
Cu Modified TiO2 Catalyst for Electrochemical Reduction of Carbon Dioxide to Methane

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
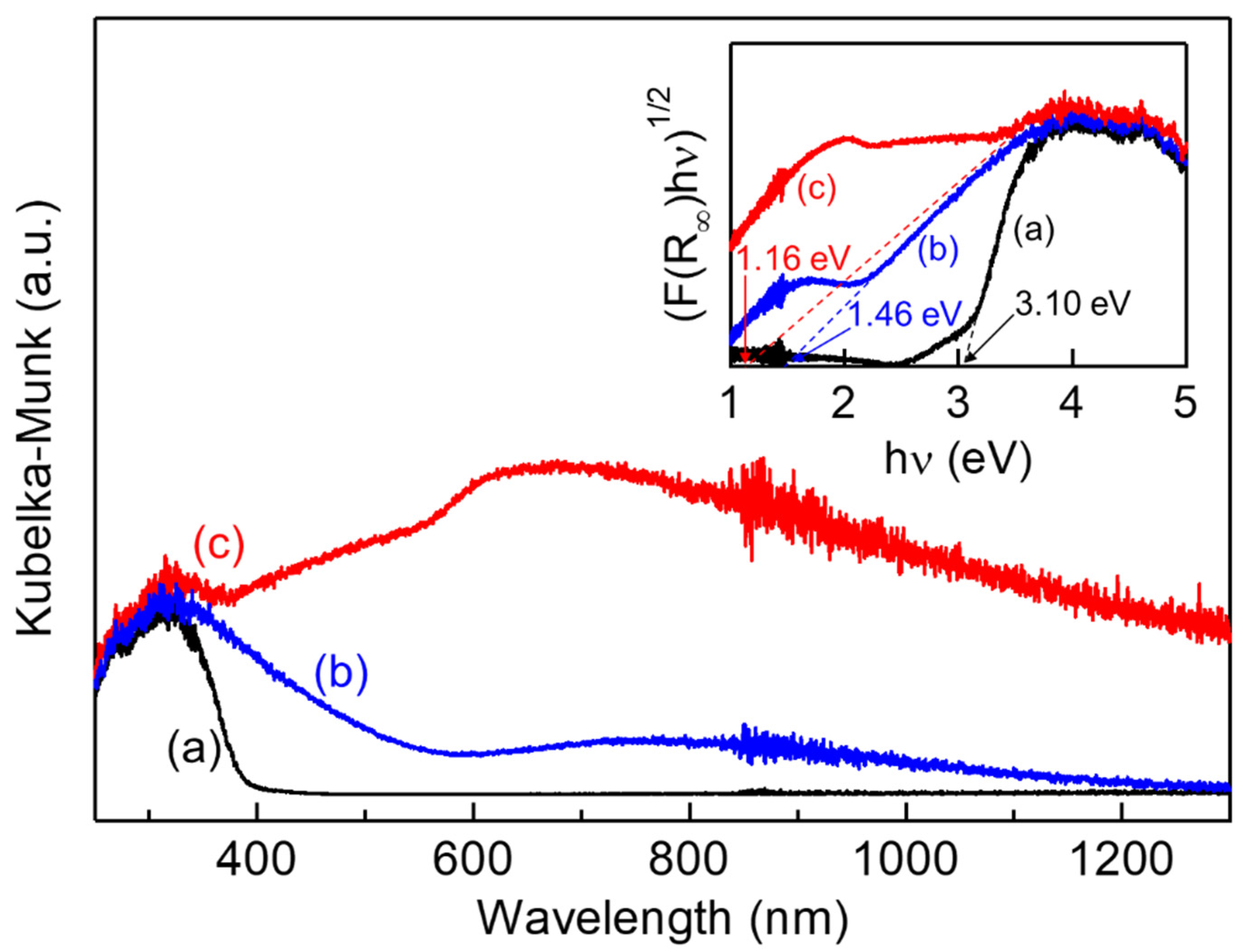
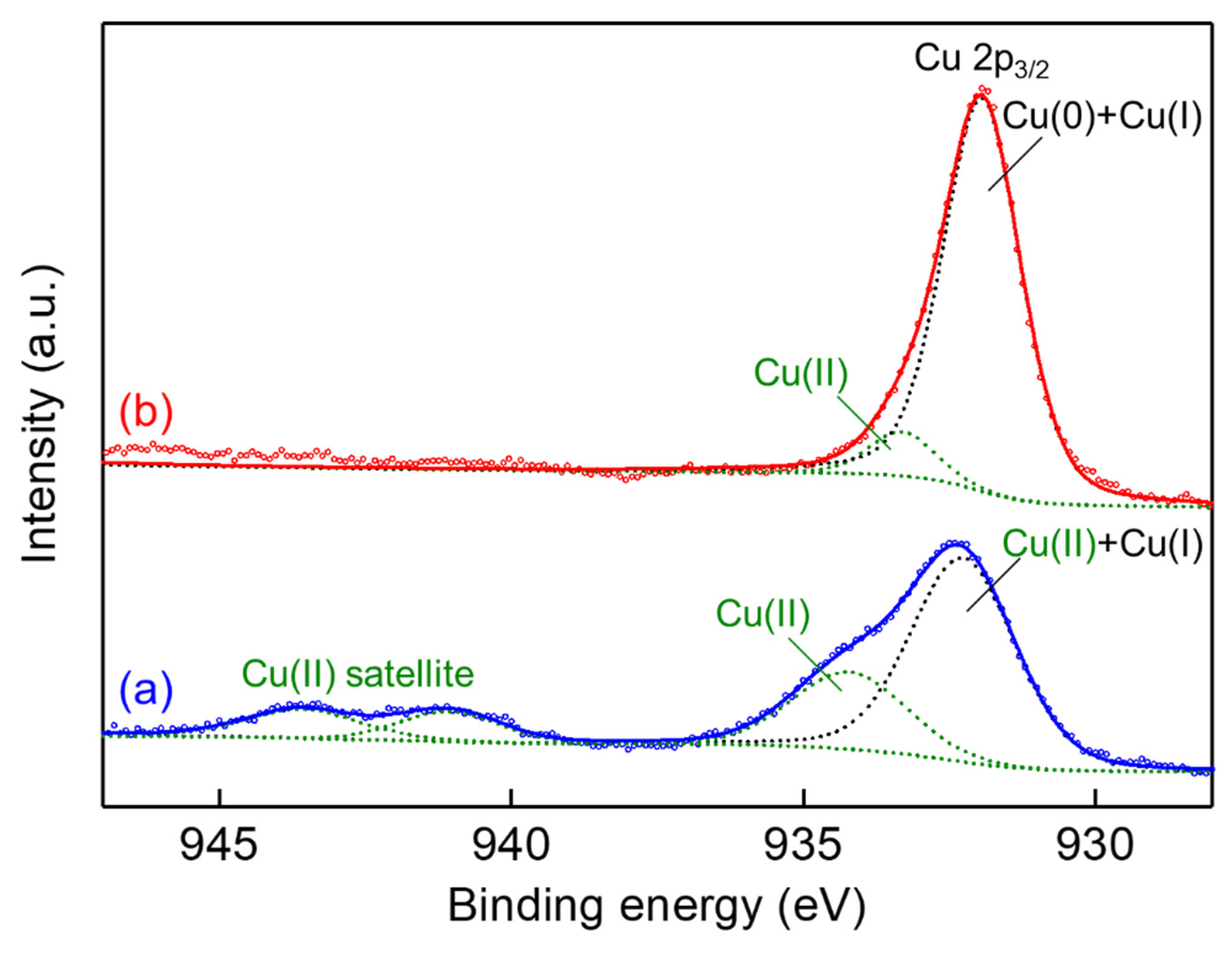
2. Results
3. Materials and Methods
3.1. Materials
3.2. Preparation of Cu-TiO2 Composite Catalysts
3.3. Catalyst Characterization
3.4. Electrode Preparation
3.5. Electrochemical Reduction of CO2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kibria, M.G.; Edwards, J.P.; Gabardo, C.M.; Dinh, C.T.; Seifitokaldani, A.; Sinton, D.; Sargent, E.H. Electrochemical CO2 Reduction into Chemical Feedstocks: From Mechanistic Electrocatalysis Models to System Design. Adv. Mater. 2019, 31, e1807166. [Google Scholar] [CrossRef] [PubMed]
- Nitopi, S.; Bertheussen, E.; Scott, S.B.; Liu, X.; Engstfeld, A.K.; Horch, S.; Seger, B.; Stephens, I.E.L.; Chan, K.; Hahn, C.; et al. Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte. Chem. Rev. 2019, 119, 7610–7672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, A.A.; Abild-Pedersen, F.; Studt, F.; Rossmeisl, J.; Nørskov, J.K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 2010, 3, 1311–1315. [Google Scholar] [CrossRef]
- Wang, Y.F.; Chen, Z.; Han, P.; Du, Y.H.; Gu, Z.X.; Xu, X.; Zheng, G.F. Single-Atomic Cu with Multiple Oxygen Vacancies on Ceria for Electrocatalytic CO2 Reduction to CH4. ACS Catal. 2018, 8, 7113–7119. [Google Scholar] [CrossRef]
- Xu, Y.; Li, F.W.; Xu, A.N.; Edwards, J.P.; Hung, S.F.; Gabardo, C.M.; O’Brien, C.P.; Liu, S.J.; Wang, X.; Li, Y.H.; et al. Low coordination number copper catalysts for electrochemical CO2 methanation in a membrane electrode assembly. Nat. Commun. 2021, 12, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Varela, A.S.; Ju, W.; Strasser, P. Molecular Nitrogen–Carbon Catalysts, Solid Metal Organic Framework Catalysts, and Solid Metal/Nitrogen-Doped Carbon (MNC) Catalysts for the Electrochemical CO2 Reduction. Adv. Energy Mater. 2018, 8, 1703614. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.L.; Gao, Z.Y.; Wei, F.Y.; Liu, C.; Gong, J.; Li, J.M.; Li, W.Z.; Li, X.; Wang, G.W.; Lu, J.T.; et al. Customizable CO2 Electroreduction to C1 or C2+ Products through Cuy/CeO2 Interface Engineering. ACS Catal. 2022, 12, 1004–1011. [Google Scholar] [CrossRef]
- Gao, D.F.; Zhang, Y.; Zhou, Z.W.; Cai, F.; Zhao, X.F.; Huang, W.; Li, Y.S.; Zhu, J.F.; Liu, P.; Yang, F.; et al. Enhancing CO2 Electroreduction with the Metal–Oxide Interface. J. Am. Chem. Soc. 2017, 139, 5652–5655. [Google Scholar] [CrossRef]
- Nakata, K.; Fujishima, A. TiO2 photocatalysis: Design and applications. J. Photochem. Photobiol. C Photochem. Rev. 2012, 13, 169–189. [Google Scholar] [CrossRef]
- Watanabe, R.; Yamauchi, M.; Sadakiyo, M.; Abe, R.; Takeguchi, T. CO2-free electric power circulation via direct charge and discharge using the glycolic acid/oxalic acid redox couple. Energy Environ. Sci. 2015, 8, 1456–1462. [Google Scholar] [CrossRef]
- Sadakiyo, M.; Hata, S.; Cui, X.; Yamauchi, M. Electrochemical Production of Glycolic Acid from Oxalic Acid Using a Polymer Electrolyte Alcohol Electrosynthesis Cell Containing a Porous TiO2 Catalyst. Sci. Rep. 2017, 7, 17032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadakiyo, M.; Hata, S.; Fukushima, T.; Juhász, G.; Yamauchi, M. Electrochemical hydrogenation of non-aromatic carboxylic acid derivatives as a sustainable synthesis process: From catalyst design to device construction. Phys. Chem. Chem. Phys. 2019, 21, 5882–5889. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, M.; Hata, S.; Eguchi, H.; Kitano, S.; Fukushima, T.; Higashi, M.; Sadakiyo, M.; Kato, K. Catalytic enhancement on Ti-Zr complex oxide particles for electrochemical hydrogenation of oxalic acid to produce an alcoholic compound by controlling electronic states and oxide structures. Catal. Sci. Technol. 2019, 9, 6561. [Google Scholar] [CrossRef]
- Fukushima, T.; Yamauchi, M. Electrosynthesis of amino acids from biomass-derivable acids on titanium dioxide. Chem. Commun. 2019, 55, 14721–14724. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, M. Inorganic Nanocatalysts for Hydrogenation Reactions Contributable to a Sustainable Material Supply. Chem. Lett. 2021, 50, 1901–1908. [Google Scholar] [CrossRef]
- Eguchi, H.; Kato, K.; Juhasz, G.; Yamauchi, M. Selectivity enhancement in the electrochemical reduction of oxalic acid over titanium dioxide nanoparticles achieved by shape and energy-state control. Catal. Sci. Technol. 2021, 11, 7592–7597. [Google Scholar] [CrossRef]
- Isegawa, M.; Staykov, A.; Yamauchi, M. Proton-Coupled Electron Transfer in Electrochemical Alanine Formation from Pyruvic Acid: Mechanism of Catalytic Reaction at the Interface between TiO2(101) and Water. J. Phys. Chem. C 2021, 125, 12603–12613. [Google Scholar] [CrossRef]
- Fukutani, K.; Yoshinobu, J.; Yamauchi, M.; Shima, T.; Orimo, S. Hydrogenomics: Efficient and Selective Hydrogenation of Stable Molecules Utilizing Three Aspects of Hydrogen. Catal. Lett. 2021. [Google Scholar] [CrossRef]
- Paola, A.D.; Bellardita, M.; Palmisano, L. Brookite, the Least Known TiO2 Photocatalyst. Catalysts 2013, 3, 36–73. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Chen, F.; Jiao, Y.; Zhang, J. Phase transition and morphological evolution of titania/titanate nanomaterials under alkalescent hydrothermal treatment. J. Mater. Chem. 2010, 20, 7990–7997. [Google Scholar] [CrossRef]
- Haider, Z.; Kang, Y.S. Facile Preparation of Hierarchical TiO2 Nano Structures: Growth Mechanism and Enhanced Photocatalytic H2 Production from Water Splitting Using Methanol as a Sacrificial Reagent. ACS Appl. Mater. Interfaces 2014, 6, 10342–10352. [Google Scholar] [CrossRef] [PubMed]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst. 1976, A32, 751–767. [Google Scholar] [CrossRef]
- Tauc, J.; Grigorovici, R.; Vancu, A. Optical Properties and Electronic Structure of Amorphous Germanium. Phys. Status Solidi 1966, 15, 627–637. [Google Scholar] [CrossRef]
- Davis, E.A.; Mott, N.F. Conduction in non-crystalline systems V. Conductivity, optical absorption and photoconductivity in amorphous semiconductors. Philos. Mag. 1970, 22, 903–922. [Google Scholar] [CrossRef]
- Reyes-Coronado, D.; Rodríguez-Gattorno, G.; Espinosa-Pesqueira, M.E.; Cab, C.; De Coss, R.; Oskam, G. Phase-pure TiO2 nanoparticles: Anatase, brookite and rutile. Nanotechnology 2008, 19, 145605. [Google Scholar] [CrossRef]
- Choudhury, B.; Dey, M.; Choudhury, A. Defect generation, d-d transition, and band gap reduction in Cu-doped TiO2 nanoparticles. Int. Nano Lett. 2013, 3, 25. [Google Scholar] [CrossRef] [Green Version]
- Pascher, T.F.; Ončák, M.; van der Linde, C.; Beyer, M.K. UV/Vis Spectroscopy of Copper Formate Clusters: Insight into Metal-Ligand Photochemistry. Chem.-A Eur. J. 2020, 26, 8286–8295. [Google Scholar] [CrossRef]
- Desario, P.A.; Pietron, J.J.; Brintlinger, T.H.; McEntee, M.; Parker, J.F.; Baturina, O.; Stroud, R.M.; Rolison, D.R. Oxidation-stable plasmonic copper nanoparticles in photocatalytic TiO2 nanoarchitectures. Nanoscale 2017, 9, 11720–11729. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, M.; Hikino, S.; Tanabe, R.; Sano, Y. Synthesis of Bicompartmental Ag/Cu Nanoparticles Using a Two-step Polyol Process. Chem. Lett. 2009, 38, 860–861. [Google Scholar] [CrossRef]
- Amendola, V.; Meneghetti, M. Laser ablation synthesis in solution and size manipulation of noble metal nanoparticles. Phys. Chem. Chem. Phys. 2009, 11, 3805–3821. [Google Scholar] [CrossRef]
- Cano, E.; Torres, C.L.; Bastidas, J.M. An XPS study of copper corrosion originated by formic acid vapour at 40% and 80% relative humidity. Mater. Corros. 2001, 52, 667–676. [Google Scholar] [CrossRef]
- Ethiraj, A.S.; Kang, D.J. Synthesis and characterization of CuO nanowires by a simple wet chemical method. Nanoscale Res. Lett. 2012, 7, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biesinger, M.C.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S.C. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Sc, Ti, V, Cu and Zn. Appl. Surf. Sci. 2010, 257, 887–898. [Google Scholar] [CrossRef]
- Idriss, H. On the wrong assignment of the XPS O1s signal at 531–532 eV attributed to oxygen vacancies in photo- and electro-catalysts for water splitting and other materials applications. Surf. Sci. 2021, 712, 121894. [Google Scholar] [CrossRef]
- Kenausis, G.L.; Vörös, J.; Elbert, D.L.; Huang, N.; Hofer, R.; Ruiz-Taylor, L.; Textor, M.; Hubbell, J.A.; Spencer, N.D. Poly(L-lysine)-g-poly(ethylene glycol) layers on metal oxide surfaces: Attachment mechanism and effects of polymer architecture on resistance to protein adsorption. J. Phys. Chem. B 2000, 104, 3298–3309. [Google Scholar] [CrossRef]
- Chalastara, K.; Guo, F.; Elouatik, S.; Demopoulos, G.P. Tunable Composition Aqueous-Synthesized. Catalyst 2020, 10, 407. [Google Scholar] [CrossRef] [Green Version]
- Itai, T.; Takahashi, Y.; Uruga, T.; Tanida, H.; Iida, A. Selective detection of Fe and Mn species at mineral surfaces in weathered granite by conversion electron yield X-ray absorption fine structure. Appl. Geochem. 2008, 23, 2667–2675. [Google Scholar] [CrossRef]
- DuBois, J.L.; Mukherjee, P.; Stack, T.D.P.; Hedman, B.; Solomon, E.I.; Hodgson, K.O. A systematic K-edge X-ray absorption spectroscopic study of Cu(III) sites. J. Am. Chem. Soc. 2000, 122, 5775–5787. [Google Scholar] [CrossRef]
- Todaro, M.; Sciortino, L.; Gelardi, F.M.; Buscarino, G. Determination of geometry arrangement of copper ions in HKUST-1 by XAFS during a prolonged exposure to air. J. Phys. Chem. C 2017, 121, 24853–24860. [Google Scholar] [CrossRef]
- Prestipino, C.; Regli, L.; Vitillo, J.G.; Bonino, F.; Damin, A.; Lamberti, C.; Zecchina, A.; Solari, P.L.; Kongshaug, K.O.; Bordiga, S. Local structure of framework Cu(II) in HKUST-1 metallorganic framework: Spectroscopic characterization upon activation and interaction with adsorbates. Chem. Mater. 2006, 18, 1337–1346. [Google Scholar] [CrossRef]
- Shinagawa, T.; Larrazábal, G.O.; Martín, A.J.; Krumeich, F.; Pérez-Ramírez, J. Sulfur-Modified Copper Catalysts for the Electrochemical Reduction of Carbon Dioxide to Formate. ACS Catal. 2018, 8, 837–844. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Dong, W.J.; Gim, S.; Sohn, W.; Park, J.Y.; Yoo, C.J.; Jang, J.Y.; Lee, J.-L. Shape-controlled bismuth nanoflakes as highly selective catalysts for electrochemical carbon dioxide reduction to formate. Nano Energy 2017, 39, 44–52. [Google Scholar] [CrossRef]
- Zheng, T.T.; Jiang, K.; Ta, N.; Hu, Y.F.; Zeng, J.; Liu, J.Y.; Wang, H.T. Large-Scale and Highly Selective CO2 Electrocatalytic Reduction on Nickel Single-Atom Catalyst. Joule 2019, 3, 265–278. [Google Scholar] [CrossRef] [Green Version]
- Xue, L.; Zhang, C.; Wu, J.; Fan, Q.Y.; Liu, Y.; Wu, Y.; Li, J.; Zhang, H.; Liu, F.; Zeng, S. Unveiling the reaction pathway on Cu/CeO2 catalyst for electrocatalytic CO2 reduction to CH4. Appl. Catal. B Environ. 2022, 304, 120951. [Google Scholar] [CrossRef]
- Varandili, S.B.; Huang, J.; Oveisi, E.; Gregorio, G.L.D.; Mensi, M.; Strach, M.; Vavra, J.; Gadiyar, C.; Bhowmik, A.; Buonsanti, R. Synthesis of Cu/CeO2−x Nanocrystalline Heterodimers with Interfacial Active Sites To Promote CO2 Electroreduction. ACS Catal. 2019, 9, 5035–5046. [Google Scholar] [CrossRef]
- Huygh, S.; Bogaerts, A.; Neyts, E.C. How Oxygen Vacancies Activate CO2 Dissociation on TiO2 Anatase (001). J. Phys. Chem. C 2016, 120, 21659–21669. [Google Scholar] [CrossRef]
- Kato, K.; Tanaka, H. Visualizing charge densities and electrostatic potentials in materials by synchrotron X-ray powder diffraction. Adv. Phys. X 2016, 1, 55–80. [Google Scholar] [CrossRef] [Green Version]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef] [Green Version]
- Niu, S.; Li, S.; Du, Y.; Han, X.; Xu, P. How to Reliably Report the Overpotential of an Electrocatalyst. ACS Energy Lett. 2020, 5, 1083–1087. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Verma, S.; Kim, S.; Fister, T.T.; Kenis, P.J.A.; Gewirth, A.A. Highly dispersed, single-site copper catalysts for the electroreduction of CO2 to methane. J. Electroanal. Chem. 2020, 875, 113862. [Google Scholar] [CrossRef]
- Guan, A.X.; Chen, Z.; Quan, Y.L.; Peng, C.; Wang, Z.Q.; Sham, T.-K.; Yang, C.; Ji, Y.L.; Qian, L.P.; Xu, X.; et al. Boosting CO2 Electroreduction to CH4 via Tuning Neighboring Single-Copper Sites. ACS Energy Lett. 2020, 5, 1044–1053. [Google Scholar] [CrossRef]
- Chen, S.H.; Wang, B.Q.; Zhu, J.X.; Wang, L.Q.; Ou, H.H.; Zhang, Z.D.; Liang, X.; Zheng, L.R.; Zhou, L.; Su, Y.-Q.; et al. Lewis Acid Site-Promoted Single-Atomic Cu Catalyzes Electrochemical CO2 Methanation. Nano Lett. 2021, 21, 7325–7331. [Google Scholar] [CrossRef] [PubMed]
- Guan, A.X.; Yang, C.; Wang, Q.H.; Qian, L.P.; Cao, J.Y.; Zhang, L.J.; Wu, M.L.; Zheng, G.F.; Zhou, L.; Su, Y.-Q.; et al. DAtomic-Level Copper Sites for Selective CO2 Electroreduction to Hydrocarbon. ACS Sustain. Chem. Eng. 2021, 9, 13536–13544. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anzai, A.; Liu, M.-H.; Ura, K.; Noguchi, T.G.; Yoshizawa, A.; Kato, K.; Sugiyama, T.; Yamauchi, M. Cu Modified TiO2 Catalyst for Electrochemical Reduction of Carbon Dioxide to Methane. Catalysts 2022, 12, 478. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12050478
Anzai A, Liu M-H, Ura K, Noguchi TG, Yoshizawa A, Kato K, Sugiyama T, Yamauchi M. Cu Modified TiO2 Catalyst for Electrochemical Reduction of Carbon Dioxide to Methane. Catalysts. 2022; 12(5):478. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12050478
Chicago/Turabian StyleAnzai, Akihiko, Ming-Han Liu, Kenjiro Ura, Tomohiro G. Noguchi, Akina Yoshizawa, Kenichi Kato, Takeharu Sugiyama, and Miho Yamauchi. 2022. "Cu Modified TiO2 Catalyst for Electrochemical Reduction of Carbon Dioxide to Methane" Catalysts 12, no. 5: 478. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12050478