar] [CrossRef]
- Huang, J.; Guo, X.; Wei, Y.; Hu, Q.; Yu, X.; Wang, L. A renewable, flexible and robust single layer nitrogen-doped graphene coating Sn foil for boosting formate production from electrocatalytic CO2 reduction. J. CO2 Util. 2019, 33, 166–170. [Google Scholar] [CrossRef]
- Lei, F.; Liu, W.; Sun, Y.; Xu, J.; Liu, K.; Liang, L.; Yao, T.; Pan, B.; Wei, S.; Xie, Y. Metallic tin quantum sheets confined in graphene toward high-efficiency carbon dioxide electroreduction. Nat. Commun. 2016, 7, 12697. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Chen, L.; Xue, M.; Williams, T.; Zhang, Y.; MacFarlane, D.R.; Zhang, J. Towards a better Sn: Efficient electrocatalytic reduction of CO2 to formate by Sn/SnS2 derived from SnS2 nanosheets. Nano Energy 2017, 31, 270–277. [Google Scholar] [CrossRef]
- Lu, X.; Tan, T.H.; Ng, Y.H.; Amal, R. Highly Selective and Stable Reduction of CO2 to CO by a Graphitic Carbon Nitride/Carbon Nanotube Composite Electrocatalyst. Eur. J. Chem. 2016, 22, 11991–11996. [Google Scholar] [CrossRef]
- Guo, S.; Zhao, S.; Wu, X.; Li, H.; Zhou, Y.; Zhu, C.; Yang, N.; Jiang, X.; Gao, J.; Bai, L.; et al. A Co3O4-CDots-C3N4 three component electrocatalyst design concept for efficient and tunable CO2 reduction to syngas. Nat. Commun. 2017, 8, 1828. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, P.; Asadi, M.; Liu, C.; Sharifi-Asl, S.; Sayahpour, B.; Behranginia, A.; Zapol, P.; Shahbazian-Yassar, R.; Curtiss, L.A.; Salehi-Khojin, A. Tailoring the Edge Structure of Molybdenum Disulfide toward Electrocatalytic Reduction of Carbon Dioxide. ACS Nano 2017, 11, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Asadi, M.; Kumar, B.; Behranginia, A.; Rosen, B.A.; Baskin, A.; Repnin, N.; Pisasale, D.; Phillips, P.; Zhu, W.; Haasch, R.; et al. Robust carbon dioxide reduction on molybdenum disulphide edges. Nat. Commun. 2014, 5, 4470. [Google Scholar] [CrossRef] [Green Version]
- Asadi, M.; Kim, K.; Liu, C.; Addepalli, A.V.; Abbasi, P.; Yasaei, P.; Phillips, P.; Behranginia, A.; Cerrato, J.M.; Haasch, R.; et al. Nanostructured transition metal dichalcogenide electrocatalysts for CO2 reduction in ionic liquid. Science 2016, 353, 467–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornienko, N.; Zhao, Y.; Kley, C.S.; Zhu, C.; Kim, D.; Lin, S.; Chang, C.J.; Yaghi, O.M.; Yang, P. Metal-organic frameworks for electrocatalytic reduction of carbon dioxide. J. Am. Chem. Soc. 2015, 137, 14129–14135. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Dai, X.; Yao, T.; Chen, W.; Wang, X.; Wang, J.; Yang, J.; Wei, S.; Wu, Y.; Li, Y. Ionic Exchange of Metal-Organic Frameworks to Access Single Nickel Sites for Efficient Electroreduction of CO2. J. Am. Chem. Soc. 2017, 139, 8078–8081. [Google Scholar] [CrossRef]
- Zhang, Z.; Xiao, J.; Chen, X.J.; Yu, S.; Yu, L.; Si, R.; Wang, Y.; Wang, S.; Meng, X.; Wang, Y.; et al. Reaction Mechanisms of Well-Defined Metal-N4 Sites in Electrocatalytic CO2 Reduction. Angew. Chem. Int. Ed. 2018, 57, 16339–16342. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Jiao, X.; Sun, Z.; Zhang, W.; Sun, Y.; Wang, C.; Hu, Q.; Zu, X.; Yang, F.; Yang, S.; et al. Ultrathin Co3O4 Layers Realizing Optimized CO2 Electroreduction to Formate. Angew. Chem. Int. Ed. 2016, 55, 698–702. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Sun, Z.; Liu, W.; Jiao, X.; Zu, X.; Hu, Q.; Sun, Y.; Yao, T.; Zhang, W.; Wei, S.; et al. Atomic layer confined vacancies for atomic-level insights into carbon dioxide electroreduction. Nature. Commun. 2017, 8, 14503. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Chen, L.; Knowles, G.P.; MacFarlane, D.R.; Zhang, J. Hierarchical Mesoporous SnO2 Nanosheets on Carbon Cloth: A Robust and Flexible Electrocatalyst for CO2 Reduction with High Efficiency and Selectivity. Angew. Chem. Int. Ed. 2017, 56, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, Z.; Han, P.; Du, Y.; Gu, Z.; Xu, X.; Zheng, G. Single-Atomic Cu with Multiple Oxygen Vacancies on Ceria for Electrocatalytic CO2 Reduction to CH4. ACS Catal. 2018, 8, 7113–7119. [Google Scholar] [CrossRef]
- Huan, T.N.; Ranjbar, N.; Rousse, G.; Sougrati, M.; Zitolo, A.; Mougel, V.; Jaouen, F.; Fontecave, M. Electrochemical Reduction of CO2 Catalyzed by Fe-N-C Materials: A Structure–Selectivity Study. ACS Catal. 2017, 7, 1520–1525. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Yang, S.; Wu, J.; Liu, M.; Yazdi, S.; Ren, M.; Sha, J.; Zhong, J.; Nie, K.; Jalilov, A.S.; et al. Electrochemical CO2 Reduction with Atomic Iron-Dispersed on Nitrogen-Doped Graphene. Adv. Energy Mater. 2018, 8, 1703487. [Google Scholar] [CrossRef]
- Zhang, H.; Li, J.; Xi, S.; Du, Y.; Hai, X.; Wang, J.; Xu, H.; Wu, G.; Zhang, J.; Lu, J.; et al. A Graphene-Supported Single-Atom FeN5 Catalytic Site for Efficient Electrochemical CO2 Reduction. Angew. Chem. Int. Ed. 2019, 58, 14871–14876. [Google Scholar] [CrossRef]
- Zhang, Z.; Ma, C.; Tu, Y.; Si, R.; Wei, J.; Zhang, S.; Wang, Z.; Li, J.-F.; Wang, Y.; Deng, D. Multiscale carbon foam confining single iron atoms for efficient electrocatalytic CO2 reduction to CO. Nano Res. 2019, 12, 2313–2317. [Google Scholar] [CrossRef]
- Gu, J.; Hsu, C.S.; Bai, L.; Chen, H.M.; Hu, X. Atomically dispersed Fe3+ sites catalyze efficient CO2 electroreduction to CO. Science 2019, 364, 1091–1094. [Google Scholar] [CrossRef]
- Yang, F.; Song, P.; Liu, X.; Mei, B.; Xing, W.; Jiang, Z.; Gu, L.; Xu, W. Highly Efficient CO2 Electroreduction on ZnN4-based Single-Atom Catalyst. Angew. Chem. Int. Ed. 2018, 57, 12303–12307. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Chen, G.; Zhou, G.; Yin, L.C.; Veder, J.P.; Johannessen, B.; Saunders, M.; Yang, S.Z.; De Marco, R.; Liu, C.; et al. A Universal Seeding Strategy to Synthesize Single Atom Catalysts on 2D Materials for Electrocatalytic Applications. Adv. Funct. Mater. 2019, 30, 1906157. [Google Scholar] [CrossRef]
- Yang, H.B.; Hung, S.F.; Liu, S.; Yuan, K.; Miao, S.; Zhang, L.; Huang, X.; Wang, H.Y.; Cai, W.; Chen, R.; et al. Atomically dispersed Ni(i) as the active site for electrochemical CO2 reduction. Nat. Energy 2018, 3, 140–147. [Google Scholar] [CrossRef]
- Liu, S.; Yang, H.B.; Hung, S.F.; Ding, J.; Cai, W.; Liu, L.; Gao, J.; Li, X.; Ren, X.; Kuang, Z.; et al. Elucidating the Electrocatalytic CO2 Reduction Reaction over a Model Single-Atom Nickel Catalyst. Angew. Chem. Int. Ed. 2020, 59, 798–803. [Google Scholar] [CrossRef]
- Gong, Y.N.; Jiao, L.; Qian, Y.; Pan, C.Y.; Zheng, L.; Cai, X.; Liu, B.; Yu, S.H.; Jiang, H.L. Regulating the Coordination Environment of MOF-Templated Single-Atom Nickel Electrocatalysts for Boosting CO2 Reduction. Angew. Chem. Int. Ed. 2020, 59, 2705–2709. [Google Scholar] [CrossRef]
- Han, J.; An, P.; Liu, S.; Zhang, X.; Wang, D.; Yuan, Y.; Guo, J.; Qiu, X.; Hou, K.; Shi, L.; et al. Reordering d Orbital Energies of Single-Site Catalysts for CO2 Electroreduction. Angew. Chem. Int. Ed. 2019, 58, 12711–12716. [Google Scholar] [CrossRef]
- Zheng, W.; Yang, J.; Chen, H.; Hou, Y.; Wang, Q.; Gu, M.; He, F.; Xia, Y.; Xia, Z.; Li, Z.; et al. Atomically Defined Undercoordinated Active Sites for Highly Efficient CO2 Electroreduction. Adv. Func. Mater. 2019, 30, 1907658. [Google Scholar] [CrossRef]
- Murata, A.; Hori, Y. Product Selectivity Affected by Cationic Species in Electrochemical Reduction of CO2 and CO at a Cu Electrode. Bull. Chem. Soc. Jpn. 1991, 64, 123–127. [Google Scholar] [CrossRef] [Green Version]
- Qin, B.; Li, Y.; Fu, H.; Wang, H.; Chen, S.; Liu, Z.; Peng, F. Electrochemical Reduction of CO2 into Tunable Syngas Production by Regulating the Crystal Facets of Earth-Abundant Zn Catalyst. ACS Appl. Mater. Interfaces 2018, 10, 20530–20539. [Google Scholar] [CrossRef]
- Jiang, K.; Huang, Y.; Zeng, G.; Toma, F.M.; Goddard, W.A.; Bell, A.T. Effects of Surface Roughness on the Electrochemical Reduction of CO2 over Cu. ACS Energy Lett. 2020, 5, 1206–1214. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Tang, S.; Mu, X.; Dai, Y.; Chen, G.; Zhou, Z.; Ruan, F.; Yang, Z.; Zheng, N. Freestanding palladium nanosheets with plasmonic and catalytic properties. Nat. Nanotechnol. 2011, 6, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Mistry, H.; Reske, R.; Zeng, Z.; Zhao, Z.J.; Greeley, J.; Strasser, P.; Cuenya, B.R. Exceptional size-dependent activity enhancement in the electroreduction of CO2 over Au nanoparticles. J. Am. Chem. Soc. 2014, 136, 16473–16476. [Google Scholar] [CrossRef]
- Yang, Q.; Shi, L.; Yu, B.; Xu, J.; Wei, C.; Wang, Y.; Chen, H. Facile synthesis of ultrathin Pt–Pd nanosheets for enhanced formic acid oxidation and oxygen reduction reaction. J. Mater. Chem. A 2019, 7, 18846–18851. [Google Scholar] [CrossRef]
- Gao, F.; Zhang, Y.; Ren, F.; Shiraishi, Y.; Du, Y. Universal Surfactant-Free Strategy for Self-Standing 3D Tremella-Like Pd–M (M = Ag, Pb, and Au) Nanosheets for Superior Alcohols Electrocatalysis. Adv. Funct. Mater. 2020, 30, 2000255. [Google Scholar] [CrossRef]
- Zhang, S.; Fan, Q.; Xia, R.; Meyer, T.J. CO2 Reduction: From Homogeneous to Heterogeneous Electrocatalysis. Acc. Chem. Res. 2020, 53, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Ma, M.; Wu, L.; Valenti, M.; Cardenas-Morcoso, D.; Hofmann, J.P.; Bisquert, J.; Gimenez, S.; Smith, W.A. Electronic Effects Determine the Selectivity of Planar Au–Cu Bimetallic Thin Films for Electrochemical CO2 Reduction. ACS Appl. Mater. Interfaces 2019, 11, 16546–16555. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.P.; Lu, X.F.; Zang, S.Q.; Lou, X.W. Non-Noble-Metal-Based Electrocatalysts toward the Oxygen Evolution Reaction. Adv. Funct. Mater. 2020, 30, 1910274. [Google Scholar] [CrossRef]
- Zhu, Y.P.; Guo, C.; Zheng, Y.; Qiao, S.Z. Surface and Interface Engineering of Noble-Metal-Free Electrocatalysts for Efficient Energy Conversion Processes. Acc. Chem. Res. 2017, 50, 915–923. [Google Scholar] [CrossRef]
- Kuhl, K.P.; Cave, E.R.; Abram, D.N.; Jaramillo, T.F. New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces. Energy Environ. Sci. 2012, 5, 7050–7059. [Google Scholar] [CrossRef]
- Kuang, Y.; Feng, G.; Li, P.; Bi, Y.; Li, Y.; Sun, X. Single-Crystalline Ultrathin Nickel Nanosheets Array from In Situ Topotactic Reduction for Active and Stable Electrocatalysis. Angew. Chem. Int. Ed. 2016, 55, 693–697. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Yang, S.; Jiang, M.; Hu, Y.; Hu, C.; Zhang, X.; Jin, Z. Nanocapillarity and Nanoconfinement Effects of Pipet-like Bismuth@Carbon Nanotubes for Highly Efficient Electrocatalytic CO2 Reduction. Nano Lett. 2021, 21, 2650–2657. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Gao, Y.; Talreja, N.; Guo, F.; Texter, J.; Yan, C.; Sun, Z. Two-dimensional nanosheets for electrocatalysis in energy generation and conversion. J. Mater. Chem. A 2017, 5, 7257–7284. [Google Scholar] [CrossRef]
- Chai, G.L.; Guo, Z.X. Highly effective sites and selectivity of nitrogen-doped graphene/CNT catalysts for CO2 electrochemical reduction. Chem. Sci. 2016, 7, 1268–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, X.; Xu, J.; Wei, Z.; Ma, J.; Guo, S.; Wang, S.; Liu, H.; Dou, S. Metal-Free Carbon Materials for CO2 Electrochemical Reduction. Adv. Mater. 2017, 29, 201701784. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, J.; Cai, Q. Pyrrolic-nitrogen doped graphene: A metal-free electrocatalyst with high efficiency and selectivity for the reduction of carbon dioxide to formic acid: A computational study. Phys. Chem. Chem. Phys. 2016, 18, 5491–5498. [Google Scholar] [CrossRef]
- Rogers, C.; Perkins, W.S.; Veber, G.; Williams, T.E.; Cloke, R.R.; Fischer, F.R. Synergistic Enhancement of Electrocatalytic CO2 Reduction with Gold Nanoparticles Embedded in Functional Graphene Nanoribbon Composite Electrodes. J. Am. Chem. Soc. 2017, 139, 4052–4061. [Google Scholar] [CrossRef] [Green Version]
- Zeng, M.; Li, Y. Recent advances in heterogeneous electrocatalysts for the hydrogen evolution reaction. J. Mater. Chem. A 2015, 3, 14942–14962. [Google Scholar] [CrossRef]
- Xie, Y.; Li, X.; Wang, Y.; Li, B.; Yang, L.; Zhao, N.; Liu, M.; Wang, X.; Yu, Y.; Liu, J.M. Reaction mechanisms for reduction of CO2 to CO on monolayer MoS2. Appl. Surf. Sci. 2020, 499, 143964. [Google Scholar] [CrossRef]
- Hong, X.; Chan, K.; Tsai, C.; Nørskov, J.K. How Doped MoS2 Breaks Transition-Metal Scaling Relations for CO2 Electrochemical Reduction. ACS Catal. 2016, 6, 4428–4437. [Google Scholar] [CrossRef]
- Chan, K.; Tsai, C.; Hansen, H.A.; Nørskov, J.K. Molybdenum Sulfides and Selenides as Possible Electrocatalysts for CO2 Reduction. ChemCatChem 2014, 6, 1899–1905. [Google Scholar] [CrossRef]
- Shao, P.; Yi, L.; Chen, S.; Zhou, T.; Zhang, J. Metal-organic frameworks for electrochemical reduction of carbon dioxide: The role of metal centers. J. Energy Chem. 2020, 40, 156–170. [Google Scholar] [CrossRef] [Green Version]
- Perfecto-Irigaray, M.; Albo, J.; Beobide, G.; Castillo, O.; Irabien, A.; Pérez-Yáñez, S. Synthesis of heterometallic metal–organic frameworks and their performance as electrocatalyst for CO2 reduction. RSC Adv. 2018, 8, 21092–21099. [Google Scholar] [CrossRef] [Green Version]
- Majidi, L.; Ahmadiparidari, A.; Shan, N.; Misal, S.N.; Kumar, K.; Huang, Z.; Rastegar, S.; Hemmat, Z.; Zou, X.; Zapol, P.; et al. 2D Copper Tetrahydroxyquinone Conductive Metal-Organic Framework for Selective CO2 Electrocatalysis at Low Overpotentials. Adv. Mater. 2021, 33, e2004393. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kanan, M.W. Tin oxide dependence of the CO2 reduction efficiency on tin electrodes and enhanced activity for tin/tin oxide thin-film catalysts. J. Am. Chem. Soc. 2012, 134, 1986–1989. [Google Scholar] [CrossRef] [PubMed]
- Li, C.W.; Kanan, M.W. CO2 reduction at low overpotential on Cu electrodes resulting from the reduction of thick Cu2O films. J. Am. Chem. Soc. 2012, 134, 7231–7234. [Google Scholar] [CrossRef]
- Liu, J.; Guo, C.; Vasileff, A.; Qiao, S. Nanostructured 2D Materials: Prospective Catalysts for Electrochemical CO2 Reduction. Small Methods 2017, 1, 1600006. [Google Scholar] [CrossRef]
- Yang, X.F.; Wang, A.Q.; Qiao, B.T.; Li, J.; Liu, J.Y.; Zhang, T. Single-Atom Catalysts: A New Frontier in Heterogeneous Catalysis. Acc. Chem. Res. 2013, 46, 1740–1748. [Google Scholar] [CrossRef]
- Wang, A.; Li, J.; Zhang, T. Heterogeneous single-atom catalysis. Nat. Rev. Chem. 2018, 2, 65–81. [Google Scholar] [CrossRef]
- Chorkendorff, I.; Niemantsverdriet, J.W. Concepts of Modern Catalysis and Kinetics; Wiley: Weinheim, Germany, 2003. [Google Scholar] [CrossRef]
- Jaramillo, T.F.; Jorgensen, K.P.; Bonde, J.; Nielsen, J.H.; Horch, S.; Chorkendorff, I. Identification of Active Edge Sites for Electrochemical H2 Evolution from MoS2 Nanocatalysts. Science 2007, 317, 100–102. [Google Scholar] [CrossRef] [Green Version]
- Seh, Z.W.; Kibsgaard, J.; Dickens, C.F.; Chorkendorff, I.; Norskov, J.K.; Jaramillo, T.F. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 2017, 355, aad4998. [Google Scholar] [CrossRef] [Green Version]
- Kyriakou, G.; Boucher, M.W.; Jewell, A.D.; Lewis, E.A.; Lawton, T.J.; Baber, A.E.; Tierney, H.L.; Flytzani-Stephanopoulos, M.; Sykes, E.C. Isolated Metal Atom Geometries as a Strategy for Selective Heterogeneous Hydrogenations. Science 2012, 335, 1209–1212. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Zhou, T.; Chen, M.; Feng, H.; Yuan, R.; Zhong, C.a.; Yan, W.; Tian, Y.; Wu, X.; Chu, W.; et al. High-purity pyrrole-type FeN4 sites as a superior oxygen reduction electrocatalyst. Energy Environ. Sci. 2020, 13, 111–118. [Google Scholar] [CrossRef]
- Yin, X.P.; Wang, H.J.; Tang, S.F.; Lu, X.L.; Shu, M.; Si, R.; Lu, T.B. Engineering the Coordination Environment of Single-Atom Platinum Anchored on Graphdiyne for Optimizing Electrocatalytic Hydrogen Evolution. Angew. Chem. Int. Ed. 2018, 57, 9382–9386. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Gao, L.; Wang, Y.; Wang, X.; Zhu, J.; Jin, Z.; Liu, C.; Chen, H.; Li, G.; Ge, J.; et al. Engineering Energy Level of Metal Center: Ru Single-Atom Site for Efficient and Durable Oxygen Reduction Catalysis. J. Am. Chem. Soc. 2019, 141, 19800–19806. [Google Scholar] [CrossRef]
- Zhang, H.; Yu, L.; Chen, T.; Zhou, W.; Lou, X.W.D. Surface Modulation of Hierarchical MoS2 Nanosheets by Ni Single Atoms for Enhanced Electrocatalytic Hydrogen Evolution. Adv. Funct. Mater. 2018, 28, 1807086. [Google Scholar] [CrossRef]
- Li, H.; Wang, L.; Dai, Y.; Pu, Z.; Lao, Z.; Chen, Y.; Wang, M.; Zheng, X.; Zhu, J.; Zhang, W.; et al. Synergetic interaction between neighbouring platinum monomers in CO2 hydrogenation. Nat. Nanotechnol. 2018, 13, 411–417. [Google Scholar] [CrossRef]
- Ye, S.; Luo, F.; Zhang, Q.; Zhang, P.; Xu, T.; Wang, Q.; He, D.; Guo, L.; Zhang, Y.; He, C.; et al. Highly stable single Pt atomic sites anchored on aniline-stacked graphene for hydrogen evolution reaction. Energy Environ. Sci. 2019, 12, 1000–1007. [Google Scholar] [CrossRef]
- Yuan, J.; Zhang, W.; Li, X.; Yang, J. A high-performance catalyst for methane conversion to methanol: Graphene supported single atom Co. Chem. Commun. 2018, 54, 2284–2287. [Google Scholar] [CrossRef]
- Ramalingam, V.; Varadhan, P.; Fu, H.C.; Kim, H.; Zhang, D.; Chen, S.; Song, L.; Ma, D.; Wang, Y.; Alshareef, H.N.; et al. Heteroatom-Mediated Interactions between Ruthenium Single Atoms and an MXene Support for Efficient Hydrogen Evolution. Adv. Mater. 2019, 31, e1903841. [Google Scholar] [CrossRef] [Green Version]
- Back, S.; Lim, J.; Kim, N.Y.; Kim, Y.H.; Jung, Y. Single-atom catalysts for CO2 electroreduction with significant activity and selectivity improvements. Chem. Sci. 2017, 8, 1090–1096. [Google Scholar] [CrossRef] [Green Version]
- Jiang, K.; Siahrostami, S.; Zheng, T.T.; Hu, Y.F.; Hwang, S.Y.; Stavitski, E.; Peng, Y.D.; Dynes, J.; Gangisetty, M.; Su, D.; et al. Isolated Ni single atoms in graphene nanosheets for high-performance CO2 reduction. Energy Environ. Sci. 2018, 11, 893–903. [Google Scholar] [CrossRef]
- Jiao, Y.; Zheng, Y.; Chen, P.; Jaroniec, M.; Qiao, S.Z. Molecular Scaffolding Strategy with Synergistic Active Centers to Facilitate Electrocatalytic CO2 Reduction to Hydrocarbon/Alcohol. J. Am. Chem. Soc. 2017, 139, 18093–18100. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.; Cai, J.; Lee, C.; Lee, H.M.; Xu, M.; Huang, Y. Intimate atomic Cu-Ag interfaces for high CO2RR selectivity towards CH4 at low over potential. Nano Res. 2021, 14, 3497–3501. [Google Scholar] [CrossRef]
- Chen, B.; Li, B.; Tian, Z.; Liu, W.; Liu, W.; Sun, W.; Wang, K.; Chen, L.; Jiang, J. Enhancement of Mass Transfer for Facilitating Industrial-Level CO2 Electroreduction on Atomic Ni-N4 Sites. Adv. Energy Mater. 2021, 11, 2102152. [Google Scholar] [CrossRef]
- Yang, Y.; Li, F. Reactor design for electrochemical CO2 conversion toward large-scale applications. Curr. Opin. Green Sustain. Chem. 2021, 27, 100419. [Google Scholar] [CrossRef]
- Jayashree, R.S.; Mitchell, M.; Natarajan, D.; Markoski, L.J.; Kenis, P.J.A. Microfluidic Hydrogen Fuel Cell with a Liquid Electrolyte. Langmuir 2007, 23, 6871–6874. [Google Scholar] [CrossRef]
- Whipple, D.T.; Finke, E.C.; Kenis, P.J.A. Microfluidic Reactor for the Electrochemical Reduction of Carbon Dioxide: The Effect of pH. Electrochem. Solid-State Lett. 2010, 13, 9. [Google Scholar] [CrossRef]
- Higgins, D.; Hahn, C.; Xiang, C.; Jaramillo, T.F.; Weber, A.Z. Gas-Diffusion Electrodes for Carbon Dioxide Reduction: A New Paradigm. ACS Energy Lett. 2018, 4, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Lees, E.W.; Mowbray, B.A.W.; Parlane, F.G.L.; Berlinguette, C.P. Gas diffusion electrodes and membranes for CO2 reduction electrolysers. Nat. Rev. Mater. 2021, 7, 55–64. [Google Scholar] [CrossRef]
- Lim, J.; Kang, P.W.; Jeon, S.S.; Lee, H. Electrochemically deposited Sn catalysts with dense tips on a gas diffusion electrode for electrochemical CO2 reduction. J. Mater. Chem. A 2020, 8, 9032–9038. [Google Scholar] [CrossRef]
- Rabiee, H.; Ge, L.; Zhang, X.; Hu, S.; Li, M.; Yuan, Z. Gas diffusion electrodes (GDEs) for electrochemical reduction of carbon dioxide, carbon monoxide, and dinitrogen to value-added products: A review. Energy Environ. Sci. 2021, 14, 1959–2008. [Google Scholar] [CrossRef]
- Gabardo, C.M.; O’Brien, C.P.; Edwards, J.P.; McCallum, C.; Xu, Y.; Dinh, C.T.; Li, J.; Sargent, E.H.; Sinton, D. Continuous Carbon Dioxide Electroreduction to Concentrated Multi-carbon Products Using a Membrane Electrode Assembly. Joule 2019, 3, 2777–2791. [Google Scholar] [CrossRef]
- Ren, S.X.; Joulié, D.; Salvatore, D.; Torbensen, K.; Wang, M.; Robert, M.; Berlinguette, C.P. Molecular electrocatalysts can mediate fast, selective CO2 reduction in a flow cell. Science 2019, 365, 367–369. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Potential electrochemical CO2 reduction pathways to form the C1 products CO, HCOOH and CH4 and the C2 products C2H4 and C2H5OH. Reprinted from [53], Copyright (2019), with permission from Wiley.
Figure 1.
Potential electrochemical CO2 reduction pathways to form the C1 products CO, HCOOH and CH4 and the C2 products C2H4 and C2H5OH. Reprinted from [53], Copyright (2019), with permission from Wiley.

Figure 2.
(a) Surface valence band X-ray photoemission spectra of the as-prepared Au, Cu and Au–Cu alloys. (b) d-band center of different samples before and after ECR at −0.7 V vs. RHE for 1 h. The gravity center of the valance band center is shown by the dotted lines compared to 0 eV. The binding energy is the value of |E − EFermi|. (c) Faradaic efficiency of and (d) partial current densities of CO. Reprinted from [117], Copyright (2018), with permission from American Chemical Society.
Figure 2.
(a) Surface valence band X-ray photoemission spectra of the as-prepared Au, Cu and Au–Cu alloys. (b) d-band center of different samples before and after ECR at −0.7 V vs. RHE for 1 h. The gravity center of the valance band center is shown by the dotted lines compared to 0 eV. The binding energy is the value of |E − EFermi|. (c) Faradaic efficiency of and (d) partial current densities of CO. Reprinted from [117], Copyright (2018), with permission from American Chemical Society.

Figure 3.
(a) Current densities of Bi nanosheets, bulk Bi and acetylene black decorated carbon paper (AB/CP) within a potential window of −0.5 to −1.2 V vs. RHE in CO2-saturated 0.1 M KHCO3 aqueous solution. (b) Faradaic efficiencies (left Y axes) and partial current densities of formate product (right Y axes) of Bi nanosheets. DFT calculated ΔG in the reaction pathways of CO2 conversion to formate from the facet sites and edge sites of the (c) (003) plane and (d) (012) plane on Bi. Reprinted from [76], Copyright (2018), with permission from Elsevier.
Figure 3.
(a) Current densities of Bi nanosheets, bulk Bi and acetylene black decorated carbon paper (AB/CP) within a potential window of −0.5 to −1.2 V vs. RHE in CO2-saturated 0.1 M KHCO3 aqueous solution. (b) Faradaic efficiencies (left Y axes) and partial current densities of formate product (right Y axes) of Bi nanosheets. DFT calculated ΔG in the reaction pathways of CO2 conversion to formate from the facet sites and edge sites of the (c) (003) plane and (d) (012) plane on Bi. Reprinted from [76], Copyright (2018), with permission from Elsevier.

Figure 4.
(a) Cyclic voltammograms of nickel–nitrogen comodified graphene (Ni–N–Gr) catalyst and (c) Ni foil in CO2-saturated 0.1 M KHCO3 and Ar-saturated 0.1 M KH2PO4/K2HPO4 (pH = 6.8) at 10 mV s−1. FEs for the reduction products by (b) Ni–N–Gr catalyst and (d) Ni foil. Reprinted from [81], Copyright (2016), with permission from Wiley.
Figure 4.
(a) Cyclic voltammograms of nickel–nitrogen comodified graphene (Ni–N–Gr) catalyst and (c) Ni foil in CO2-saturated 0.1 M KHCO3 and Ar-saturated 0.1 M KH2PO4/K2HPO4 (pH = 6.8) at 10 mV s−1. FEs for the reduction products by (b) Ni–N–Gr catalyst and (d) Ni foil. Reprinted from [81], Copyright (2016), with permission from Wiley.

Figure 5.
(a) Current density as a function of dopant percentage for Nb doped and Ta doped MoS2 samples. (b) Calculated CO formation TOF at different applied overpotentials for 0.5% Nb-doped MoS2, pristine VA-MoS2 and Ag nanoparticles. Reprinted from [87], Copyright (2017), with permission from American Chemical Society.
Figure 5.
(a) Current density as a function of dopant percentage for Nb doped and Ta doped MoS2 samples. (b) Calculated CO formation TOF at different applied overpotentials for 0.5% Nb-doped MoS2, pristine VA-MoS2 and Ag nanoparticles. Reprinted from [87], Copyright (2017), with permission from American Chemical Society.

Figure 6.
(a) Linear-sweep voltametric (LSV) tests in CO2-saturated electrolyte. (b) CO current density of MePcs at various potentials. (c) Faradaic efficiency of CO and H2 formation at different potentials for the CoPc sample. (d) Faradaic efficiency of CO formation at −0.8 V versus RHE for the different MePc samples. (e) The calculated free-energy diagram for all the MePc electrodes. The inset shows the adsorption energy of *COOH (unit eV) on different sites of CoPc. (f) Fitted *CO desorption and *COOH formation trends over the five MePc electrodes. Reprinted from [92], Copyright (2018), with permission from Wiley.
Figure 6.
(a) Linear-sweep voltametric (LSV) tests in CO2-saturated electrolyte. (b) CO current density of MePcs at various potentials. (c) Faradaic efficiency of CO and H2 formation at different potentials for the CoPc sample. (d) Faradaic efficiency of CO formation at −0.8 V versus RHE for the different MePc samples. (e) The calculated free-energy diagram for all the MePc electrodes. The inset shows the adsorption energy of *COOH (unit eV) on different sites of CoPc. (f) Fitted *CO desorption and *COOH formation trends over the five MePc electrodes. Reprinted from [92], Copyright (2018), with permission from Wiley.

Figure 7.
The calculated DOS for (a) Co3O4 atomic layer slab with a thickness of 1.72 nm and (b) bulk Co3O4 slab. The yellow shaded parts represent the increased DOS at the conduction band edges of Co3O4 atomic layer. The partial charge density around the conduction band minimum of (c) Co3O4 atomic layer slab and (d) bulk Co3O4 slab, in which yellow refers to charge density contour with the isovalue of 0.0001 e/bohr3. Cyan represents Co and red represents O atoms. Electroreduction of CO2 to formate by Co3O4 with different thicknesses: (e) LSV curves in the CO2-saturated (solid line) and N2-saturated (dashed line) 0.1 M KHCO3 aqueous solutions. (f) Faradaic efficiencies for formate at each applied potential for 4 h. (g) Charging current density differences plotted against scan rates. (h) ECSA—corrected current densities vs. applied potentials. Reprinted from [93], Copyright (2016), with permission from Wiley.
Figure 7.
The calculated DOS for (a) Co3O4 atomic layer slab with a thickness of 1.72 nm and (b) bulk Co3O4 slab. The yellow shaded parts represent the increased DOS at the conduction band edges of Co3O4 atomic layer. The partial charge density around the conduction band minimum of (c) Co3O4 atomic layer slab and (d) bulk Co3O4 slab, in which yellow refers to charge density contour with the isovalue of 0.0001 e/bohr3. Cyan represents Co and red represents O atoms. Electroreduction of CO2 to formate by Co3O4 with different thicknesses: (e) LSV curves in the CO2-saturated (solid line) and N2-saturated (dashed line) 0.1 M KHCO3 aqueous solutions. (f) Faradaic efficiencies for formate at each applied potential for 4 h. (g) Charging current density differences plotted against scan rates. (h) ECSA—corrected current densities vs. applied potentials. Reprinted from [93], Copyright (2016), with permission from Wiley.

Figure 8.
(a,b) The scanning electron microscope (SEM) images of porous SnO2 nanosheets on carbon cloth (SnO2/CC). (c) Current density of the SnO2/CC electrode in CO2-saturated 0.5 M NaHCO3 solution at different applied potentials. (d) Corresponding FE for formate, CO and H2. (e) i–t curve and FE for formate of SnO2/CC electrode in CO2-saturated 0.5 M NaHCO3 solution at an applied potential of −1.6 V for 24 h. (f) i–t curves of the SnO2/CC electrode at −1.6 V after folding or twisting 10 times. Reprinted from [95], Copyright (2017), with permission from Wiley.
Figure 8.
(a,b) The scanning electron microscope (SEM) images of porous SnO2 nanosheets on carbon cloth (SnO2/CC). (c) Current density of the SnO2/CC electrode in CO2-saturated 0.5 M NaHCO3 solution at different applied potentials. (d) Corresponding FE for formate, CO and H2. (e) i–t curve and FE for formate of SnO2/CC electrode in CO2-saturated 0.5 M NaHCO3 solution at an applied potential of −1.6 V for 24 h. (f) i–t curves of the SnO2/CC electrode at −1.6 V after folding or twisting 10 times. Reprinted from [95], Copyright (2017), with permission from Wiley.

Figure 9.
Free energy change in the first protonation step in ECR and HER on different SACs. Catalysts below the dotted parity line are ECR selective. Reprinted from [152], Copyright (2017), with permission from Royal Society of Chemistry.
Figure 9.
Free energy change in the first protonation step in ECR and HER on different SACs. Catalysts below the dotted parity line are ECR selective. Reprinted from [152], Copyright (2017), with permission from Royal Society of Chemistry.

Figure 10.
(a) Typical high-angle annular dark-field scanning transmission electron microscope (HAADF-STEM) image of the optimal ZnNx/C catalyst. The circles indicate the individual Zn atoms. (b) A typical fitting curve of the EXAFS signal in R-space for the adsorbed ZnNx/C catalyst. (c) FEs of CO and H2 at various applied potentials on ZnNx/C catalyst. (d) Long-term stability of ZnNx/C at a potential load of −0.43 V and the corresponding FEs of CO and H2. Reprinted from [102] Copyright (2018), with permission from Wiley.
Figure 10.
(a) Typical high-angle annular dark-field scanning transmission electron microscope (HAADF-STEM) image of the optimal ZnNx/C catalyst. The circles indicate the individual Zn atoms. (b) A typical fitting curve of the EXAFS signal in R-space for the adsorbed ZnNx/C catalyst. (c) FEs of CO and H2 at various applied potentials on ZnNx/C catalyst. (d) Long-term stability of ZnNx/C at a potential load of −0.43 V and the corresponding FEs of CO and H2. Reprinted from [102] Copyright (2018), with permission from Wiley.

Figure 11.
(a) Transmission electron microscope (TEM) and (b) HAADF-STEM images of NiSA–N2–C. (c) EDS mapping of Ni, N and C elements in NiSA–N2–C. (d) LSV curves in CO2-saturated 0.5 M KHCO3 electrolyte. (e) FEs of CO at different applied potentials. (f) The corresponding TOFs of CO production over NiSA–Nx–C. (g) Stability of NiSA–N2–C at −0.8 V during 10 h. (h) Proposed reaction paths for ECR with NiSA–N2–C as an example. (i) Free-energy diagram of CO2 reduction to CO over NiSA–Nx–C catalysts. Reprinted from [106], Copyright (2020), with permission from Wiley.
Figure 11.
(a) Transmission electron microscope (TEM) and (b) HAADF-STEM images of NiSA–N2–C. (c) EDS mapping of Ni, N and C elements in NiSA–N2–C. (d) LSV curves in CO2-saturated 0.5 M KHCO3 electrolyte. (e) FEs of CO at different applied potentials. (f) The corresponding TOFs of CO production over NiSA–Nx–C. (g) Stability of NiSA–N2–C at −0.8 V during 10 h. (h) Proposed reaction paths for ECR with NiSA–N2–C as an example. (i) Free-energy diagram of CO2 reduction to CO over NiSA–Nx–C catalysts. Reprinted from [106], Copyright (2020), with permission from Wiley.

Figure 12.
(a) Fourier transformed (FT)-EXAFS fitting results of Cu–N2/GN. (b) CO FE of Cu–N4/GN–700, Cu–N4/GN–800 and Cu–N2/GN. Reprinted from [108], Copyright 2020, with permission from Wiley. (c) Nitrogen K-edge NEXAFS of Cu–C3N4 and pure g–C3N4. Arrows show the weak shoulders in the N K-edge assigned to the Cu–N interaction. Dotted lines show the channels of photon energy in the two samples. (d) Measured Faradaic efficiencies of various products on Cu–C3N4. Reprinted from [154], Copyright (2017), with permission from American Chemical Society.
Figure 12.
(a) Fourier transformed (FT)-EXAFS fitting results of Cu–N2/GN. (b) CO FE of Cu–N4/GN–700, Cu–N4/GN–800 and Cu–N2/GN. Reprinted from [108], Copyright 2020, with permission from Wiley. (c) Nitrogen K-edge NEXAFS of Cu–C3N4 and pure g–C3N4. Arrows show the weak shoulders in the N K-edge assigned to the Cu–N interaction. Dotted lines show the channels of photon energy in the two samples. (d) Measured Faradaic efficiencies of various products on Cu–C3N4. Reprinted from [154], Copyright (2017), with permission from American Chemical Society.


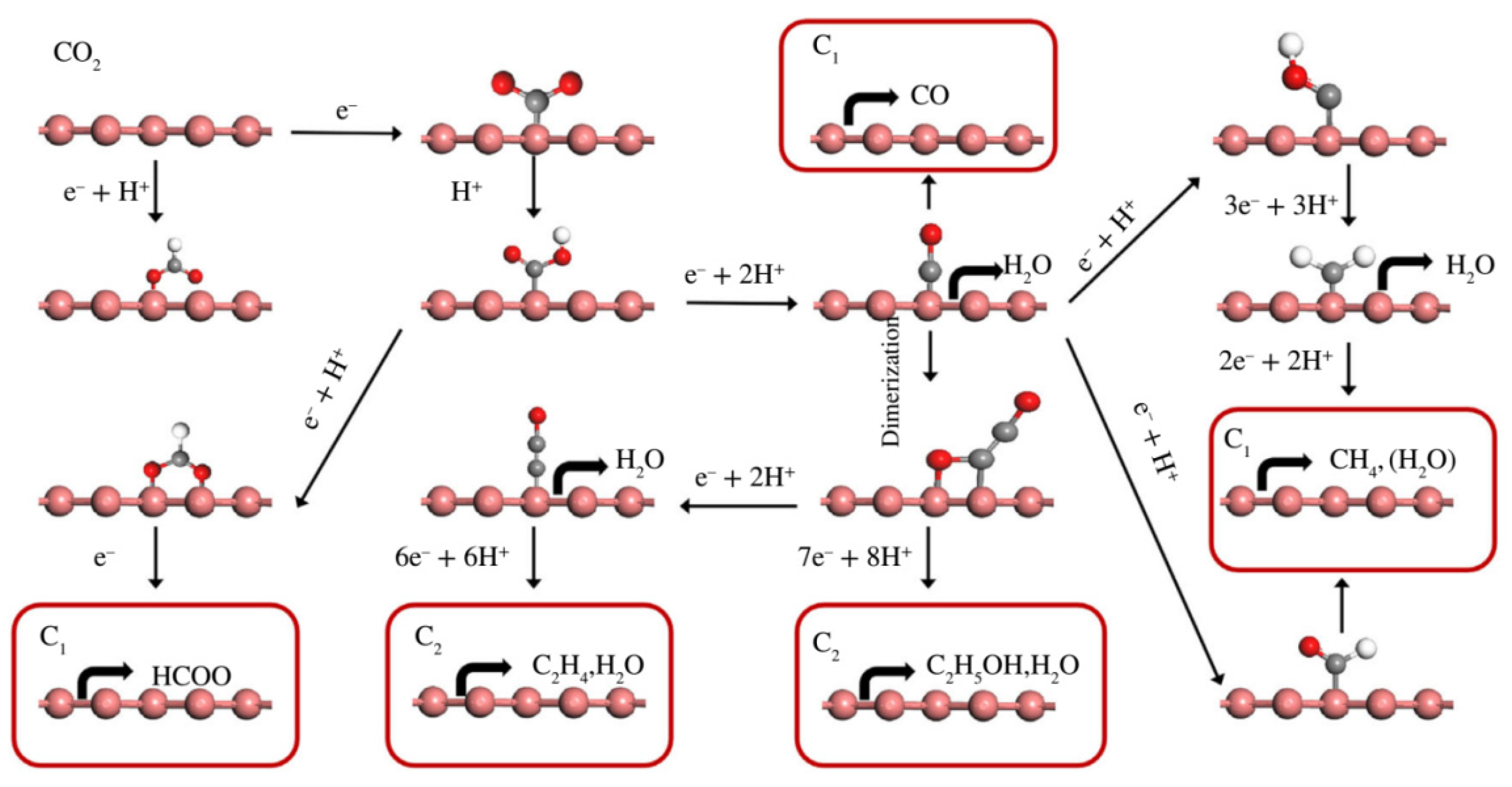{kind=link}
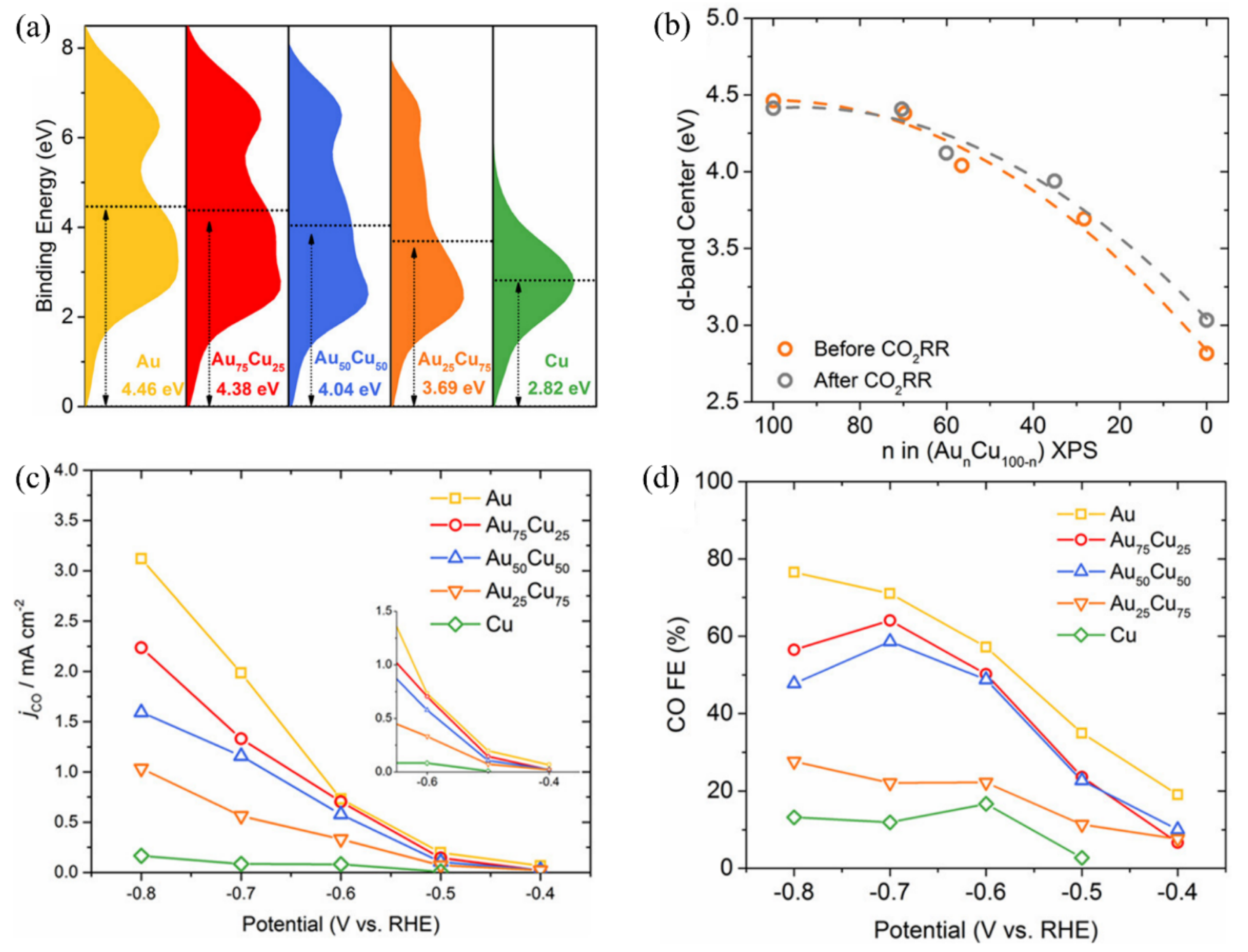{kind=link}
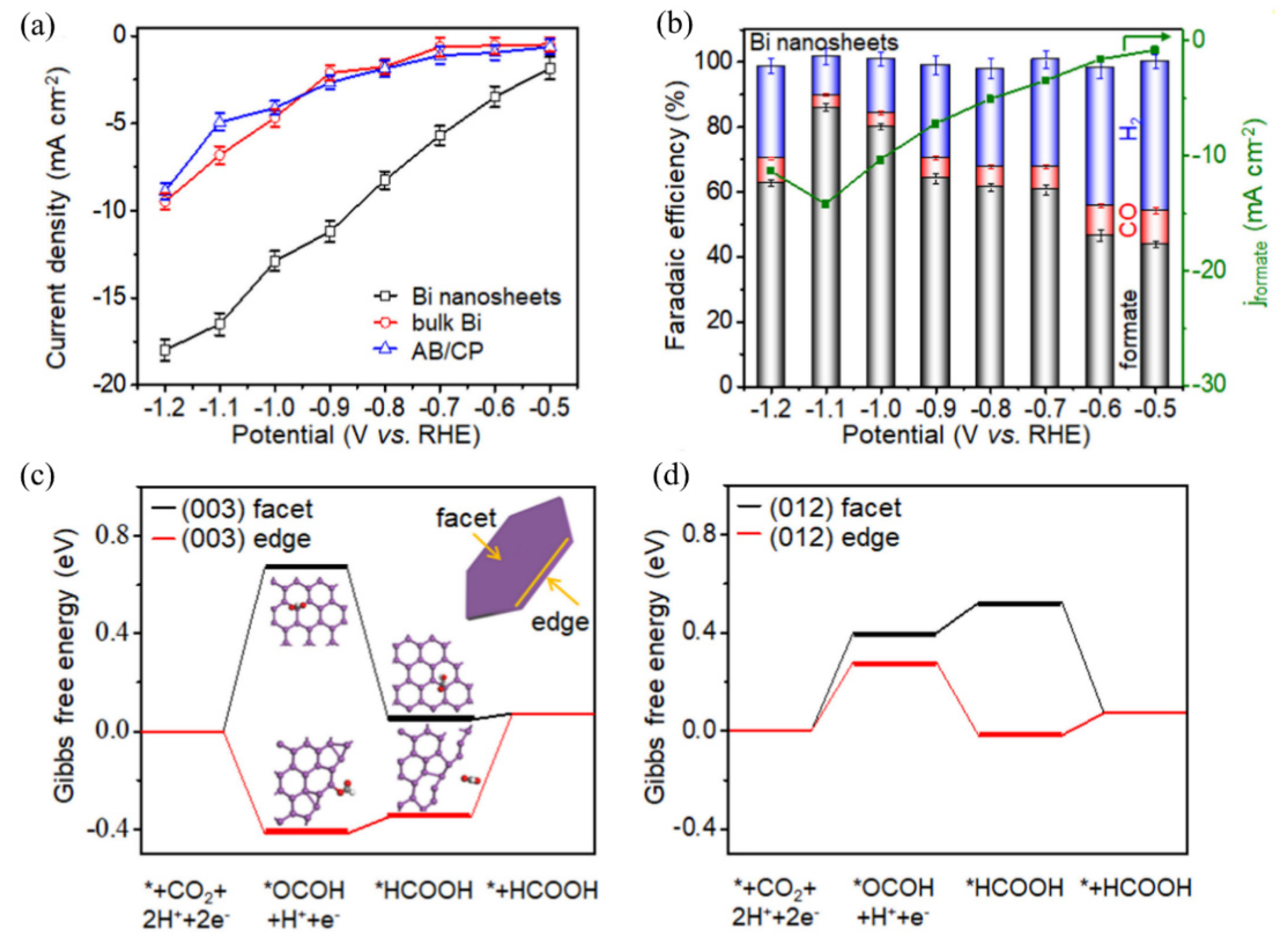{kind=link}
{kind=link}
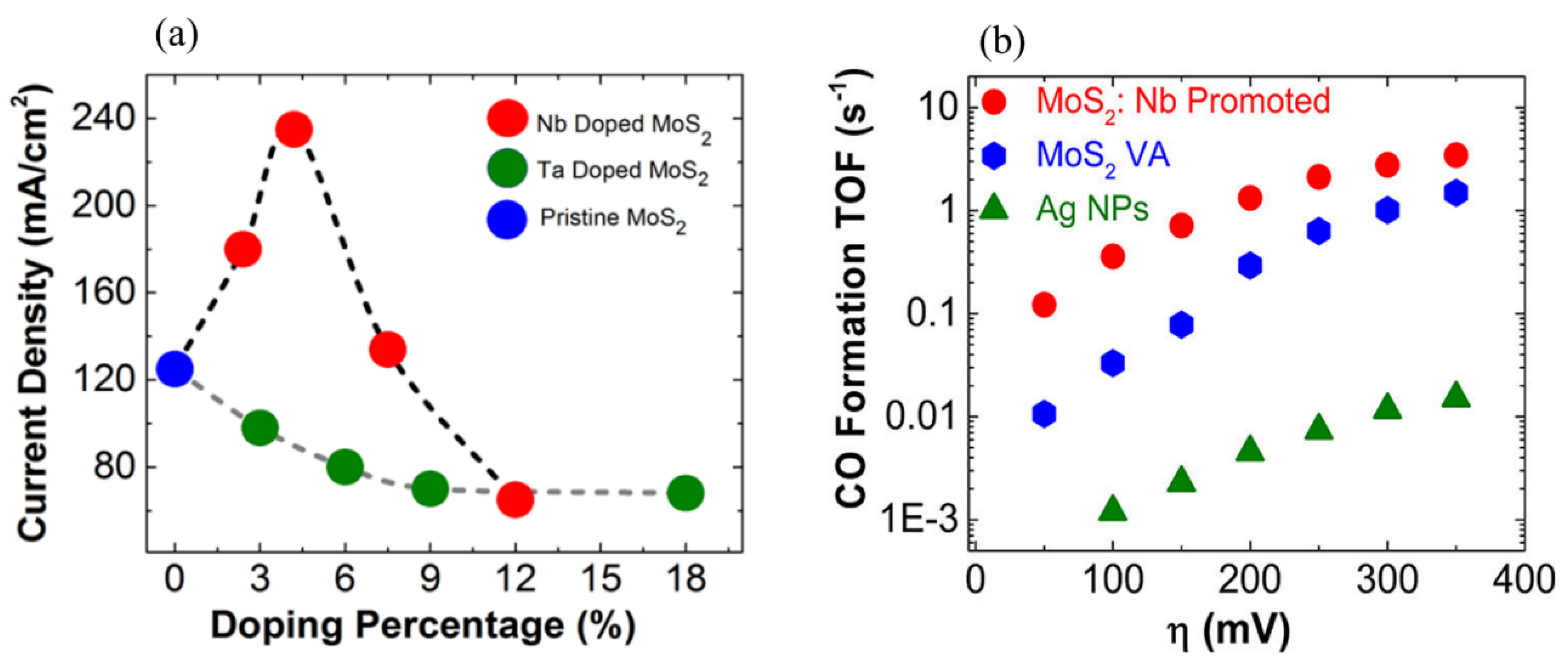{kind=link}
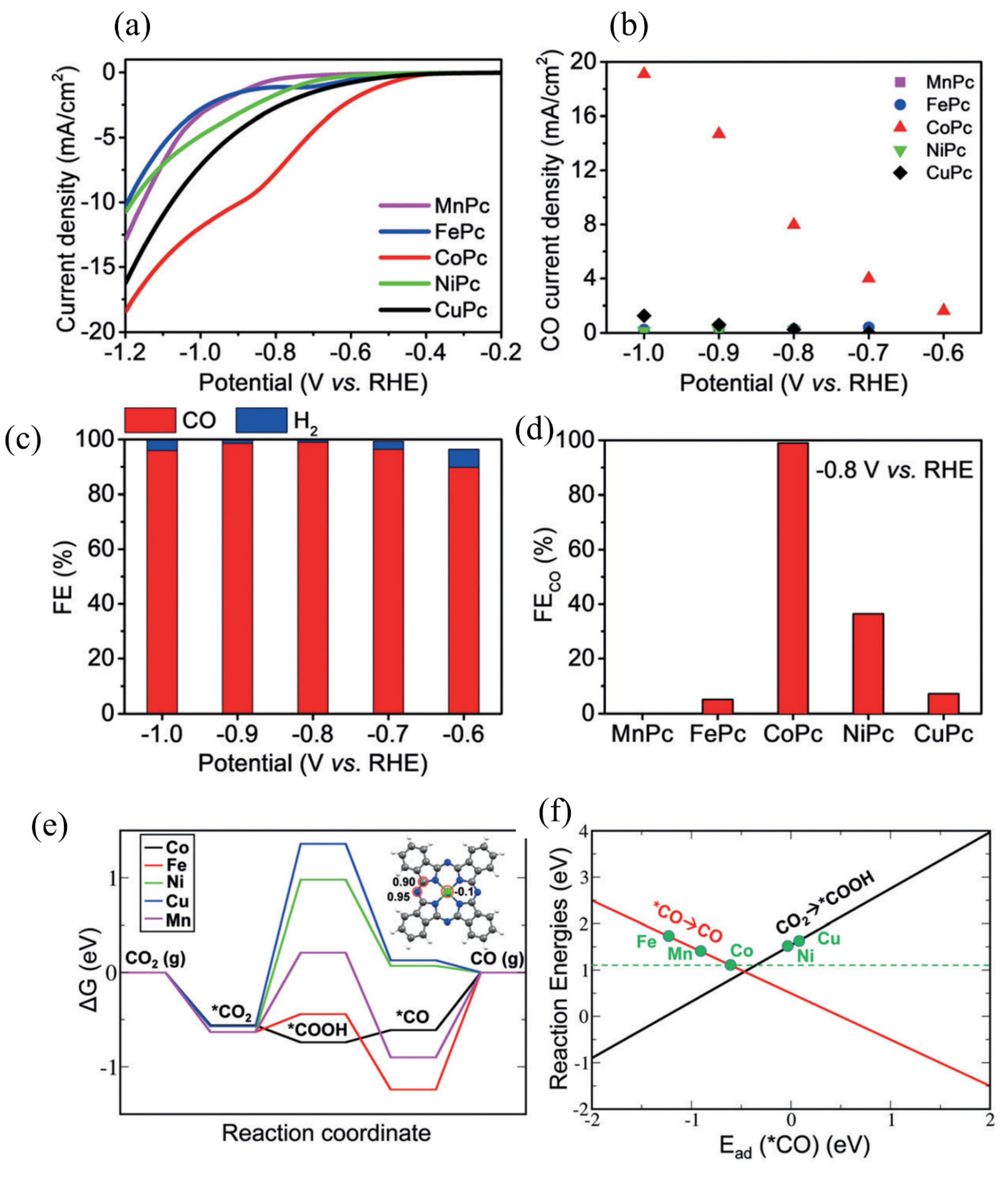{kind=link}
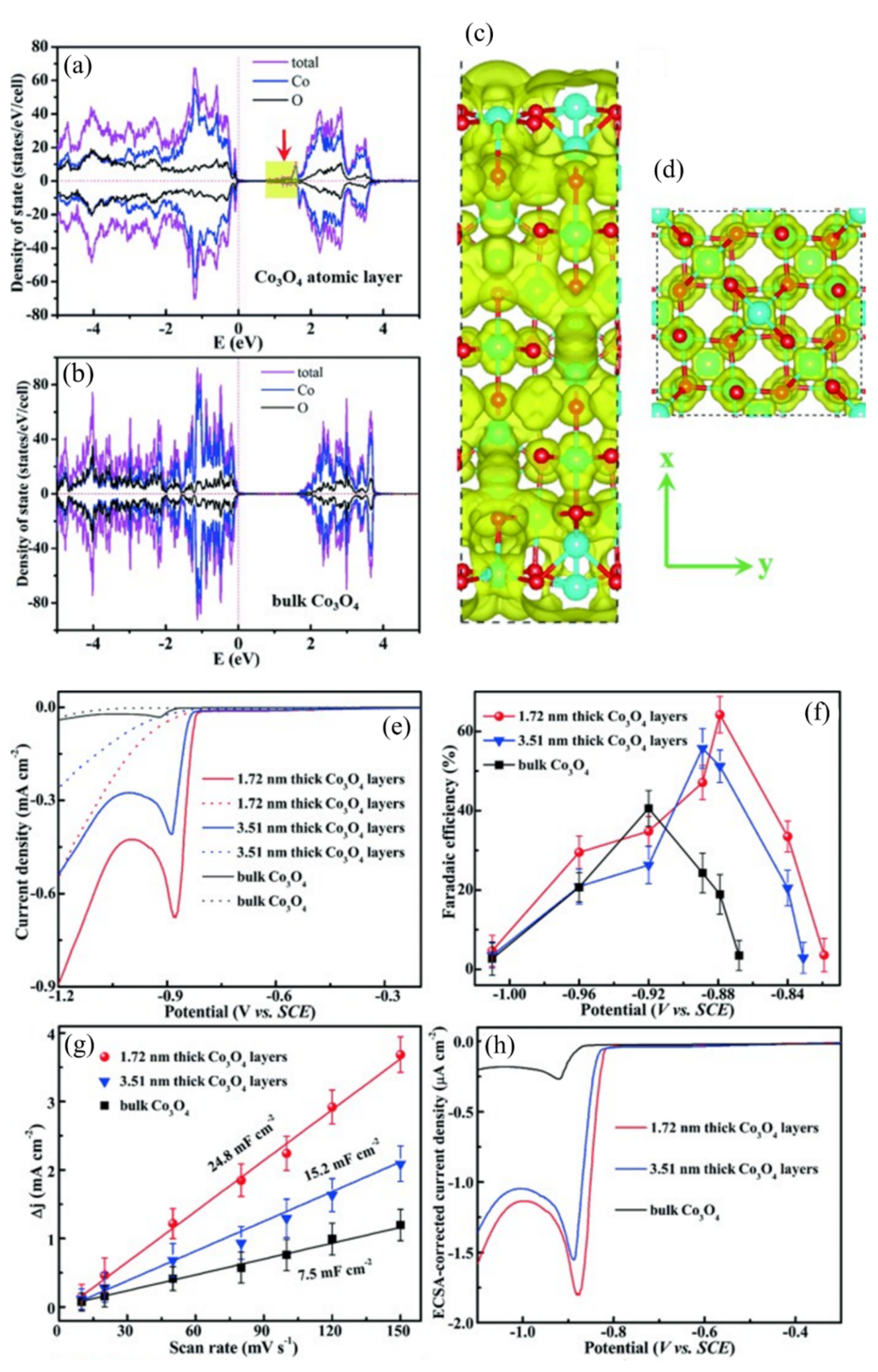{kind=link}
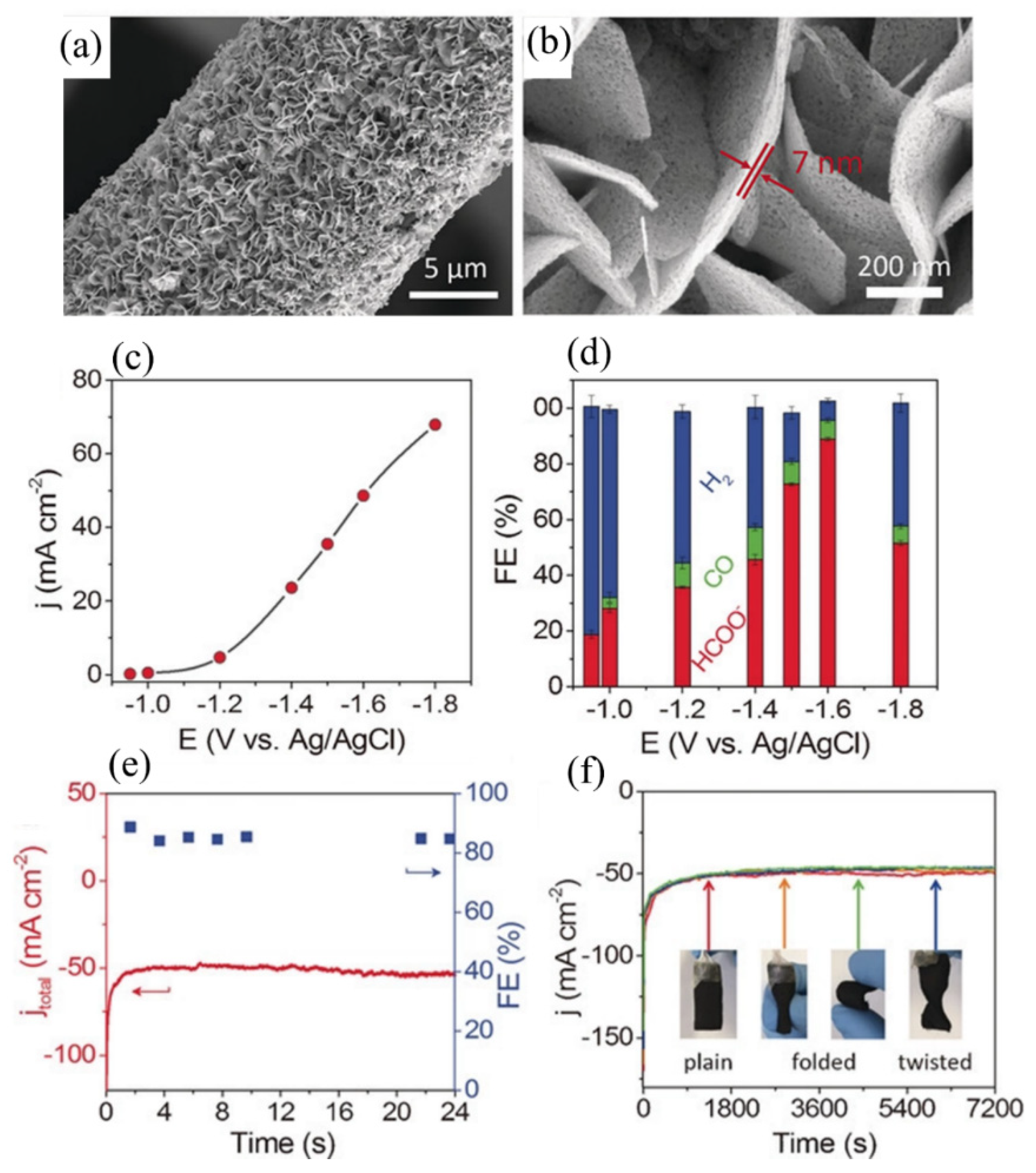{kind=link}
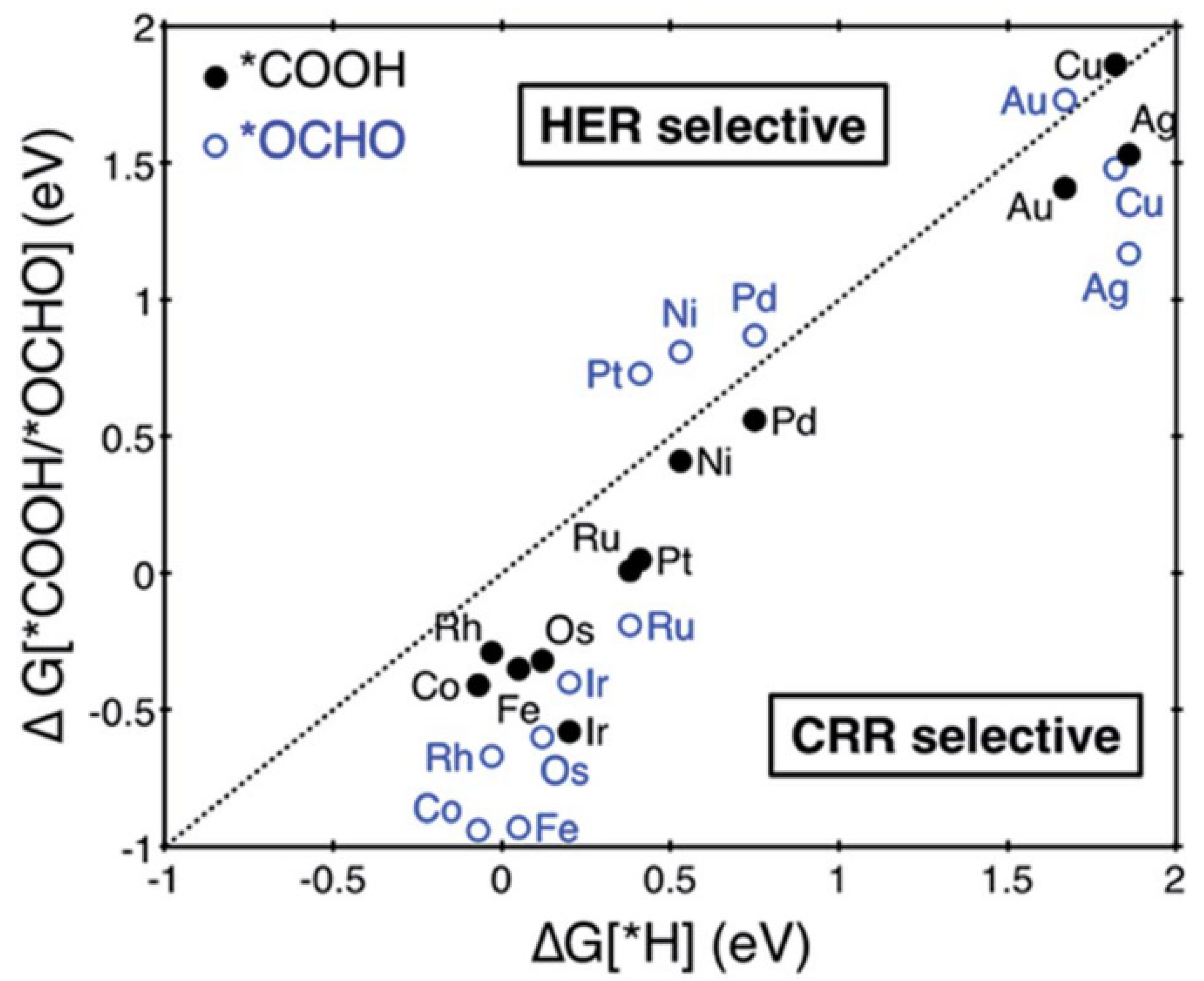{kind=link}
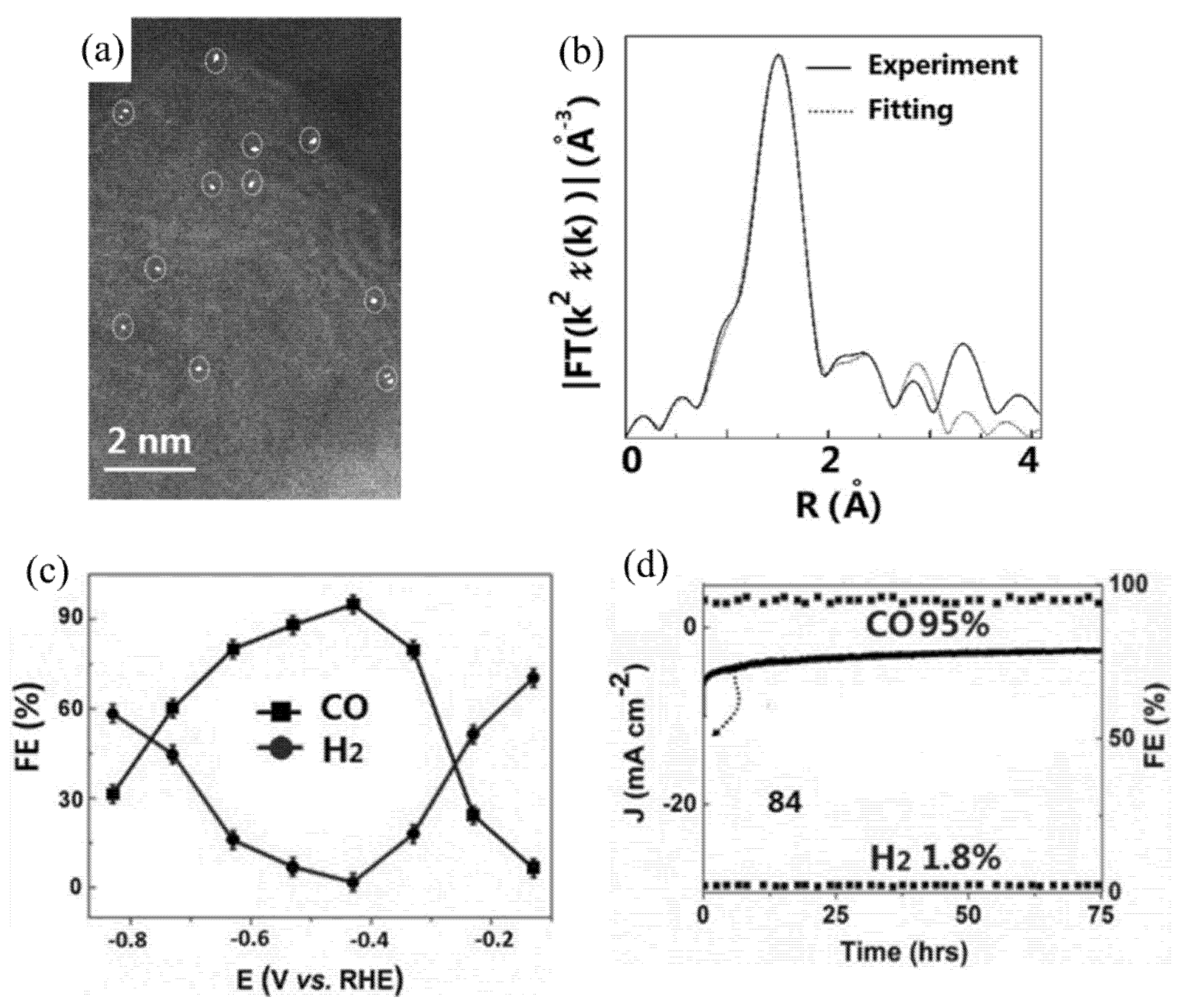{kind=link}
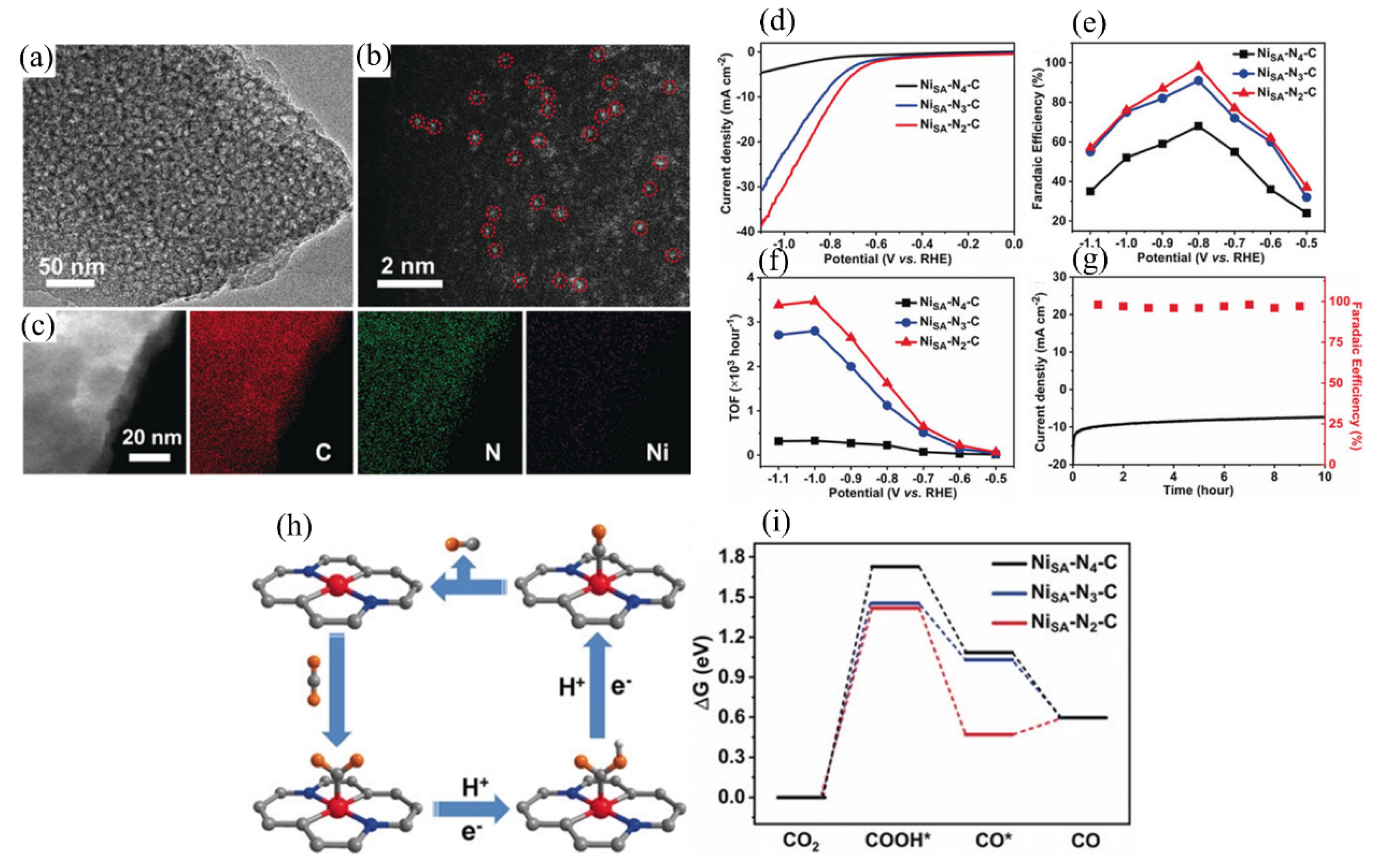{kind=link}
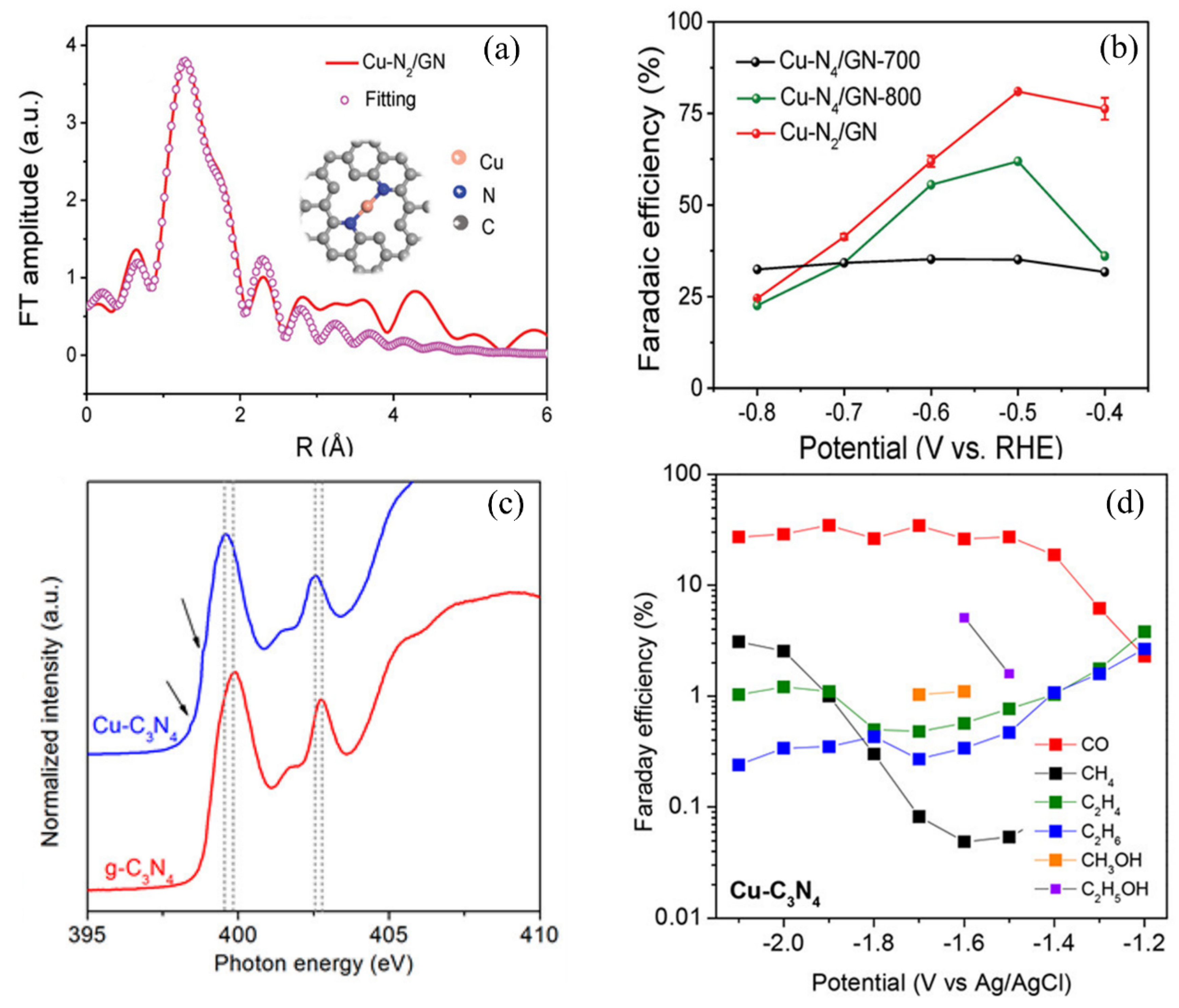{kind=link}
Table 1.
Standard electrochemical potentials for reducing CO2 into different products.
| CO2 Reduction Reactions | Standard Electrode Potentials vs. SHE (V) |
|---|---|
| −1.900 | |
| → HCOOH | −0.610 |
| → CO + H2O | −0.530 |
| → H2C2O4 | −0.913 |
| → HCHO + H2O | −0.480 |
| → CH3OH + H2O | −0.380 |
| → CH4 + 2H2O | −0.240 |
| → C2H4 + 4H2O | −0.349 |
| → C2H5OH + 3H2O | −0.329 |
| → C2H6 + 4H2O | −0.270 |
| → C3H7OH + 5H2O | −0.310 |
Table 2.
Typical electrocatalysts in ECR.
| Electrocatalyst | Product | FE (%) | Potential | Reference |
|---|---|---|---|---|
| Pd-Pt/C | HCOOH | 88 | −0.40 V vs. RHE | [16] |
| nanoporous silver | CO | 92 | −0.50 V vs. RHE | [30] |
| copper foam | CO | 80 | −1.00 V vs. Ag/AgCl | [66] |
| Cu-Al | C2H4 | 80 | −1.50 V vs. RHE | [68] |
| GN/ZnO/Cu2O | C3H7OH | 30 | −0.90 V vs. Ag/AgCl. | [69] |
| graphene-supported MoSx | CO | 85.1 | −0.65 V vs. RHE | [71] |
| Au NW | CO | 94 | −0.35 V vs. RHE | [72] |
| ultrathin Pd nanosheets | CO | 94 | −0.50 V vs. RHE | [73] |
| Pd-Au | CO | 80 | −0.50 V vs. RHE | [74] |
| nitrogen-doped 3D graphene foam | CO | 85 | −0.58 V vs. RHE | [75] |
| ultrathin Bi | HCOOH | 86 | −1.10 V vs. RHE | [76] |
| ultrathin porous Cu nanosheets | CO | 74.1 | −1.00 V vs. RHE | [77] |
| Cu nanocubes | C2H4 | 60 | −0.50 V vs. RHE | [78] |
| B-doped graphene | HCOOH | 66 | −1.40 V vs. SCE | [79] |
| N-doped graphene | HCOOH | 73 | −0.84 vs. RHE | [38] |
| N-doped graphene quantum dots | C2H5OH + C2H4 | 45 | −0.75 V vs. RHE | [59] |
| Au NPs x-embedded graphene nanoribbon | CO | 92 | −0.66 V vs. RHE | [80] |
| Ni-N modified graphene | CO | 90 | −0.70 V vs. RHE | [81] |
| N-doped graphene monolayer-coated Sn foil | HCOOH | 92 | −1.00 V vs. RHE | [82] |
| Sn quantum sheets confined in graphene | HCOOH | 86 | −1.80 V vs. SCE | [83] |
| SnS2 supported on graphene oxide | HCOOH | 84.5 | −1.40 V vs. Ag/AgCl | [84] |
| g–C3N4/MWCNTs | CO | 60 | −0.75 V vs. RHE | [85] |
| Co3O4-CDots-C3N4 | CO | 89 | −0.60 V vs. RHE | [86] |
| Nb-doped vertically aligned MoS2 | CO | 82 | −0.80 V vs. RHE | [87] |
| MoS2 | CO | 98 | −0.764 V vs. RHE | [88] |
| WSe2 nanoflake | CO | 24 | −0.164 V vs. RHE | [89] |
| Cobalt–porphyrin MOF | CO | 76 | −0.70 V vs. RHE | [90] |
| Zn MOF | CO | 71.9 | −0.89 V vs. RHE | [91] |
| Co-based phthalocyanine | CO | 99 | −0.80 V vs. RHE | [92] |
| ultrathin Co3O4 | HCOOH | 64.3 | −0.88 V vs. SCE | [93] |
| oxygen-deficient Co3O4 | HCOOH | 87.6 | −0.87 V vs. SCE | [94] |
| atomic Co3O4 | HCOOH | 90 | −0.85 V vs. SCE | [28] |
| mesoporous SnO2 nanosheets | HCOOH | 87 | −1.60 V vs. Ag/AgCl | [95] |
| Cu-CeO2 | CH4 | 58 | −1.80 V vs. RHE | [96] |
| Fe–N4–C | CO | 80 | −0.50 V vs. RHE | [97] |
| Fe–N4–C | CO | 80 | −0.57 V vs. RHE | [98] |
| Fe–N5–C | CO | 97 | −0.46 V vs. RHE | [99] |
| Fe–N4–C | CO | 94.9 | −0.5 V vs. RHE | [100] |
| Fe3+–N–C | CO | 90 | −0.45 V vs. RHE | [101] |
| Zn–N–C | CO | 95 | −0.43 V vs. RHE | [102] |
| Ni-graphene oxide | CO | 96.5 | −0.63 V vs. RHE | [103] |
| Ni(I)–N4–C | CO | 97 | −0.61 V vs. RHE | [104] |
| Ni–N4–C | CO | 90 | −0.65 V vs. RHE | [105] |
| Ni–N2–C | CO | 98 | −0.80 V vs. RHE | [106] |
| STPyP–Co | CO | 96 | −0.62 V vs. RHE | [107] |
| Cu–N2/GN nanosheets | CO | 81 | −0.50 V vs. RHE | [108] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lu, S.; Lou, F.; Yu, Z. Recent Progress in Two-Dimensional Materials for Electrocatalytic CO2 Reduction. Catalysts 2022, 12, 228. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12020228
AMA Style
Lu S, Lou F, Yu Z. Recent Progress in Two-Dimensional Materials for Electrocatalytic CO2 Reduction. Catalysts. 2022; 12(2):228. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12020228
Chicago/Turabian StyleLu, Song, Fengliu Lou, and Zhixin Yu. 2022. "Recent Progress in Two-Dimensional Materials for Electrocatalytic CO2 Reduction" Catalysts 12, no. 2: 228. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12020228
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.