
Biosynthesis of Pyrrole-2-carbaldehyde via Enzymatic CO2 Fixation


Manchester Institute of Biotechnology, University of Manchester, 131 Princess Street, Manchester M1 7DN, UK
*
Author to whom correspondence should be addressed.
†
Current address: Chemistry Research Laboratory, University of Oxford, Oxford OX1 3TA, UK.
Catalysts 2022, 12(5), 538; https://0-doi-org.brum.beds.ac.uk/10.3390/catal12050538
Submission received: 15 March 2022
/
Revised: 29 April 2022
/
Accepted: 6 May 2022
/
Published: 14 May 2022
(This article belongs to the Special Issue CO2 Catalytic Conversion and Utilization)
Abstract
:The use of CO2 as a chemical building block is of considerable interest. To achieve carbon fixation at ambient conditions, (de)carboxylase enzymes offer an attractive route but frequently require elevated [CO2] levels to yield the acid product. However, it has recently been shown that the coupling of a UbiD-type decarboxylase with carboxylic acid reductase yields the corresponding aldehyde product at near ambient [CO2]. Here, we show this approach can be expanded to different UbiD and CAR enzymes to yield alternative products, in this case, the production of pyrrole-2-carbaldehyde from pyrrole, using Pseudomonas aeruginosa HudA/PA0254 in combination with Segniliparus rotundus CAR. This confirms the varied substrate range of the respective UbiD and CAR enzymes can be harnessed in distinct combinations to support production of a wide range of aldehydes via enzymatic CO2 fixation.
1. Introduction
The traditional organic chemistry synthesis required for the manufacture of pharmaceuticals and fine chemicals often relies on the application of metal-based catalysis as well as petrochemical feedstocks. These resources are often energy intensive and environmentally damaging, both in their application and their extraction [1]. The use of (waste) CO2 as a building block to produce high value compounds and pharmaceutical precursors is of considerable interest, given the potential green chemistry credentials of such processes. However, the high-activation-energy barriers of C-H activation, and subsequent carboxylation, present a formidable challenge, requiring high pressures and harsh reaction conditions [2].
In contrast, biological CO2 fixation offers a sustainable route to a wide range of compounds, and recent work has identified several enzyme classes capable of C-H activation and (de)carboxylation [3]. One such class is the UbiD family, a group of enzymes that rely on the modified flavin cofactor prFMN to perform the reversible (de)carboxylation of a range of aromatic and aliphatic poly-unsaturated carboxylic acids [4]. While the inherent thermodynamics usually strongly favor the decarboxylative reaction [5,6], at increased [CO2] these enzymes can be used to achieve C-H activation through carboxylation at ambient conditions.
In an alternative, bioinspired approach, it has been demonstrated that by coupling this prFMN-dependent class of UbiD (de)carboxylases with carboxylic acid reductase (CAR), it is possible to overcome the inherent thermodynamic bias towards decarboxylation, even at ambient [CO2]. Indeed, combining fungal Fdc1 with CAR yields carboxylation of terminal alkenes and concomitant reduction of the intermediate carboxylic acids, to produce value-added aldehydes [7].
However, Fdc1 type enzymes have limited substrate specificity compared to that seen across the collective UbiD family, with a substrate preference largely consisting of cinnamic acid derivatives, or short chain polyunsaturated aliphatic acids [5]. The varied substrate range of the UbiD family suggests that coupling distinct family members to CAR can offer a putative route to other aldehyde products. The recently characterised Pseudomonas aeruginosa PA0254 has been shown to catalyse the interconversion of pyrrole and pyrrole-2-carboxylic acid. With the introduction of a single point mutation, it is possible to broaden the substrate scope of PA0254 to include furan and thiophene type substrates [8].
Five-member heterocyclic aldehydes such as furfural, thiophene-, and pyrrole-2-carbaldehyde have been found to be useful building blocks in the synthesis of high value compounds such as polymers, biochemical dyes, and pharmaceuticals [9,10,11]. For example, pyrrole-2-carbaldehyde has been utilised in the production of the BODIPY family of fluorescent dyes [9], while furfural has applications in resin production, herbicides, and hypergolic fuels [10]. Thiophene-2-carbaldehyde has been used in the purification of palladium from aqueous solutions [11].
Hence, a system coupling PA0254 with a suitable CAR could, in principle, yield valuable heteroaromatic aldehydes and present an attractive route to this class of compounds. However, to our knowledge, no CAR enzyme has been reported to reduce pyrrole-2-carboxylic acid or the furan and thiophene counterparts [12,13,14,15,16].
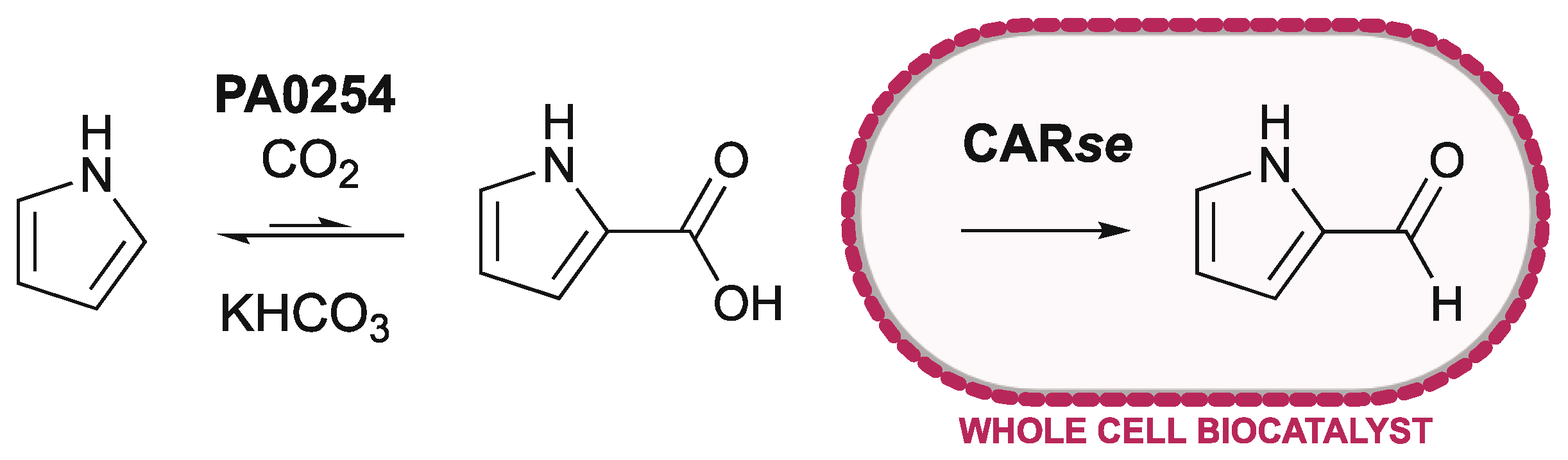
We report that carboxylic acid reductase from Segniliparus rotundus (CARse) is capable of the reduction of pyrrole-, furan- and thiophene-2-carboxylic acids to their corresponding aldehydes. Furthermore, we demonstrate that coupling with PA0254 supports a one-pot biocatalytic method for the carboxylation and subsequent reduction of pyrrole to form pyrrole-2-carbaldehyde (Figure 1).
2. Results and Discussion
2.1. Screening of Carboxylic Acid Reductases for Activity towards Five-Membered Heterocyclic Carboxylic Acids
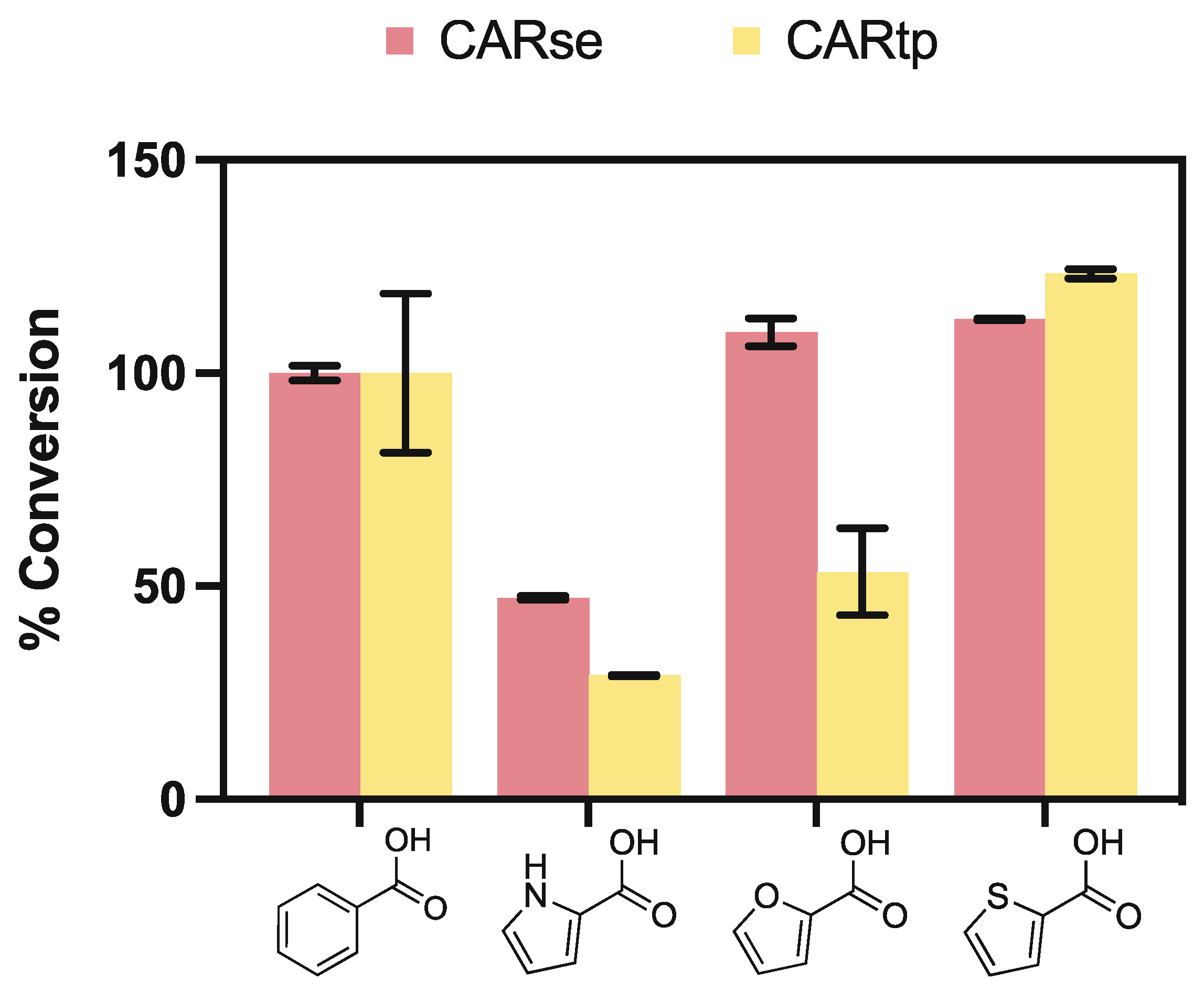
To obtain a suitable CAR with activity with pyrrole-, furan-, and thiophene-2-carboxylic acids, we tested two specific CARs. CARse was selected for its previously described activity towards heteroaromatic carboxylic acids, functionalised at the third position [16]. In addition, Tsukamurella paurometabola CAR (CARtp) was selected, as previously utilised in UbiD-CAR-linked reactions [7]. Both enzymes exhibited activity with all three carboxylic acid substrates, when assayed with 5 mM substrate in the presence of 10 mM each of ATP and NADPH in 100 mM Tris buffer pH 7.5 (Figure 2). The comparative activity observed with furan-2-carboxylic acid resembled that previously observed with the 3-carboxylic acid isomer [16], with CARse displaying a relative 56% increase in furfural yield compared to CARte.
However, comparative substrate specificity differed for isomers of the pyrrole and thiophene class compounds, with CARse providing an ~18% higher yield of pyrrole-2-carbaldehyde over CARtp, while ~11% less thiophene-2-carboxaldehyde was produced by CARse versus CARtp. This appears to be the reverse of previously reported data for isomers carboxylated at the third position, where CARse displayed markedly lower activity in comparison to CARtp, while providing higher levels of reduction for thiophene-3-carboxylic acid [16]. We suspect that the differing substrate specificity observed between these two homologues is due to minor amino acid changes in the enzyme active site or substrate access tunnel, however, additional investigation may clarify this further.
2.2. Coupling of Purified PA0254 and CARse
To test the initial feasibility of the proposed one-pot carboxylation of heteroaromatics, we combined purified PA0254 and CARse and performed a pyrrole carboxylation and reduction reaction at a 500 µL scale in the presence of 2x excess of ATP and NADPH cofactors and 500 mM KHCO3. This resulted in an average production of 0.89 ± 0.10 mM of pyrrole-2-carbaldehyde from 10 mM pyrrole substrate, equating to, approximately, 9% conversion.
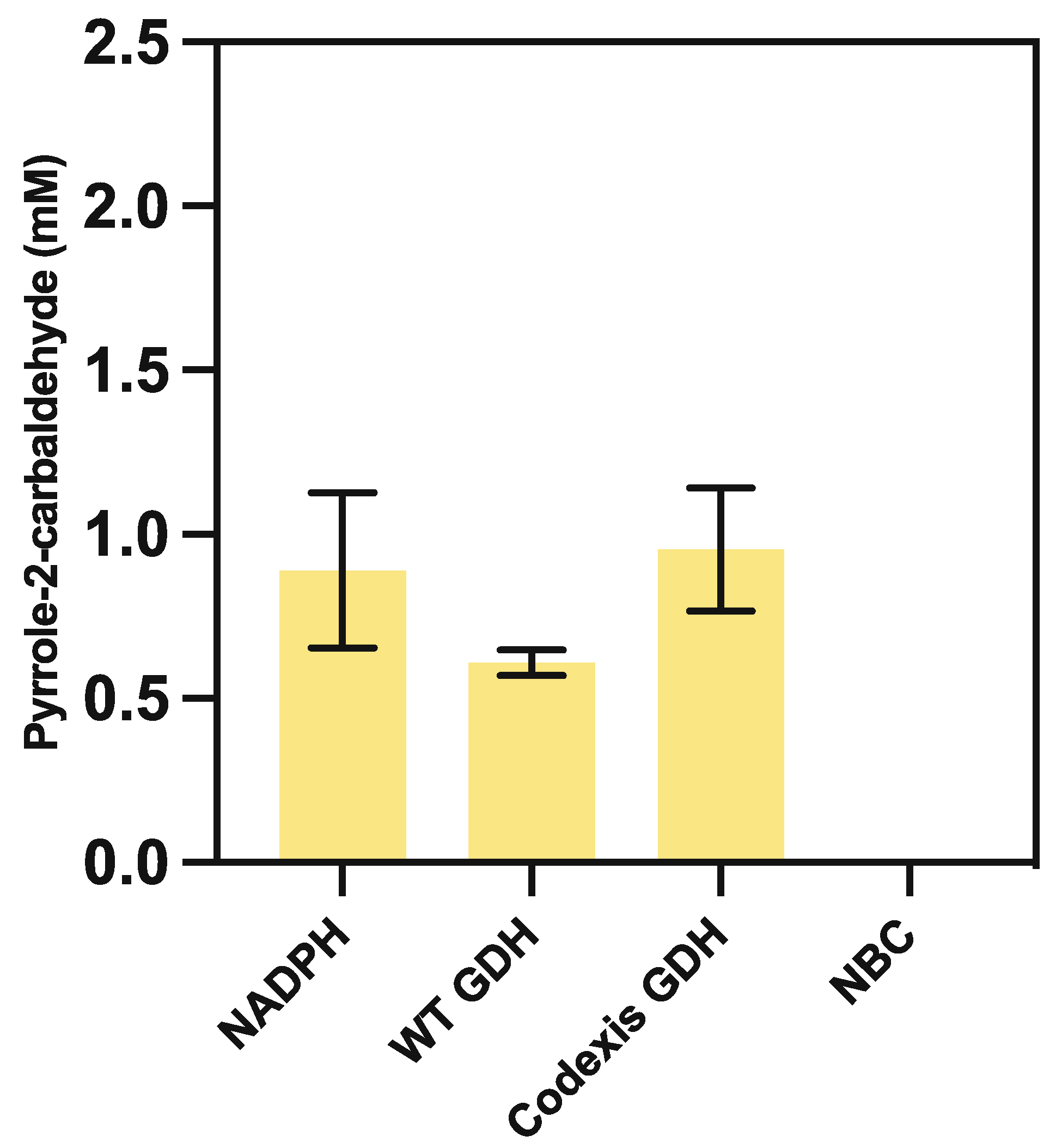
Following the initial successful production of our target aldehyde, we performed a series of iterative optimisation assays, with the aim of improved yield and lowered reaction cost. Our initial target was the reliance of CARse on the reduced nicotinamide NADPH cofactor, as this poses the largest reagent cost factor within this system. As such, we tested the introduction of a nicotinamide recycling system, using glucose dehydrogenase (GDH) to reduce NADP+ at the expense of glucose. This showed that a comparable average product yield of 0.93 ± 0.11 mM could be obtained via supplementation of 0.25 mM NADPH in the presence of a commercially available GDH enzyme obtained from Codexis Inc. (Figure 3). In comparison, similar trials using a wildtype GDH from Bacillus subtilis only yielded just 0.60 ± 0.11 mM of product.
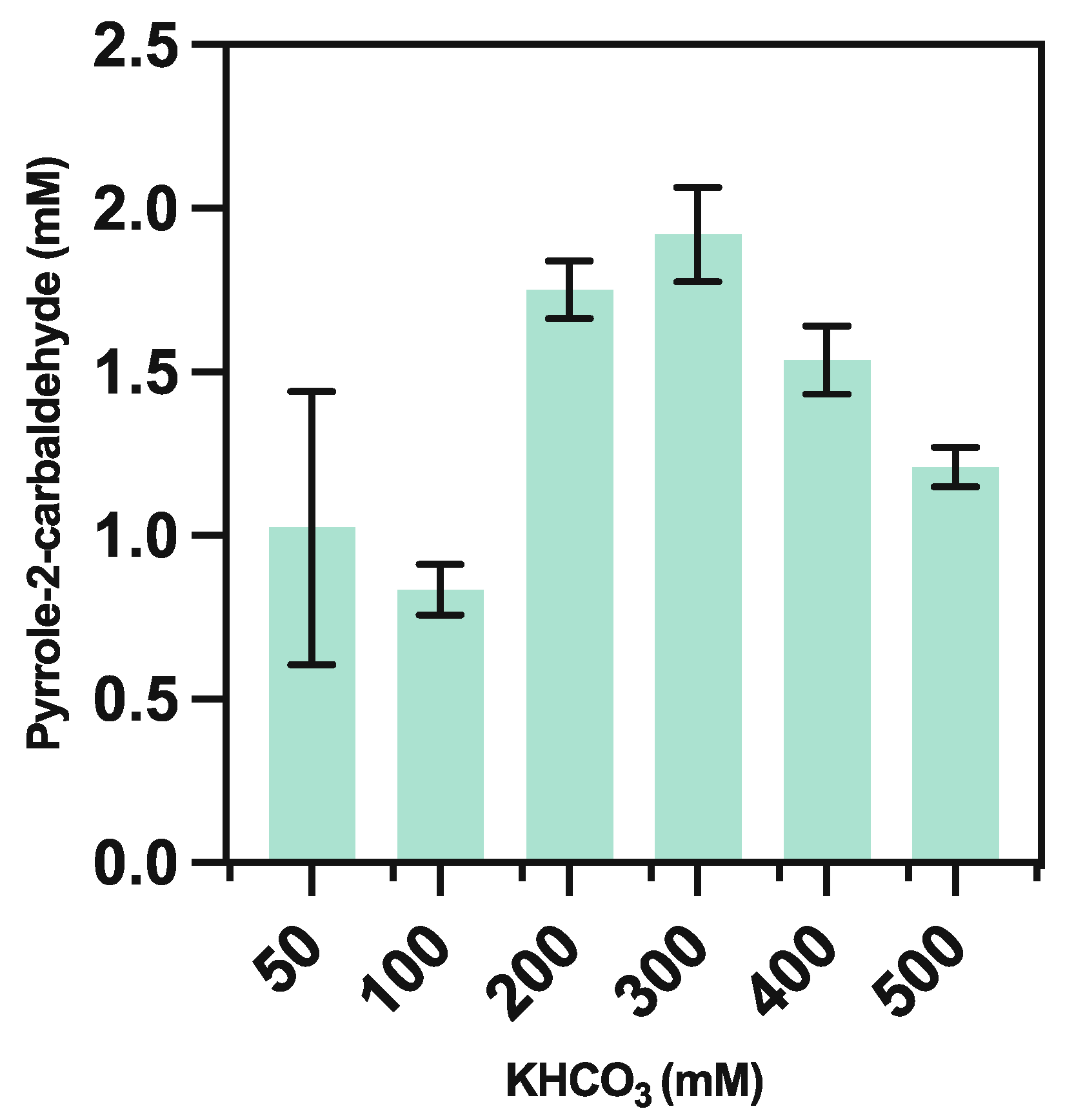
We next focused the optimization of the supplemented bicarbonate, as prior work has shown that the optimum bicarbonate concentration is highly specific to the carboxylase enzyme in use [3]. We found that altering the concentration of KHCO3 improved the previously obtained yield by a factor of ~2, equating to 1.90 ± 0.15 mM of the target aldehyde at 300 mM of bicarbonate salt (Figure 4).
We surmise that the decreased activity at an even higher bicarbonate concentration may be due to an inhibitory effect on CARse-mediated catalysis or by the accompanying pH change resultant from the addition. Finally, the introduction of an ATP recycling system derived from Acinetobacter johnsonii [17], later trailed to further reduce the reliance of the reaction on supplemented cofactors, resulted in a 9% reduction in aldehyde yield (Figure 5).
2.3. Using Whole-Cell CARse Biocatalysts
While the introduction of a nicotinamide recycling system enabled a significant reduction in reaction cost, the <20% average yield of aldehyde obtained with a purified biocatalyst system limits the cost effectiveness of this approach, due to the expense incurred to produce the purified enzyme. Therefore, as an alternative approach, we trailed the replacement of purified CARse biocatalyst with whole E. coli cells, expressing both CARse and the phosphopantetheine-transferase BsSfp. This not only served to eliminate the requirement for both supplemented NADPH and ATP cofactors and their associated recycling systems, but also resulted in a comparable yield of 2.10 ± 0.17 mM of pyrrole-2-carbaldehyde after 18 h (Figure 5). The implementation of whole-cell biocatalyst also reduces the complexity and manufacturing costs associated with the used of purified CARse enzyme.
2.4. Pyrrole-2-carbaldehyde Is Unstable
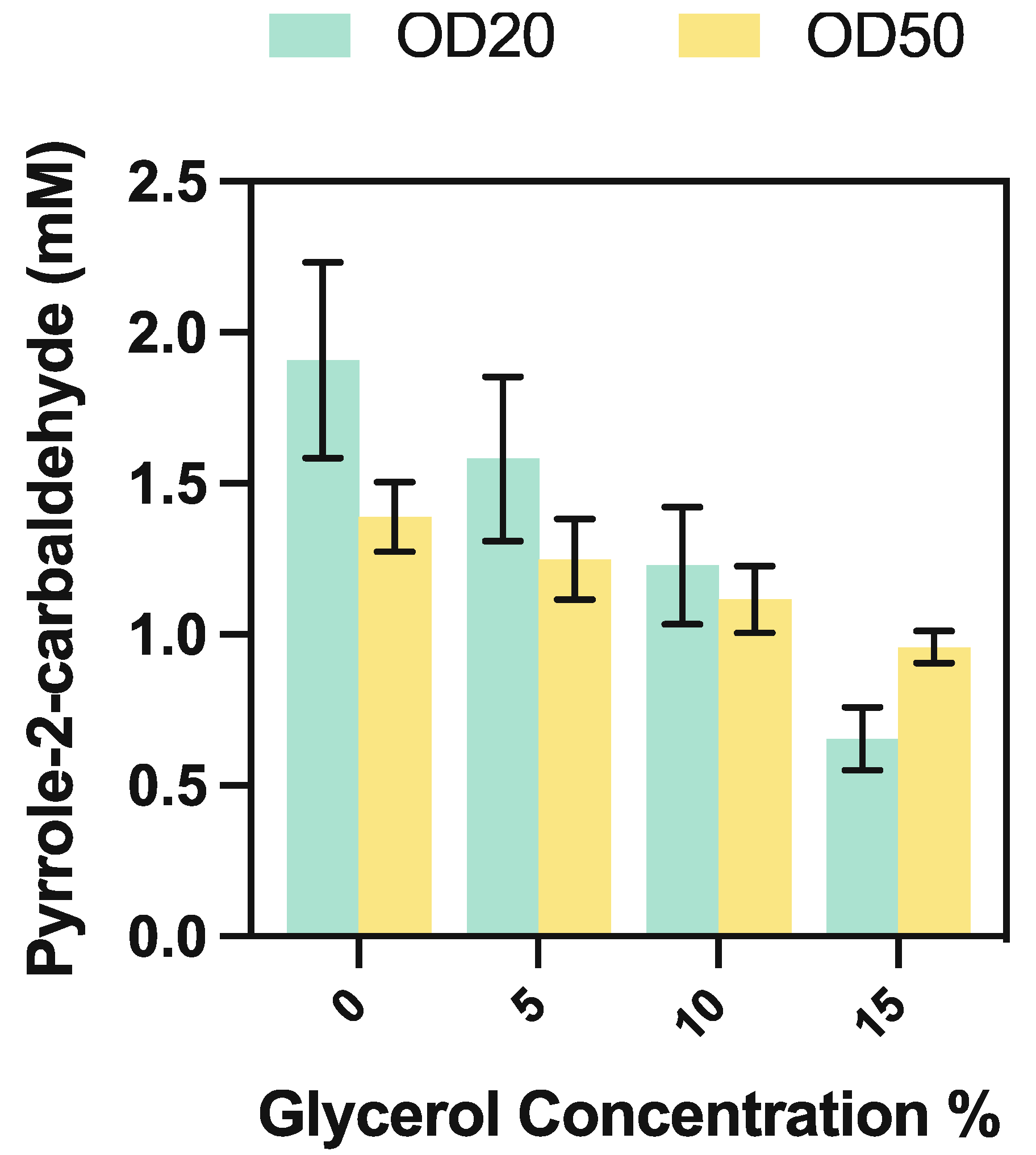
Further attempts to optimise the production of pyrrole-2-carbaldehyde, via the increase in final whole-cell biocatalyst concentration from OD600 = 20 to OD600 = 50, resulted in a reduced observable product yield. Simultaneous supplementation of glycerol, as a cheap carbon feedstock for whole-cell biocatalysts, to final concentrations between 5 and 15% v/v further resulted in decreased aldehyde yield (Figure 6).
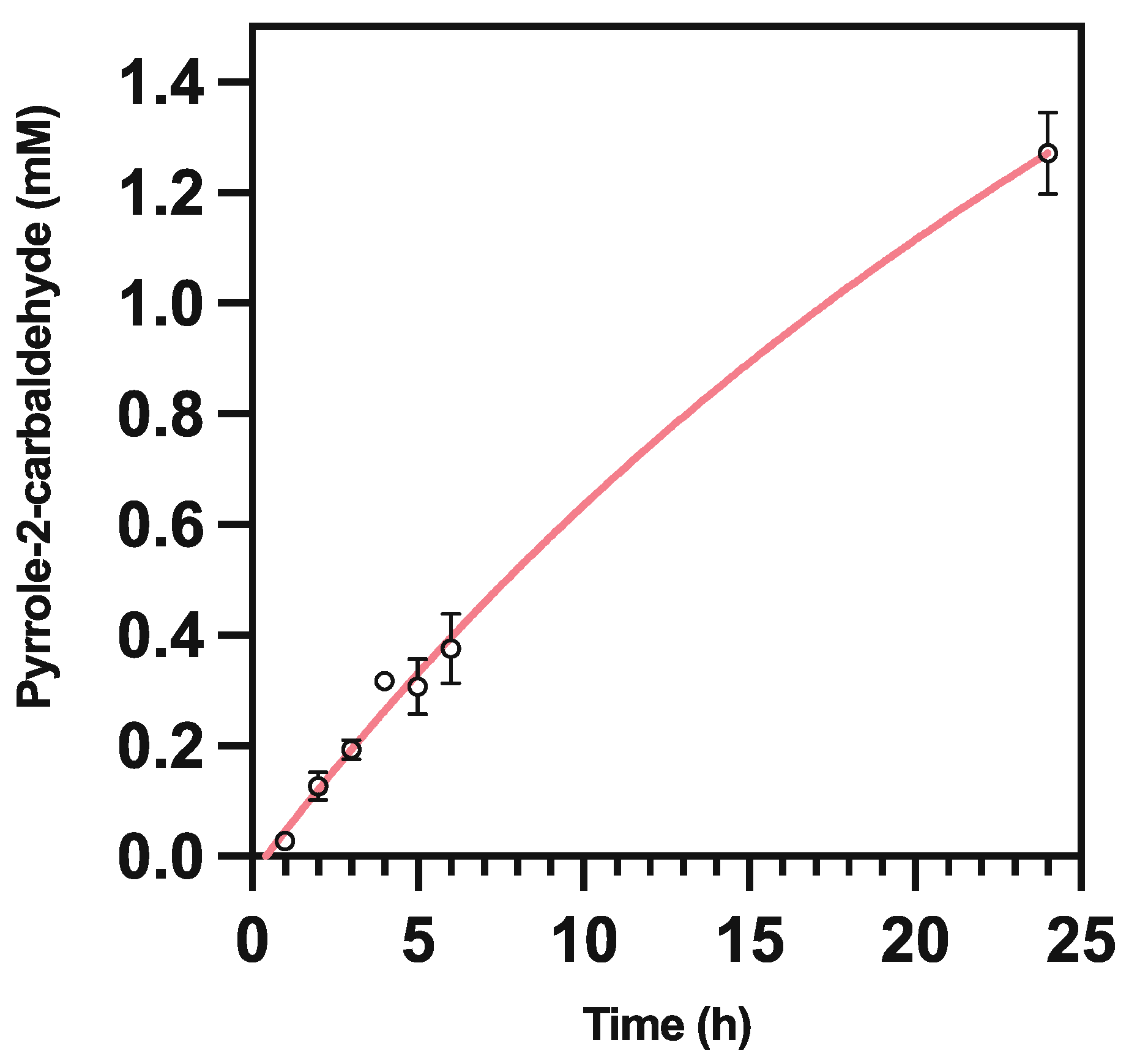
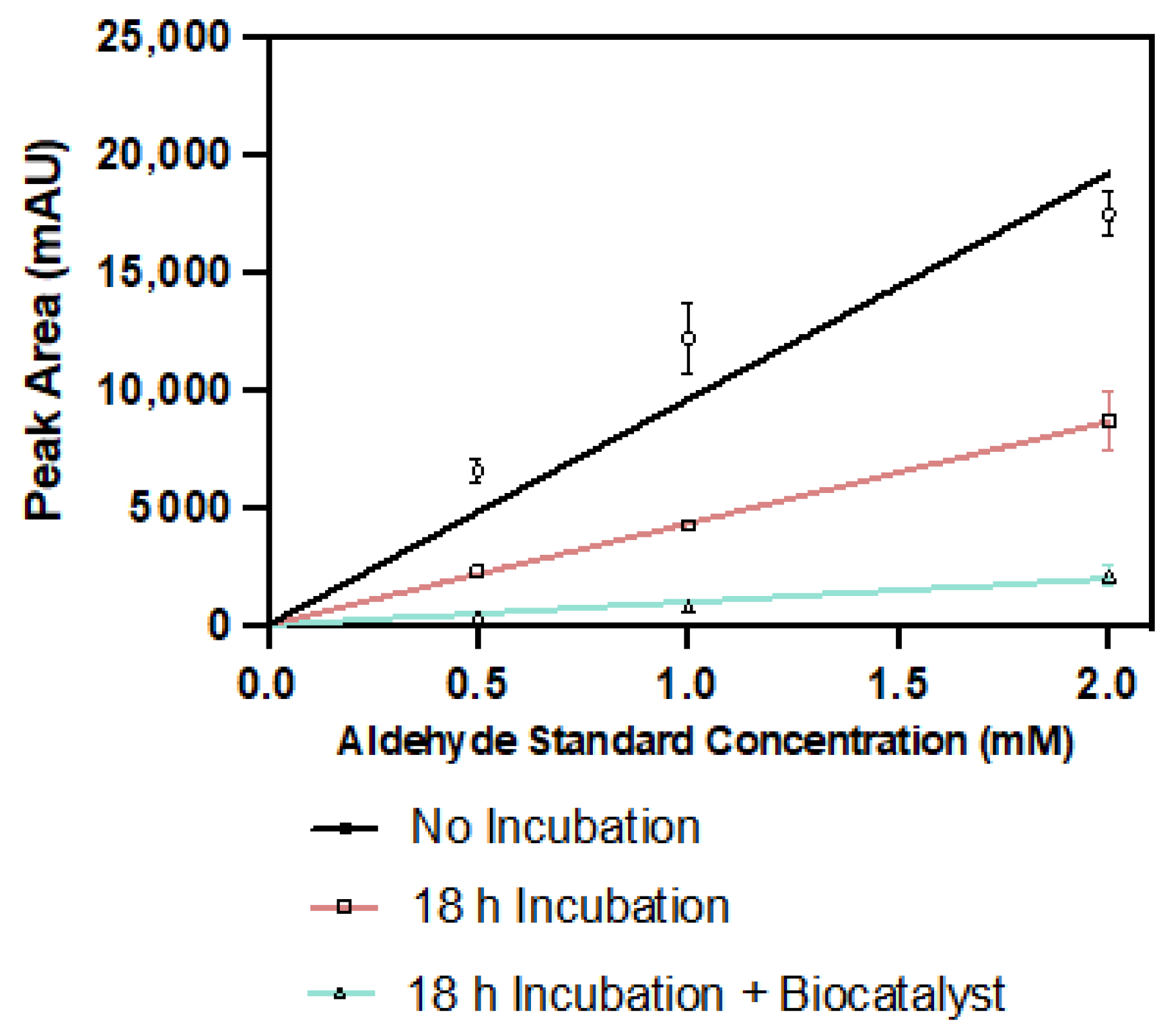
This may be in line with recent observations that increased buffer viscosity reduces product yield in UbiD enzymes, via inhibition of domain motion [18]. A short-time course analysis (Figure 7) indicated that the biocatalysts remained active following 24 h of incubation, so were unlikely to be undergoing significant degradation during the course of the reaction; we surmised that the reactive aldehyde product may be degrading in the presence of our biocatalyst. To test this hypothesis, we compared the peak area response of a range of freshly prepared pyrrole-2-carboxaldehyde standards (as measured on HPLC at 290 nm) to equivalent concentrations incubated in under reaction conditions with and without the presence of biocatalyst over 18 h (Figure 8).
We observed an average two-fold decrease in the peak area response versus the freshly prepared standard solutions, when pyrrole-2-carbaldehyde was incubated in solution for 18 h at 30 °C, which dropped, further, to a 90% reduction of the peak area when incubated for the same period in the presence of a biocatalyst. Given the inherent reactivity of aldehydes, this indicates the desired product is likely converted to other species (due to condensation and/or redox reactions) over the course of the reaction. Previous work has noted that in the presence of whole-cell biocatalysts, aldehyde products may be further reduced to form the corresponding alcohol [7,19,20], perhaps contributing to the reduction in product we observe here, so this possibility is a subject for potential further study. Hence, the present observed maximal product yield of 2.14 ± 0.16 mM likely is indicative of significantly higher production levels.
2.5. Neither Furan or Thiophene Class Substrates Yield Product
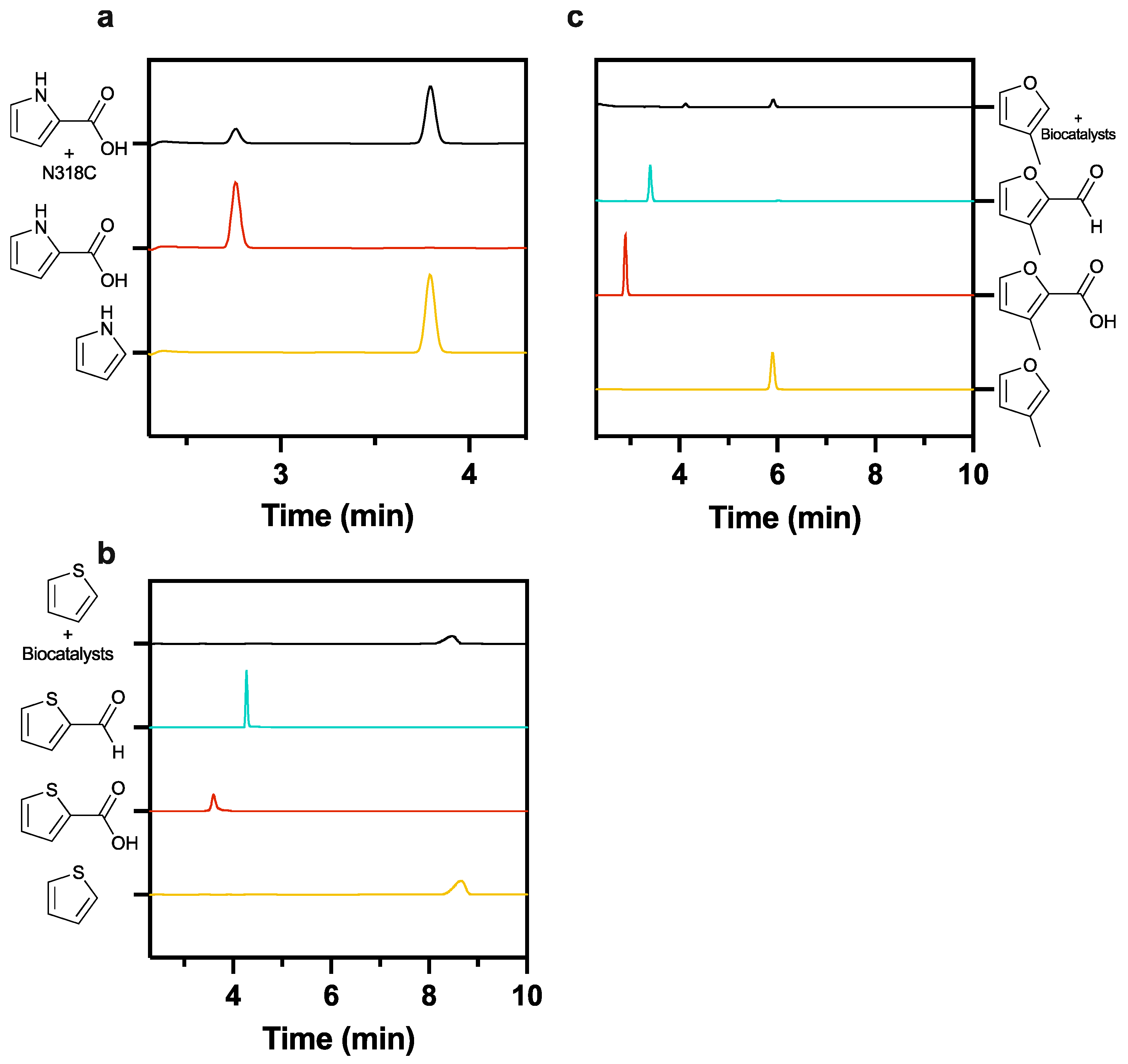
Prior work has shown that a point mutation of N318C in PA0254 broadens the substrate specificity to include furan- and thiophene-2-carboxylic acids, so we tested whether a CARse-linked reaction using N318C variant would act on these substrates. Given that the low boiling point of furan (approx. 31 °C) renders it an unattractive substrate for our reaction conditions, 3-methylfuran was utilised in these trials, as PA0254 can tolerate a 3-methyl substitution [8]. Unfortunately, the active (Figure 9a) N318C enzyme combined with our optimised whole-cell CARse system yielded no production of thiophene-2-carbaldehyde (Figure 9b) or 3-methylfuran-2-carbaldehyde (Figure 9c).
3. Materials and Methods
3.1. Production and Preparation of Active PA0254 Biocatalysts
Heterologous expression and purification of PA0254 was performed as described previously [8]. Briefly, PA0254 was co-transformed with Pseudomonas aeruginosa UbiX in HMS174(DE3) chemically competent E. coli (Merck) and expressed in liquid culture via IPTG-mediated induction. The obtained cells were lysed via constant flow cell-disruption, and the active PA0254UbiX was purified from the obtained lysate via nickel-affinity chromatography performed using gravity flow. The purified biocatalyst was desalted to remove imidazole and flash frozen prior to storage at −80 °C. SDS-Page characterisation of purified biocatalysts may be found in Figure S1.
3.2. Production and Preparation of CARse Biocatalysts
3.2.1. Cloning and Expression
Cloning and expression of CARse was performed, as previously described [16]. In short, CARse was co-expressed in HMS174(DE3) (Merck), with a sfp-type 4′-phosphopantetheinyl transferase from Bacillus subtilis in the same manner described for PA0254.
3.2.2. Preparation of Whole-Cell Biocatalyst
Harvested cells were washed by resuspension in sterile chilled phosphate buffered saline (PBS) solution, via gently repeated aspiration with a 50 mL serological pipette, and were transferred to 50 mL conical centrifuge tubes. Suspension was then centrifuged at 3500× g for 15 min, and the supernatant was discarded. This wash step was repeated two further times. The final cell suspension was split into 1 mL aliquots in 1.5 mL microcentrifuge tubes, and cells were harvested via a final centrifugation step and the supernatant discarded. Whole-cell biocatalyst was then either immediately frozen at −20 °C for storage or resuspended in 800 µL of chilled (PBS).
3.2.3. Preparation of Purified Biocatalyst
Harvested cells were resuspended at a 1:4 (wet cell mass:buffer) ratio in 50 mM Tris pH 7.0, 300 mM NaCl, 10 mM imidazole, and 1 mM MgCl2, supplemented with complete EDTA-free protease inhibitors (Roche), DNase, and RNase (Sigma). Cells were lysed via constant flow cell disruption at 10 kPSi and 20 kPsi, sequentially. Cell lysate was centrifuged at 20,000× g for 1 h, and the supernatant filter was with a 0.45 µm filter, before application to a gravity flow column containing Ni-NTA (Qiagen) equilibrated in resuspension buffer. Following loading, the column was washed with 10 column volumes of resuspension buffer, followed by 10 column volumes of 50 mM Tris pH 7.0, 300 mM NaCl, and 40 mM imidazole. The desired protein was eluted from the column in the above buffer supplemented with imidazole to 250 mM, analysed for purity via SDS-PAGE, and desalted into 50 mM Tris pH 7.0 and 200 mM NaCl, using a CentriPure P100 gel filtration column (empBiotech). SDS-Page characterisation of purified biocatalysts may be found in Figure S1.
3.3. Carboxylic Acid Reductase Enzyme Screening
Qualitive screening of CARse against heteroaromatic substrates was performed using purified enzyme at a 500 µL scale. Reactions contained 0.25 mg purified CARse, 5 mM of carboxylic acid substrate, 10 mM each NADPH and ATP, 100 mM MgCl2, and 100 mM Tris pH 7.5. Samples were incubated at 30 °C for 18 h and analysed via HPLC for the desired aldehyde product. Screening was performed in triplicate against pyrrole-2-carboxylic acid, furan-2-carboxylic acid, thiophene-2-carboxylic acid, and benzoic acid substrates.
3.4. Initial PA0254-CARse Coupling Reactions
Reactions contained 100 mM Potassium phosphate buffer pH 6.0, 500 mM Potassium bicarbonate, 10 mM Pyrrole, 0.27 mg/mL and 0.50 mg/mL purified PA0254 and CARse biocatalysts, respectively, of 20 mM ATP (pH 7.0), 20 mM NADPH, and 40 mM MgCl2. Final assay pH was measured at pH 7.4. Assays were performed in triplicate against a biocatalyst free control incubated at 30 °C for 18 h.
3.5. Optimisation of Assay Conditions
3.5.1. Nicotinamide Recycling
Comparison of nicotinamide cofactor recycling was performed as described in the initial coupling reaction, with a reduced NADPH concentration to 0.25 mM, and the addition of 20 mM glucose and 0.30 mg/mL of either purified wildtype glucose dehydrogenase from B. subtilis or a commercially available alternative (CDX-901-CODEXIS).
3.5.2. Bicarbonate Optimization
Bicarbonate concentration assays were performed by incorporating the lowered NADPH concentration of 0.25 mM and the addition of 0.30 mg/mL CDX-901 GDH (Codexis) and 20 mM glucose to the initial coupling reaction conditions. The concentration of potassium bicarbonate was then varied in 100 mM increments between 100 mM and 500 mM, in addition to one condition at 50 mM. The final pH of each condition was measured, as shown in Table 1. All assays were performed in triplicate in comparison to biocatalyst-free controls and assayed via HPLC.
3.5.3. ATP Recycling
Assays to characterise the effect of the introduction of a two-enzyme recycling system were performed at a 500 µL scale containing 100 mM potassium phosphate pH 6.0, 300 mM KHCO3, 40 mM MgCl2, 20 mM Glucose, 0.27 mg/mL PA0254, 0.50 mg/mL CARse, 0.3 mL CDX-901, and 10 mM pyrrole. In addition, the recycling system consisted of 4.00 mg/mL sodium hexametaphosphate, 0.5 mM ATP (pH 7.0), and 0.25 mg/mL, each purified adenylate kinase (AdK) and polyphosphate:AMP transferase (PAP) from Acinetobacter johnsonii. Assays were performed in triplicate vs an ATP-excess control, which replaced the recycling system with 20 mM ATP (pH 7.0). The final assay pH was measured at pH 7.22.
3.5.4. Whole-Cell CARse Biocatalyst Assays
The effect of the use of CARse whole-cell biocatalysts to enable economical in situ ATP and NADPH production was assessed in the following conditions at a 500 µL scale: 100 mM potassium phosphate pH 6.0, 300 mM KHCO3, 50 mM Glucose, 40 mM MgCl2, 10 mM Pyrrole, 0.27 mg/mL PA0254, and CAR9 whole cells at a final OD600 of 20 or 50. Glycerol supplementation was performed via the addition of 50% sterile glycerol to 5%, 10%, or 15% final volume.
3.5.5. Pyrrole-2-carboxaldehyde Degradation Assay
To assess the stability of pyrrole-2-carboxaldehyde over time, freshly prepared 0.5, 1.0, and 2.0 mM stocks of the aldehyde were prepared in 100 mM KPi pH 6.0, 300 mM KHCO3, 50 mM Glucose, and 40 mM MgCl2, and the peak response area was determined via HPLC. Identical stocks were, concurrently, prepared in the same buffer and incubated at 30 °C for 18 h, both with and without the addition of 0.27 mg/mL purified PA0254 and whole-cell CARse biocatalyst, to a final OD600 of 20. Peak area response was then determined via HPLC, as previously performed. All conditions were prepared in triplicate.
3.6. Analytical Procedures
All samples were analysed on an Agilent 1260 Infinity HPLC equipped with an Ascentis C18 581325-U (Supelco) or a Kinetex 00G-4601-E0 column (Phenomenex). The mobile phase consisted of varying ratios of water and acetonitrile modified with 0.1% v/v TFA, as described below. The quantification of aldehyde products was assessed via comparison to peak area response of analytical standard at varying concentration.
PA0254-CAR coupling reactions utilising pyrrole were extracted via the addition of 500 µL of MTBE, brief vortexing, incubation at 30 °C for 5 min, and centrifugation at 15,000× g for 30 min at 4 °C. The organic layer was then removed and analysed via HPLC, utilising a 50:50 ACN:H2O isocratic method for 6 min at a 1 mL/min flow rate monitored at 290 nm.
Reactions utilising thiophene and 3-methylfuran substrates were quenched via the addition of 1 reaction volume of acetonitrile supplemented with 0.1% v/v trifluoracetic acid. Samples were then incubated at 30 °C for 5 min, before centrifugation at 15,000× g for 30 min at 4 °C. Thiophene samples were analysed using a 10 min 60:40 ACN:H2O isocratic method and monitored at 240 nm, while 3-methylfuran was analysed at 50:50 ACN:H2O over the same time period and monitored at 220 nm.
4. Conclusions
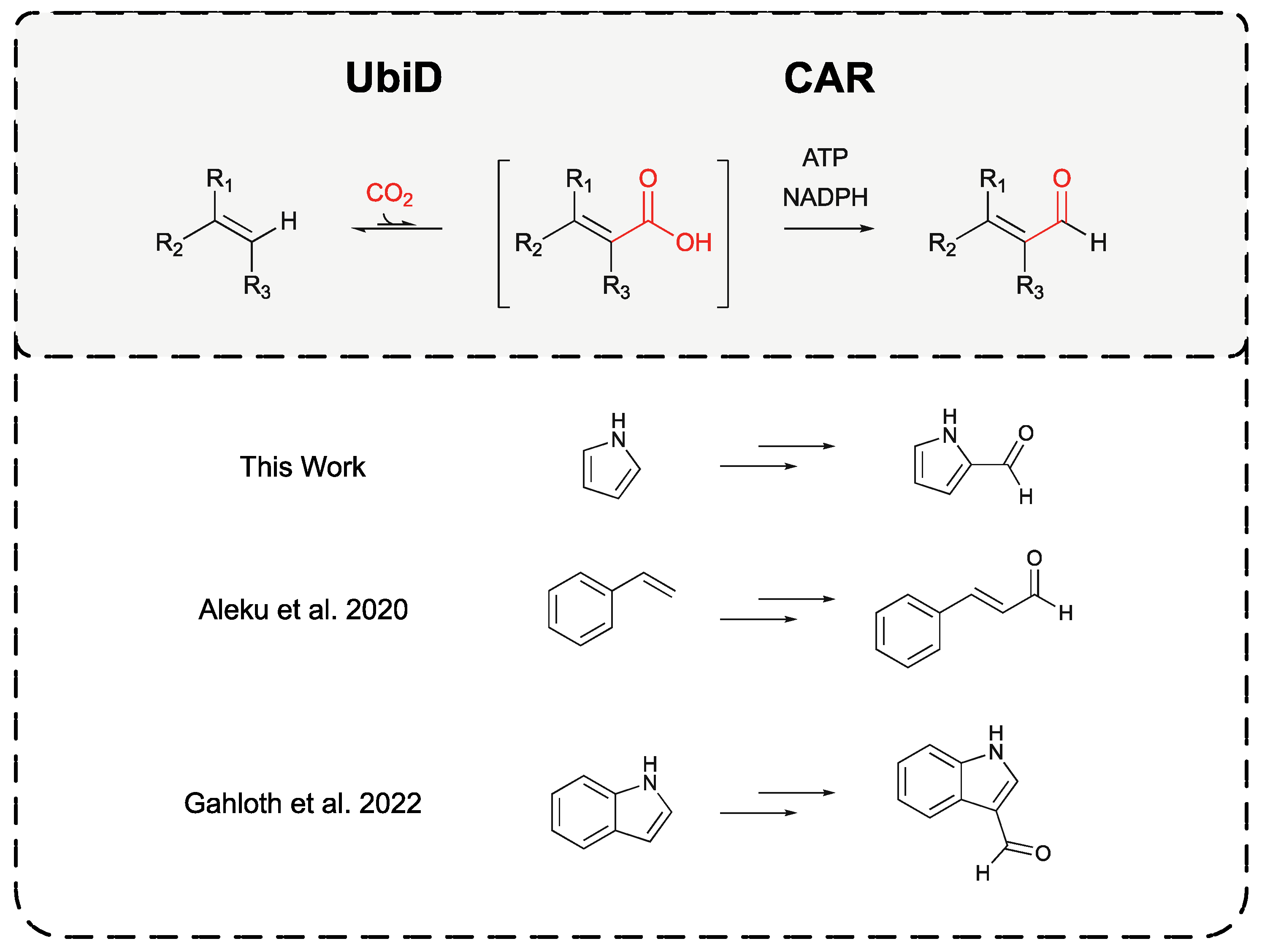
We conclude that an optimised PA0254-CARse one-pot reaction system is capable of carboxylation of pyrrole to pyrrole-2-carbaldehyde, but it suffers from low yield due to the instability of the target product. Similar observations of aldehyde instability in biocatalytic systems have been made, when coupling CAR enzymes with the prFMN-dependent indole-3-decarboxylase from Arthrobacter nicotianae [21], which noted the degredation of indole-3-carboxaldehyde product following extended incubaiton. This further highlights the applicability of a linked UbiD-CAR system, though future work may improve on this system in the stabilization or scavenging of the aldehyde, either via enzymatic means such as linkage to an imine reductase for the production of allylic amines, as previously described in the further modification of UbiD-CAR-produced cinnamaldehyde with Cystobacter ferrugineus imine reductase to produce N-cinnamylcyclopropylamine [7], or via methods of in situ product recovery such as biphasic reactions [22]. However, this work provides proof of principle of an efficient biosynthetic toolkit to produce heteroaromatic aldehydes and may be applied in future in the biological synthesis of their derivatives (Figure 10). Furthermore, it demonstrates that the relatively broad natural substrate specificity of the UbiD, and CAR enzyme families can be harnessed to deliver a route to a range of aldehydes, by tailoring the enzyme combination to ensure efficient CO2 fixation.
Supplementary Materials
The following supporting information can be downloaded at: https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/catal12050538/s1, Figure S1: SDS-Page analysis of purified biocatalysts.
Author Contributions
Conceptualization, G.R.T. and D.L.; methodology, G.R.T.; investigation, G.R.T., S.A.M. and H.M.; formal data analysis, G.R.T.; resources, D.L.; writing—original draft preparation, G.R.T.; writing—review and editing, D.L.; visualization, G.R.T.; supervision, D.L.; project administration, G.R.T. and D.L.; funding acquisition, G.R.T. and D.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Biotechnology and Biological Sciences Research Council (BBSRC), grant numbers BB/R01034X/1 (D.L.) and BB/M011208/1 (D.L. and G.R.T.), and by the European Research Council (ERC) grant pre-FAB ADG_695013 (D.L.). The APC was funded by the ERC grant pre-FAB ADG_695013.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hughes, G.; Lewis, J.C. Introduction: Biocatalysis in Industry. Chem. Rev. 2018, 118, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabral, S.; Schaub, T. The Use of Carbon Dioxide (CO2) as a Building Block in Organic Synthesis from an Industrial Perspective. Adv. Synth. Catal. 2019, 361, 223–246. [Google Scholar] [CrossRef] [Green Version]
- Aleku, G.A.; Roberts, G.W.; Titchiner, G.R.; Leys, D. Synthetic Enzyme-Catalyzed CO2 Fixation Reactions. ChemSusChem 2021, 14, 1781–1804. [Google Scholar] [CrossRef] [PubMed]
- Marshall, S.A.; Payne, K.A.P.; Leys, D. The UbiX-UbiD system: The biosynthesis and use of prenylated flavin (prFMN). Arch. Biochem. Biophys. 2017, 632, 209–221. [Google Scholar] [CrossRef] [Green Version]
- Aleku, G.A.; Prause, C.; Bradshaw-Allen, R.T.; Plasch, K.; Glueck, S.M.; Bailey, S.S.; Payne, K.A.P.; Parker, D.A.; Faber, K.; Leys, D. Terminal Alkenes from Acrylic Acid Derivatives via Non-Oxidative Enzymatic Decarboxylation by Ferulic Acid Decarboxylases. ChemCatChem 2018, 10, 3736–3745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloor, S.; Michurin, I.; Titchiner, G.; Leys, D. Prenylated Flavins: Structures and Mechanisms. FEBS J. 2022. [Google Scholar] [CrossRef]
- Aleku, G.A.; Saaret, A.; Bradshaw-Allen, R.T.; Derrington, S.R.; Titchiner, G.R.; Gostimskaya, I.; Gahloth, D.; Parker, D.A.; Hay, S.; Leys, D. Enzymatic C-H activation of aromatic compounds through CO2 fixation. Nat. Chem. Biol. 2020, 16, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Payne, K.A.P.; Marshall, S.A.; Fisher, K.; Rigby, S.E.J.; Cliff, M.J.; Spiess, R.; Cannas, D.M.; Larrosa, I.; Hay, S.; Leys, D. Structure and Mechanism of Pseudomonas aeruginosa PA0254/HudA, a prFMN-Dependent Pyrrole-2-carboxylic Acid Decarboxylase Linked to Virulence. ACS Catal. 2021, 11, 2865–2878. [Google Scholar] [CrossRef]
- Wu, L.; Burgess, K. A new synthesis of symmetric boraindacene (BODIPY) dyes. Chem. Commun. 2008, 40, 4933–4935. [Google Scholar] [CrossRef]
- Eseyin, A.E.; Steele, P.H. An overview of the applications of furfural and its derivatives. Int. J. Adv. Chem. 2015, 3, 42–47. [Google Scholar] [CrossRef]
- Jamali, M.R.; Assadi, Y.; Shemirani, F.; Salavati-Niasari, M. Application of thiophene-2-carbaldehyde-modified mesoporous silica as a new sorbent for separation and preconcentration of palladium prior to inductively coupled plasma atomic emission spectrometric determination. Talanta 2007, 71, 1524–1529. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Yao, P.; Chen, X.; Liu, X.; Zhang, R.; Feng, J.; Wu, Q.; Zhu, D. Exploring the synthetic applicability of a new carboxylic acid reductase from Segniliparus rotundus DSM 44985. J. Mol. Catal. B Enzym. 2015, 115, 1–7. [Google Scholar] [CrossRef]
- Qu, G.; Guo, J.; Yang, D.; Sun, Z. Biocatalysis of carboxylic acid reductases: Phylogenesis, catalytic mechanism and potential applications. Green Chem. 2018, 20, 777–792. [Google Scholar] [CrossRef]
- Finnigan, W.; Thomas, A.; Cromar, H.; Gough, B.; Snajdrova, R.; Adams, J.P.; Littlechild, J.A.; Harmer, N.J. Characterization of Carboxylic Acid Reductases as Enzymes in the Toolbox for Synthetic Chemistry. ChemCatChem 2017, 9, 1005–1017. [Google Scholar] [CrossRef] [Green Version]
- Moura, M.; Pertusi, D.; Lenzini, S.; Bhan, N.; Broadbelt, L.J.; Tyo, K.E. Characterizing and predicting carboxylic acid reductase activity for diversifying bioaldehyde production. Biotechnol. Bioeng. 2016, 113, 944–952. [Google Scholar] [CrossRef] [PubMed]
- Gahloth, D.; Dunstan, M.S.; Quaglia, D.; Klumbys, E.; Lockhart-Cairns, M.P.; Hill, A.M.; Derrington, S.R.; Scrutton, N.S.; Turner, N.J.; Leys, D. Structures of carboxylic acid reductase reveal domain dynamics underlying catalysis. Nat. Chem. Biol. 2017, 13, 975–981. [Google Scholar] [CrossRef] [Green Version]
- Resnick, S.M.; Zehnder, A.J. In vitro ATP regeneration from polyphosphate and AMP by polyphosphate:AMP phosphotransferase and adenylate kinase from Acinetobacter johnsonii 210A. Appl. Environ. Microbiol. 2000, 66, 2045–2051. [Google Scholar] [CrossRef] [Green Version]
- Marshall, S.A.; Payne, K.A.P.; Fisher, K.; Titchiner, G.R.; Levy, C.; Hay, S.; Leys, D. UbiD domain dynamics underpins aromatic decarboxylation. Nat. Commun. 2021, 12, 5065. [Google Scholar] [CrossRef]
- Kunjapur, A.M.; Prather, K.L. Microbial engineering for aldehyde synthesis. Appl. Environ. Microbiol. 2015, 81, 1892–1901. [Google Scholar] [CrossRef] [Green Version]
- Horvat, M.; Winkler, M. In Vivo Reduction of Medium- to Long-Chain Fatty Acids by Carboxylic Acid Reductase (CAR) Enzymes: Limitations and Solutions. ChemCatChem 2020, 12, 5076–5090. [Google Scholar] [CrossRef]
- Gahloth, D.; Fisher, K.; Payne, K.A.P.; Cliff, M.; Levy, C.; Leys, D. Structural and biochemical characterisation of the prenylated flavin mononucleotide-dependent indole-3-carboxylic acid decarboxylase. J. Biol. Chem. 2022, 101771. [Google Scholar] [CrossRef] [PubMed]
- Schmolzer, K.; Madje, K.; Nidetzky, B.; Kratzer, R. Bioprocess design guided by in situ substrate supply and product removal: Process intensification for synthesis of (S)-1-(2-chlorophenyl)ethanol. Bioresour. Technol. 2012, 108, 216–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
One-pot production of pyrrole-2-carbaldehyde, via a linked UbiD-CAR system, utilising whole-cell biocatalysts for improved cofactor economy.
Figure 1.
One-pot production of pyrrole-2-carbaldehyde, via a linked UbiD-CAR system, utilising whole-cell biocatalysts for improved cofactor economy.
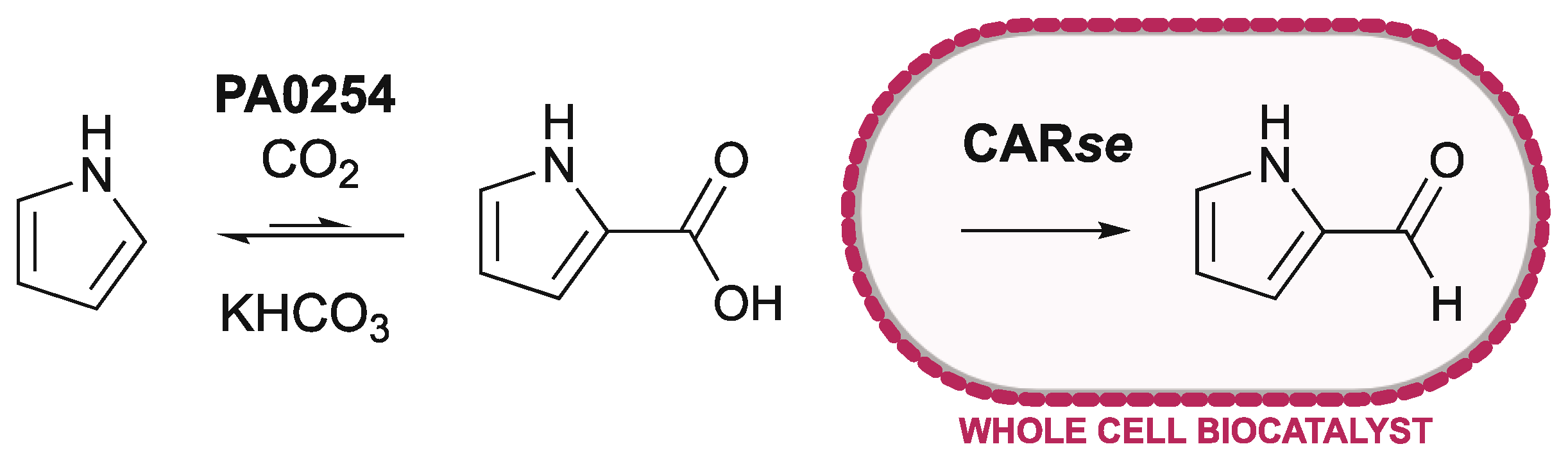
Figure 2.
Relative percent conversion of CARse and CARtp against pyrrole-, furan-, and thiophene-2-carboxylic acids normalised against reduction of benzoic acid. Benzoic acid conversion (4.21 ± 0.06 mM and 2.88 ± 0.05 mM for CARse and CARtp, respectively) normalised at 100%. Performed at a 500 μL scale in triplicate and analysed via HPLC, following 18 h of incubation at 30 °C.
Figure 2.
Relative percent conversion of CARse and CARtp against pyrrole-, furan-, and thiophene-2-carboxylic acids normalised against reduction of benzoic acid. Benzoic acid conversion (4.21 ± 0.06 mM and 2.88 ± 0.05 mM for CARse and CARtp, respectively) normalised at 100%. Performed at a 500 μL scale in triplicate and analysed via HPLC, following 18 h of incubation at 30 °C.
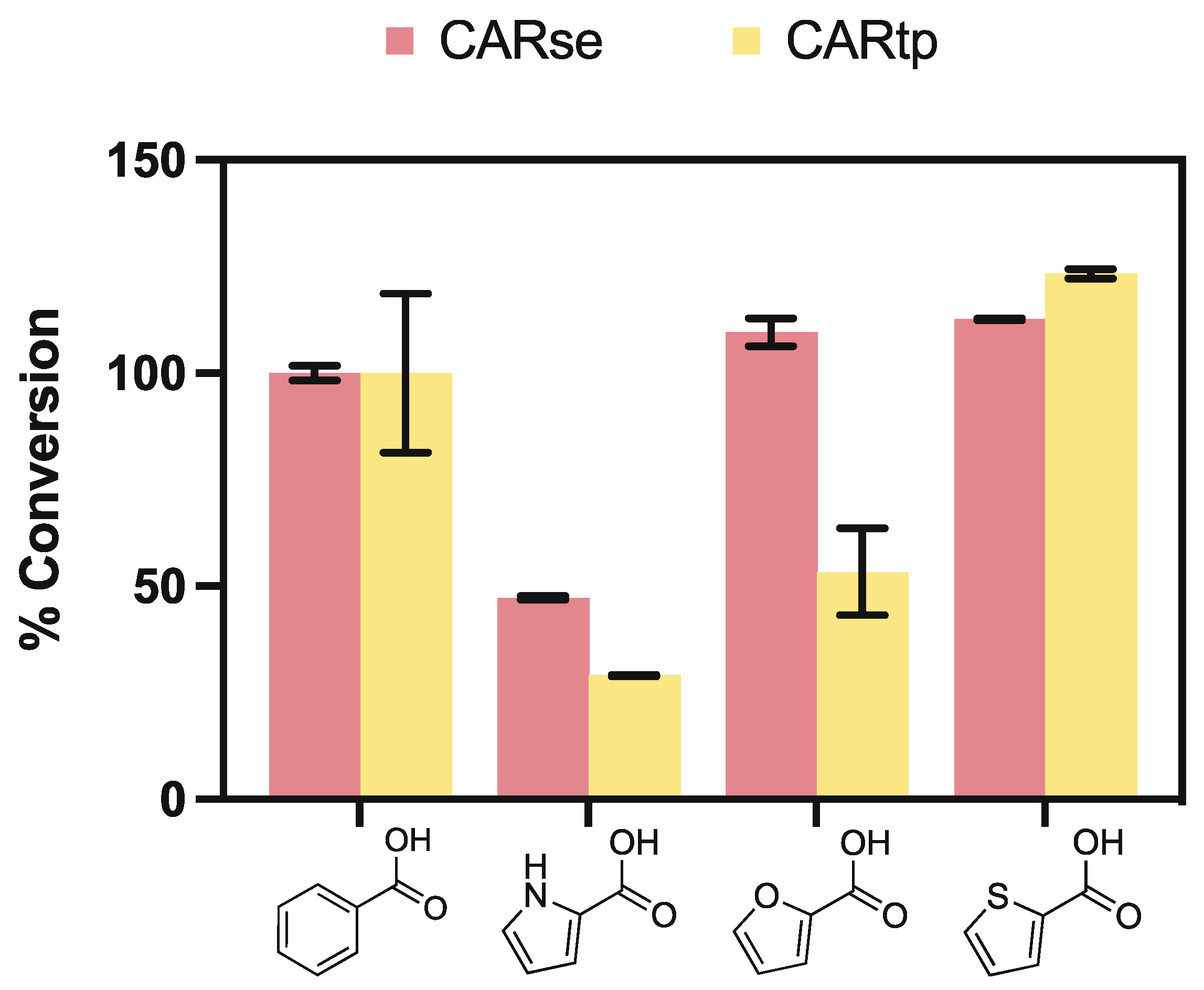
Figure 3.
Comparative pyrrole-2-carbaldehyde yield, obtained from coupling purified PA0254 and CARse enzymes in the presence of 500 mM KHCO3, as analysed via HPLC from replacement of 20 mM NADPH with wildtype bsGDH or commercially available CODEXIS CDX901-GDH at 0.30 mg/mL in the presence of 50 mM glucose. Reactions were incubated at 30 °C for 18 h. No observable carboxylation occurred in the no-biocatalyst controls (NBC).
Figure 3.
Comparative pyrrole-2-carbaldehyde yield, obtained from coupling purified PA0254 and CARse enzymes in the presence of 500 mM KHCO3, as analysed via HPLC from replacement of 20 mM NADPH with wildtype bsGDH or commercially available CODEXIS CDX901-GDH at 0.30 mg/mL in the presence of 50 mM glucose. Reactions were incubated at 30 °C for 18 h. No observable carboxylation occurred in the no-biocatalyst controls (NBC).
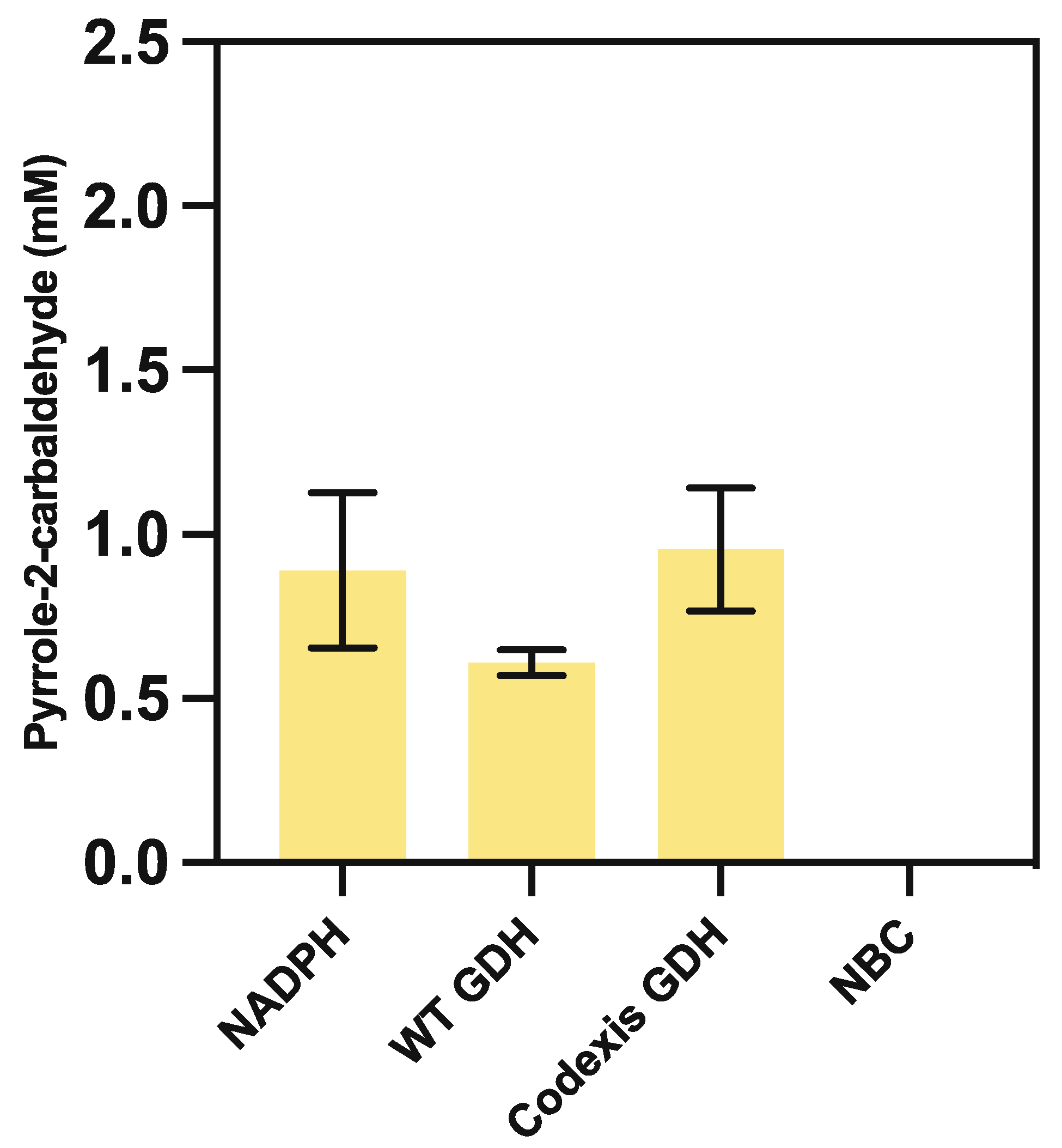
Figure 4.
Effect of bicarbonate salt concentration on yield of pyrrole-2-carbaldehyde from 10 mM pyrrole, at conditions ranging between 50–500 mM KHCO3 utilising purified PA0254 and CARse biocatalysts. No carboxylation of pyrrole was observed in controls lacking biocatalyst at any concentration of bicarbonate.
Figure 4.
Effect of bicarbonate salt concentration on yield of pyrrole-2-carbaldehyde from 10 mM pyrrole, at conditions ranging between 50–500 mM KHCO3 utilising purified PA0254 and CARse biocatalysts. No carboxylation of pyrrole was observed in controls lacking biocatalyst at any concentration of bicarbonate.
Figure 5.
Comparative aldehyde yield obtained by the reduction of supplemented ATP from 20 mM to 0.25 mM in the presence of 0.25 mg/mL each of AjPAP and AjAdK enzymes and 4 mg/mL sodium hexametaphosphate using purified PA0254 and CARse enzymes. Compared against the yield obtained with whole-cell CARse biocatalyst at OD600 = 20, supplemented with glucose to 50 mM in the presence of purified PA0254. No carboxylation of pyrrole was observed in controls lacking biocatalysts for any condition.
Figure 5.
Comparative aldehyde yield obtained by the reduction of supplemented ATP from 20 mM to 0.25 mM in the presence of 0.25 mg/mL each of AjPAP and AjAdK enzymes and 4 mg/mL sodium hexametaphosphate using purified PA0254 and CARse enzymes. Compared against the yield obtained with whole-cell CARse biocatalyst at OD600 = 20, supplemented with glucose to 50 mM in the presence of purified PA0254. No carboxylation of pyrrole was observed in controls lacking biocatalysts for any condition.
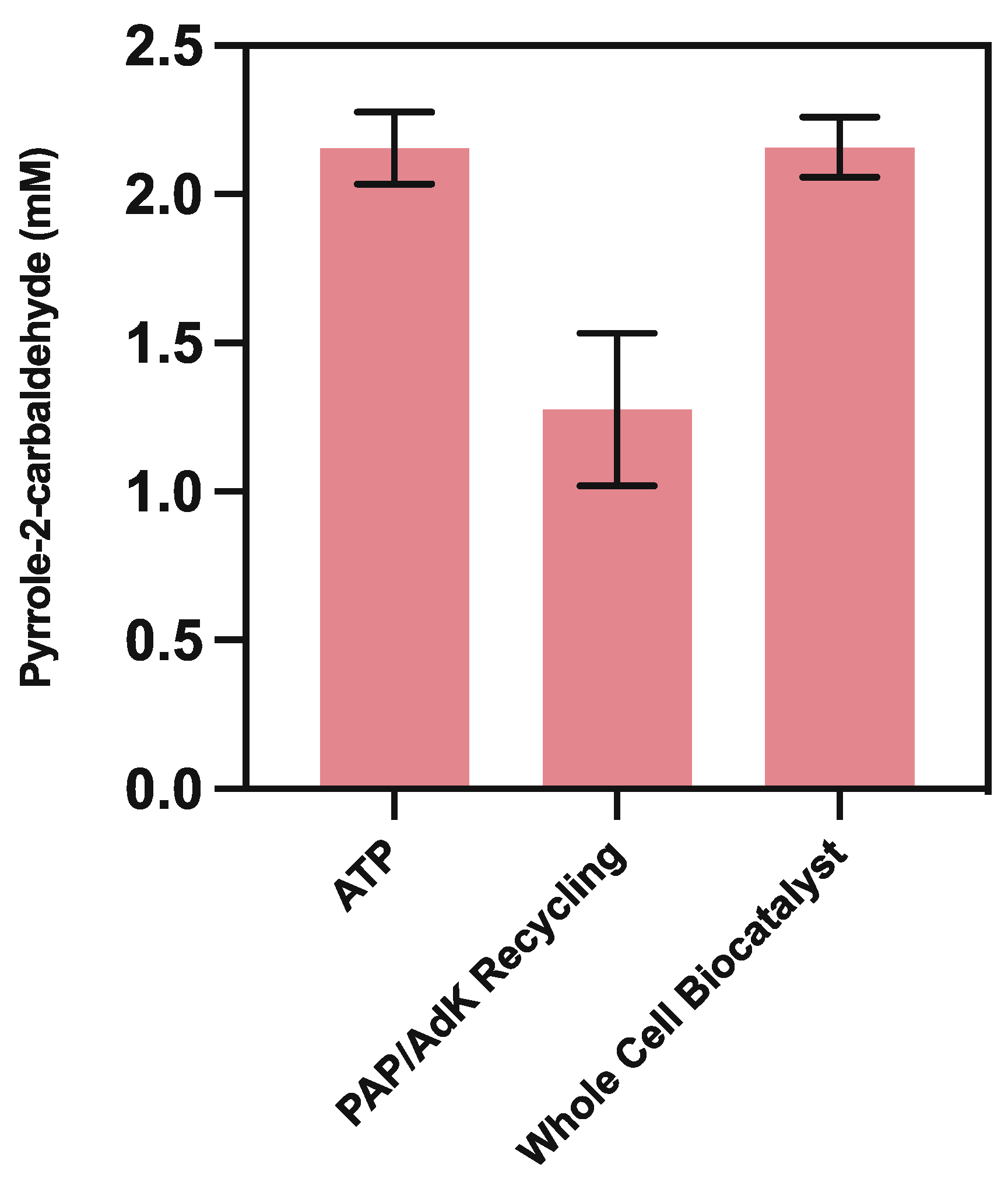
Figure 6.
Pyrrole-2-carbaldehyde yield obtained from 10 mM pyrrole by increasing final CARse whole-cell biocatalyst concentration from OD600 = 20 to OD600 = 50, and from supplementation of glycerol in 5% v/v increments in the presence of purified PA0254. No product formation was observed in biocatalyst-free controls at any condition.
Figure 6.
Pyrrole-2-carbaldehyde yield obtained from 10 mM pyrrole by increasing final CARse whole-cell biocatalyst concentration from OD600 = 20 to OD600 = 50, and from supplementation of glycerol in 5% v/v increments in the presence of purified PA0254. No product formation was observed in biocatalyst-free controls at any condition.
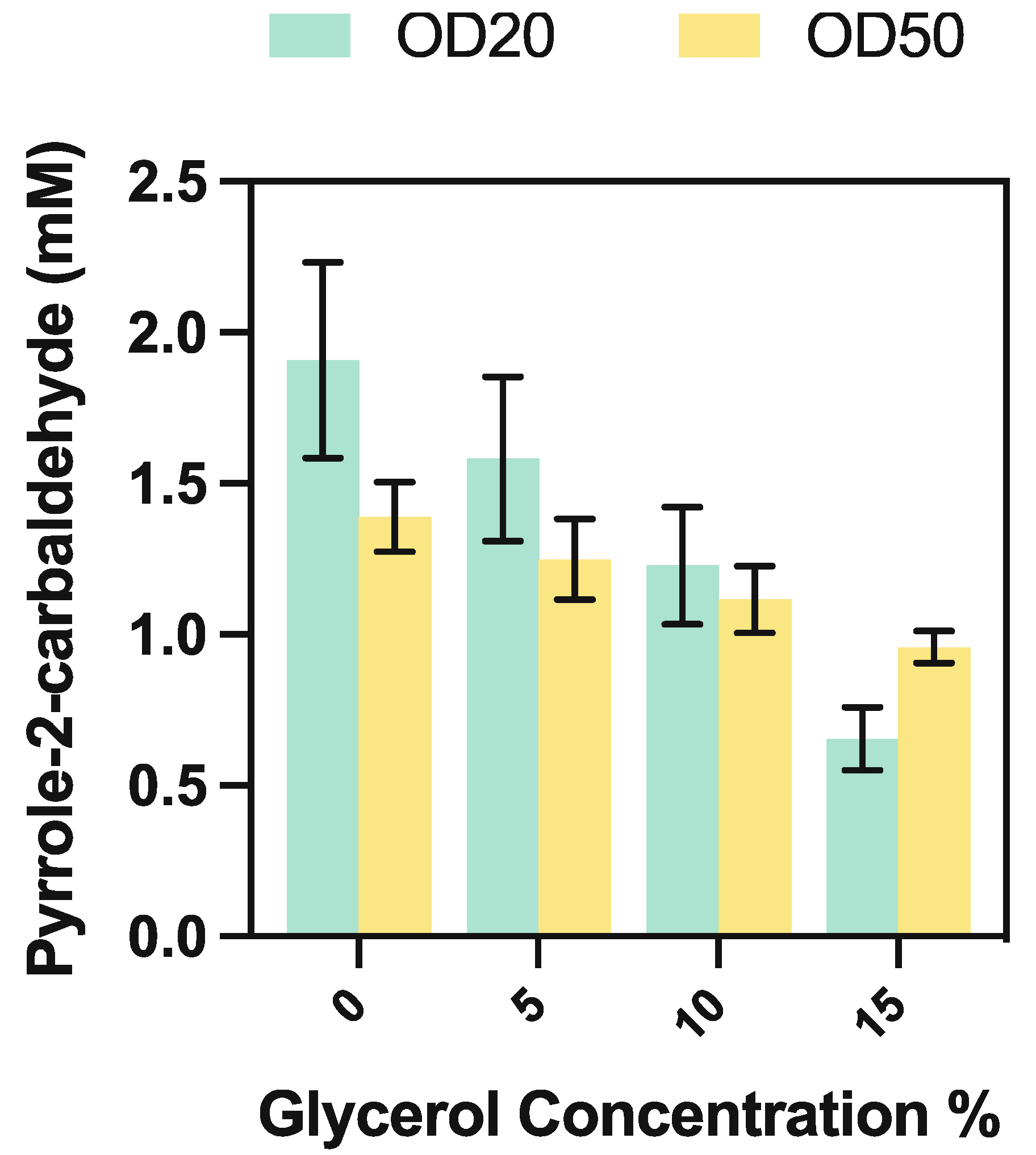
Figure 7.
Time course analysis of pyrrole-2-carbaldehyde production from whole-cell CARse and Purified PA0254 supplied with 10 mM pyrrole, performed over a period of 6 h. Comparative data points from 24 h of incubation displayed for clarity. No carboxylation of pyrrole was observed in controls lacking biocatalyst at any time point.
Figure 7.
Time course analysis of pyrrole-2-carbaldehyde production from whole-cell CARse and Purified PA0254 supplied with 10 mM pyrrole, performed over a period of 6 h. Comparative data points from 24 h of incubation displayed for clarity. No carboxylation of pyrrole was observed in controls lacking biocatalyst at any time point.
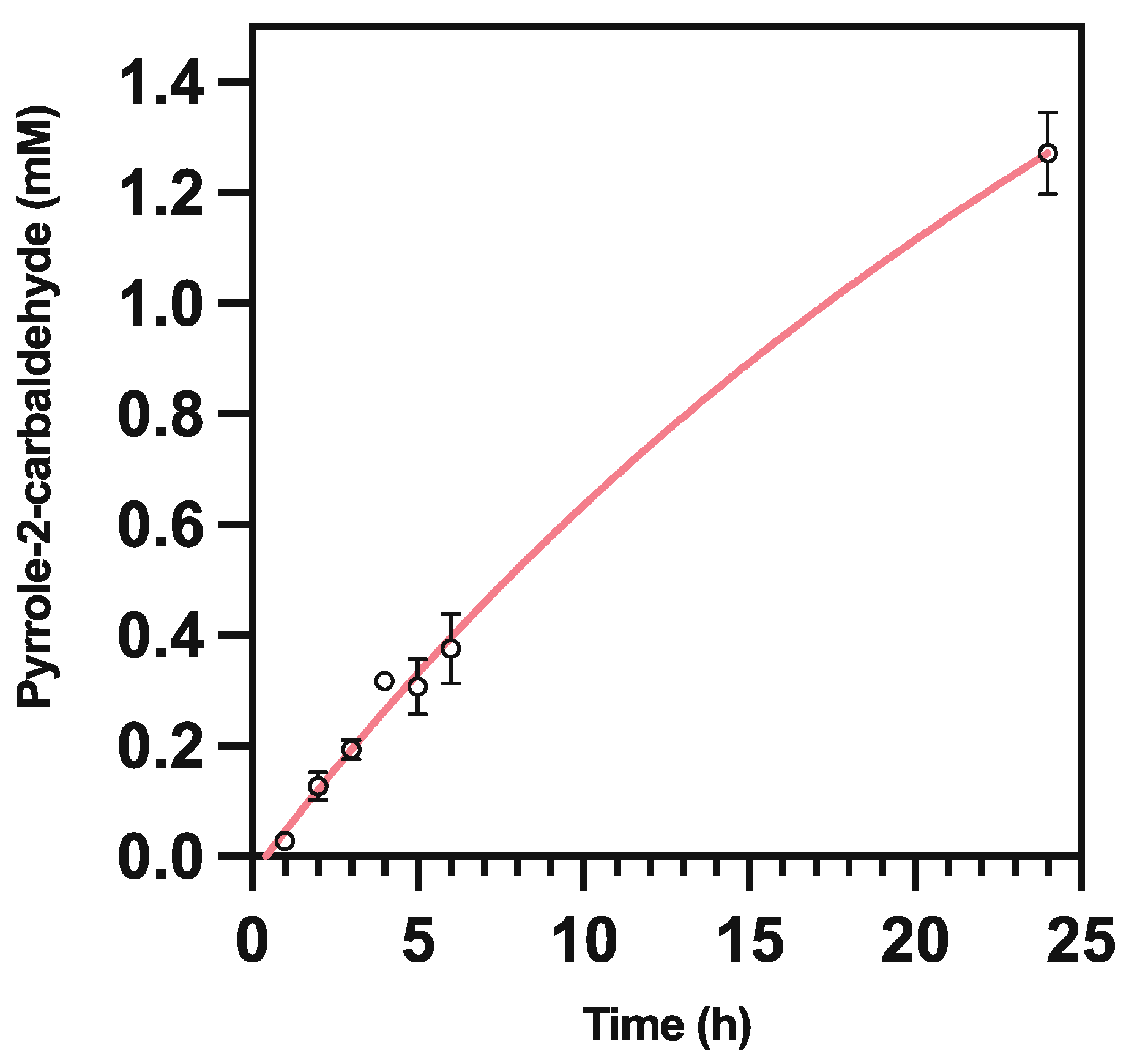
Figure 8.
Peak area response of varying concentrations of pyrrole-2-carbaldehyde prepared fresh in 100 mM KPi pH 6.0, 300 mM KHCO3, 50 mM Glucose, and 40 mM MgCl2, compared to that obtained when incubated in the same conditions for 18 h at 30 °C and when incubated for 18 h at 30 °C in the presence of 0.27 mg/mL purified PA0254 and OD600 = 20 whole-cell CARse biocatalysts. Measured via HPLC at 290 nm.
Figure 8.
Peak area response of varying concentrations of pyrrole-2-carbaldehyde prepared fresh in 100 mM KPi pH 6.0, 300 mM KHCO3, 50 mM Glucose, and 40 mM MgCl2, compared to that obtained when incubated in the same conditions for 18 h at 30 °C and when incubated for 18 h at 30 °C in the presence of 0.27 mg/mL purified PA0254 and OD600 = 20 whole-cell CARse biocatalysts. Measured via HPLC at 290 nm.
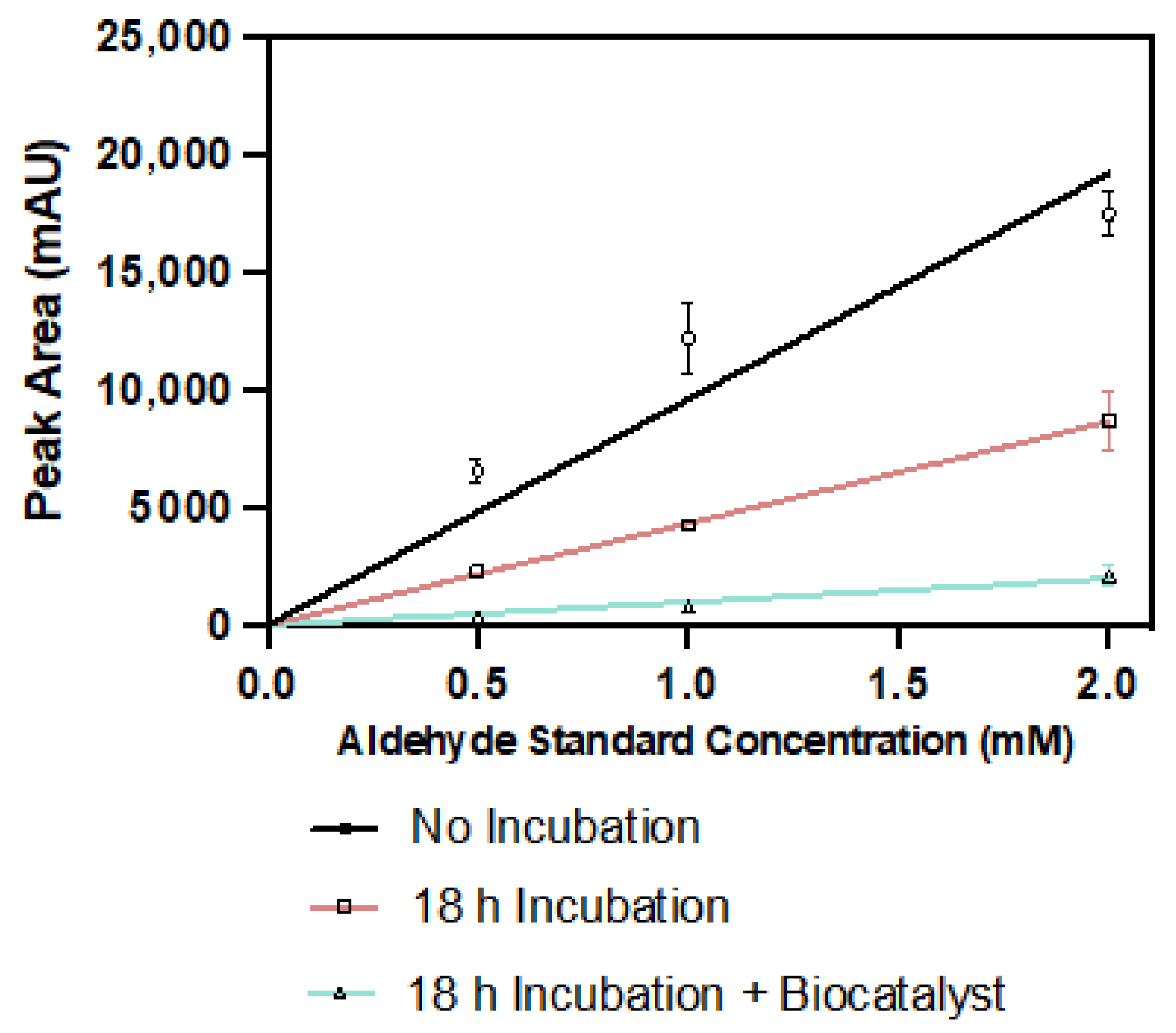
Figure 9.
Comparative HPLC trace analysis of biocatalytic reactions utilising purified PA0254-N318C mutant against known compound standards. Incubation of 0.27 mg/mL of N318C with 1 mM pyrrole-2-carboxylic acid yields the formation of pyrrole following incubation at 30 °C (a). However, no aldehyde is observed upon the application of purified N318C and whole-cell CARse biocatalysts with either thiophene (b) or 3-methylfuran (c) as starting substrates.
Figure 9.
Comparative HPLC trace analysis of biocatalytic reactions utilising purified PA0254-N318C mutant against known compound standards. Incubation of 0.27 mg/mL of N318C with 1 mM pyrrole-2-carboxylic acid yields the formation of pyrrole following incubation at 30 °C (a). However, no aldehyde is observed upon the application of purified N318C and whole-cell CARse biocatalysts with either thiophene (b) or 3-methylfuran (c) as starting substrates.
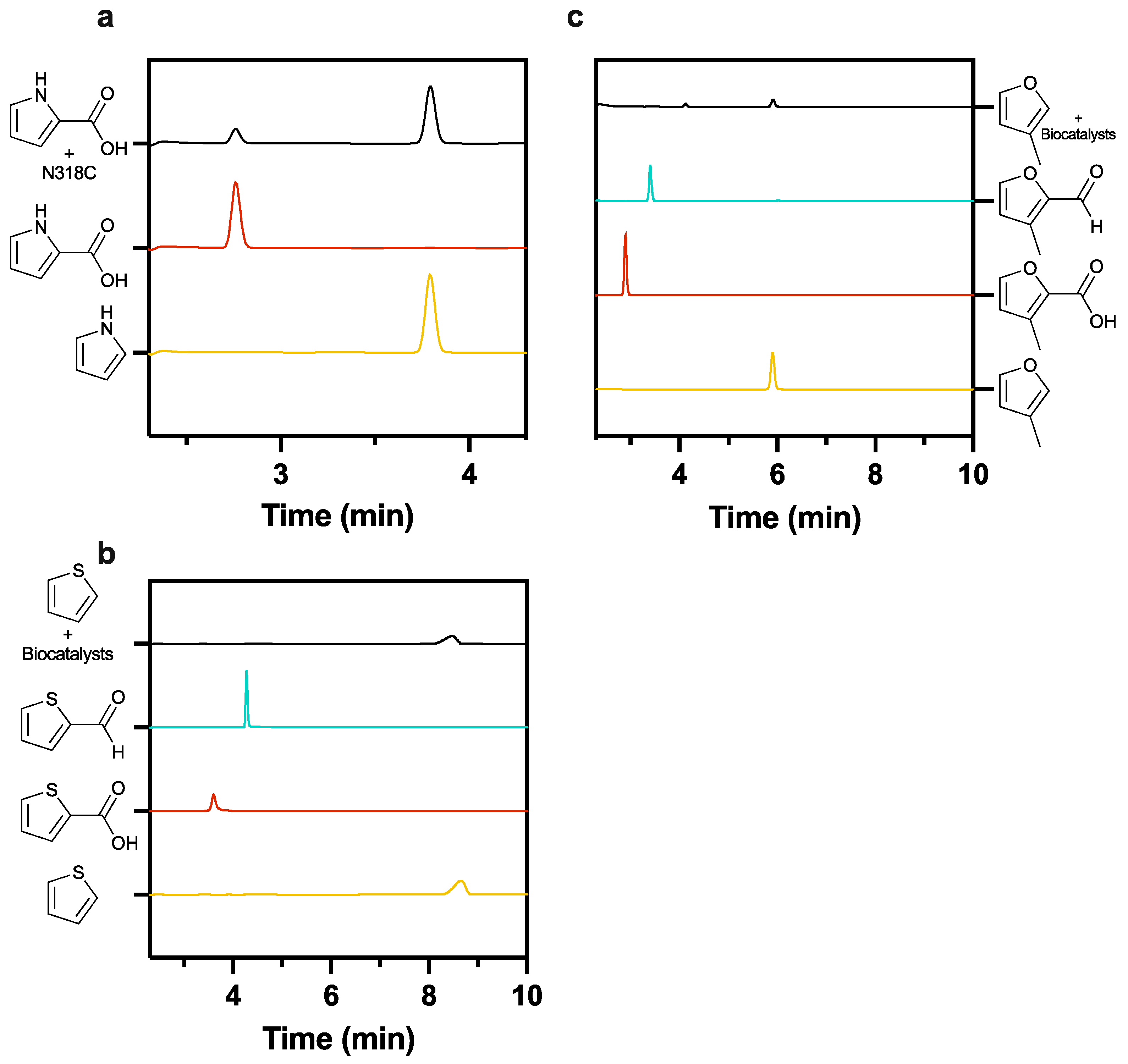
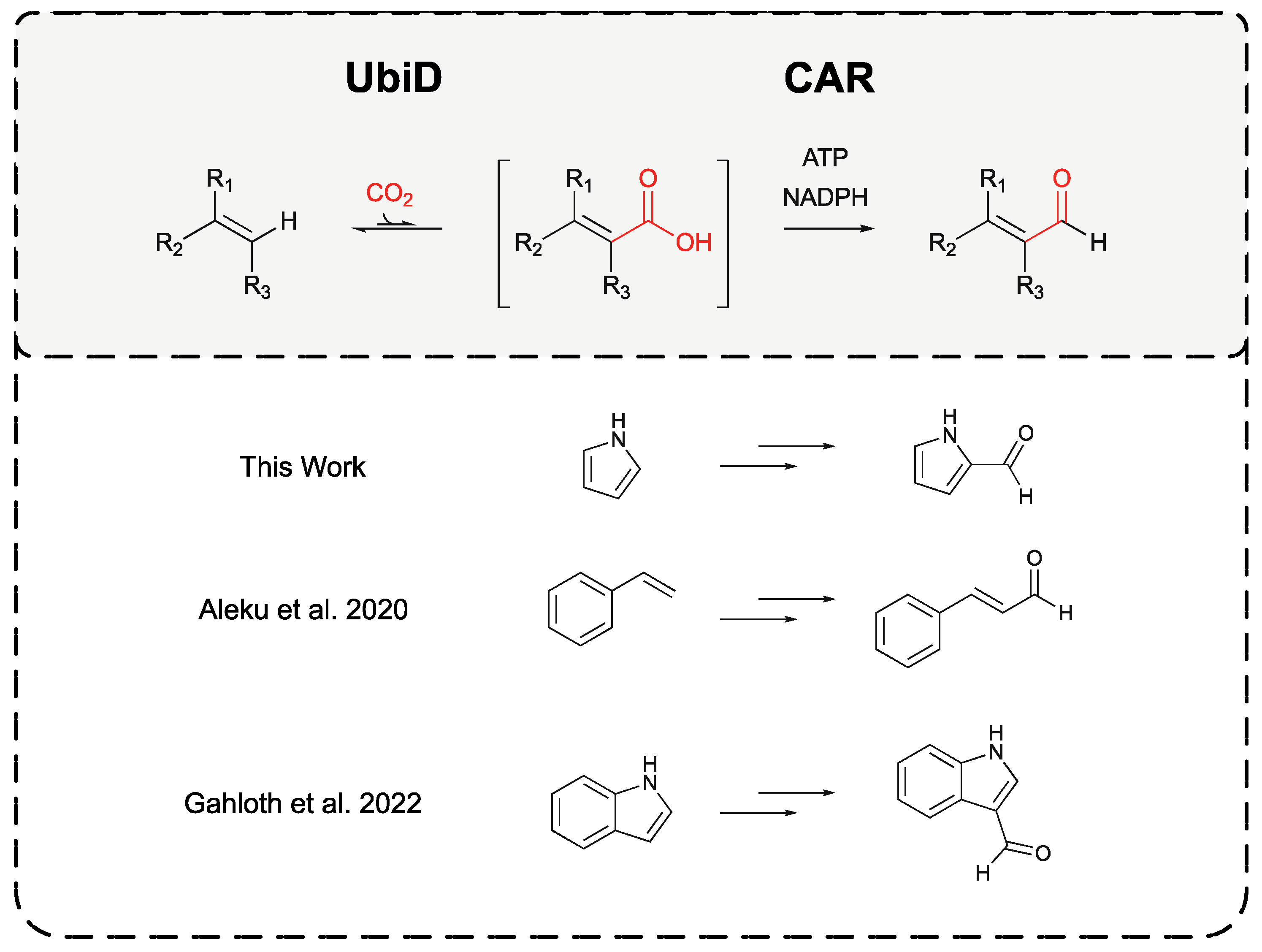
Figure 10.
Application of combined UbiD-CAR biocatalytic reactions for the production of aldehydes from terminal alkanes and heteroaromatics. Highlighting successes to date in utilising this system in the production of pyrrole-2-carbaldehyde from pyrrole, as presented within this work, the production of cinnamaldehyde from styrene [7], and the production of indole-3-carbaldehyde from indole [21].
Figure 10.
Application of combined UbiD-CAR biocatalytic reactions for the production of aldehydes from terminal alkanes and heteroaromatics. Highlighting successes to date in utilising this system in the production of pyrrole-2-carbaldehyde from pyrrole, as presented within this work, the production of cinnamaldehyde from styrene [7], and the production of indole-3-carbaldehyde from indole [21].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Effect of concentration of KHCO3 on final assay pH.
| Concentration of KHCO3 (mM) | Measured pH |
|---|---|
| 50 | 6.49 |
| 100 | 6.74 |
| 200 | 7.01 |
| 300 | 7.16 |
| 400 | 7.35 |
| 500 | 7.44 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Titchiner, G.R.; Marshall, S.A.; Miscikas, H.; Leys, D. Biosynthesis of Pyrrole-2-carbaldehyde via Enzymatic CO2 Fixation. Catalysts 2022, 12, 538. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12050538
AMA Style
Titchiner GR, Marshall SA, Miscikas H, Leys D. Biosynthesis of Pyrrole-2-carbaldehyde via Enzymatic CO2 Fixation. Catalysts. 2022; 12(5):538. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12050538
Chicago/Turabian StyleTitchiner, Gabriel R., Stephen A. Marshall, Herkus Miscikas, and David Leys. 2022. "Biosynthesis of Pyrrole-2-carbaldehyde via Enzymatic CO2 Fixation" Catalysts 12, no. 5: 538. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12050538
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.