
Expanding Access to Optically Active Non-Steroidal Anti-Inflammatory Drugs via Lipase-Catalyzed KR of Racemic Acids Using Trialkyl Orthoesters as Irreversible Alkoxy Group Donors


Abstract
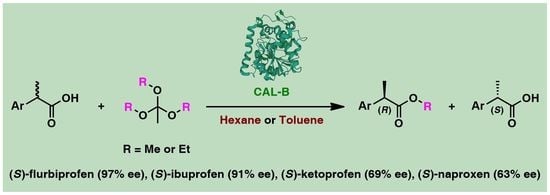
:
1. Introduction
2. Results and Discussion

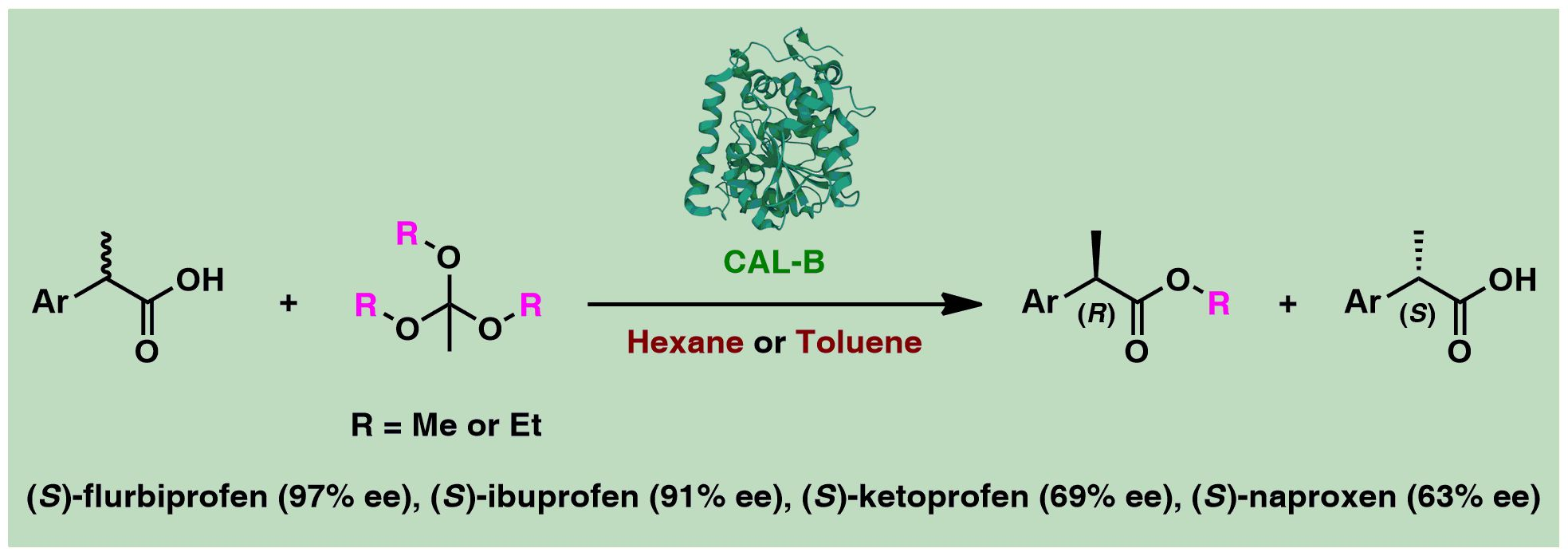{kind=link}
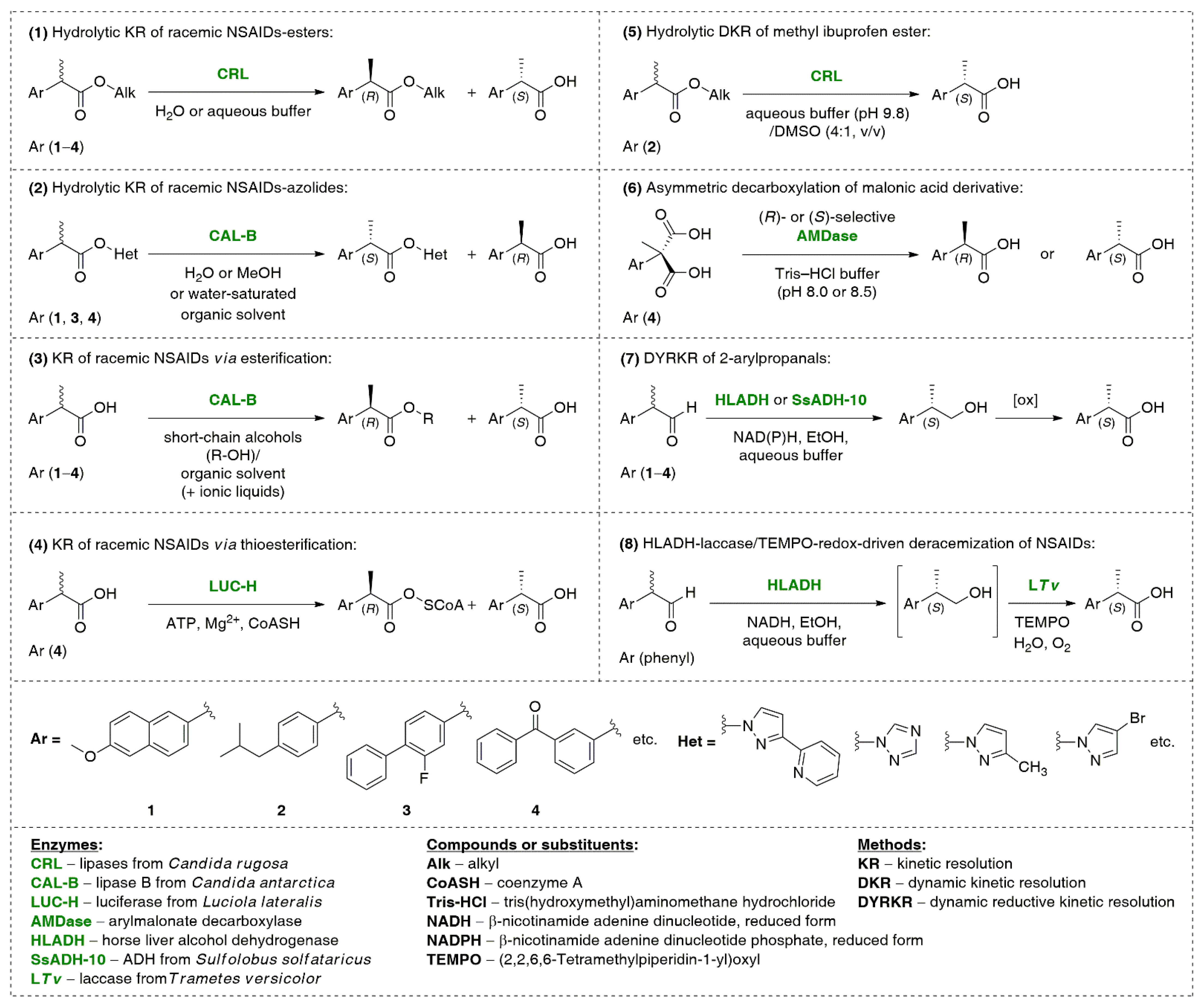{kind=link}
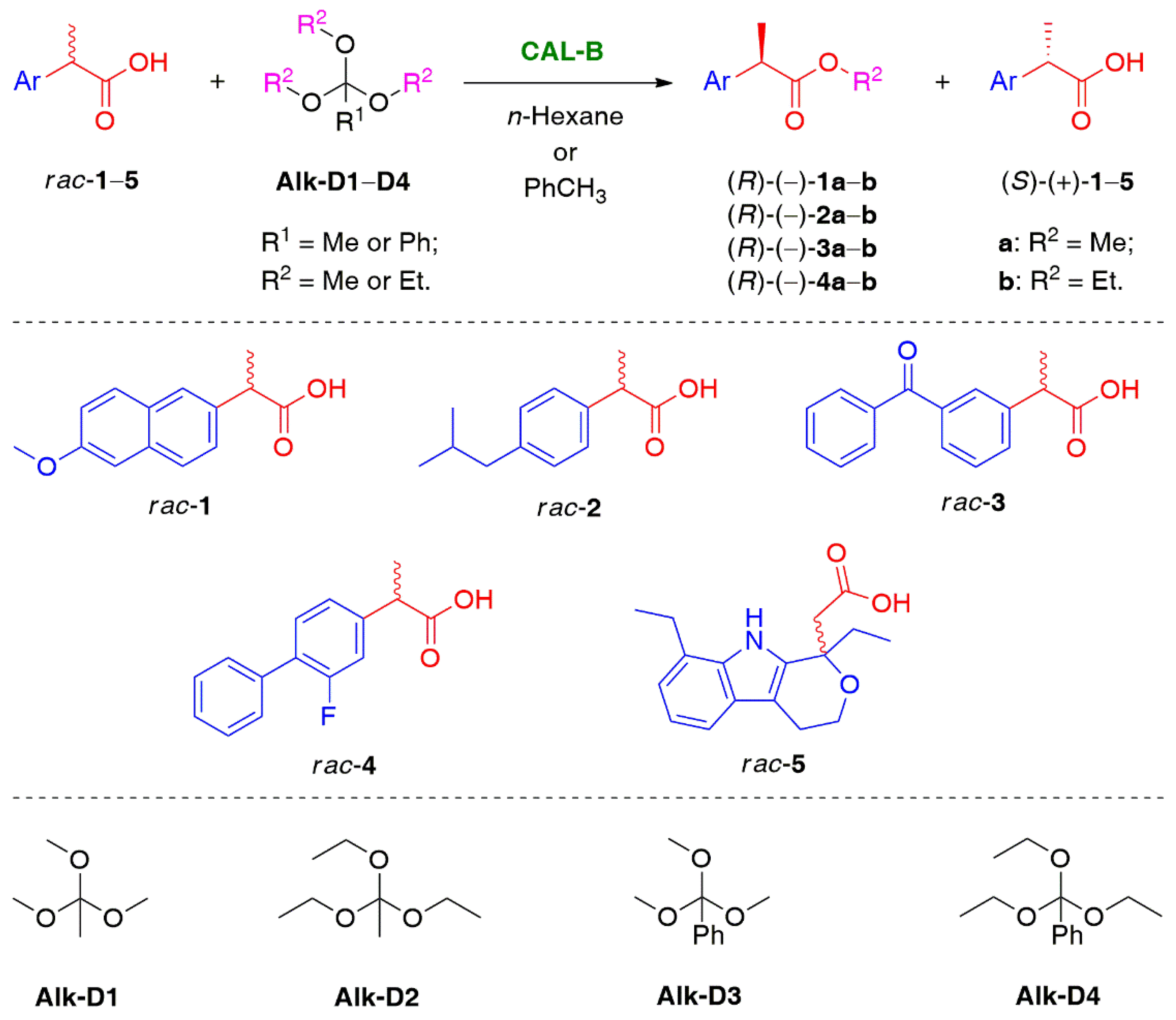{kind=link}

| Entry | Substrate a | CAL-B Preparation | Time (h) | Conv. (%) b | ees (%) c | eep (%) c | Ed |
|---|---|---|---|---|---|---|---|
| 1 |  | Novozym 435 | 72 | 7 | 6 | 75 | 7 |
| 2 | Lipozyme 435 | 72 | 13 | 13 | 86 | 15 | |
| 3 | Chirazyme L-2, C-2 | 72 | 13 | 11 | 74 | 7 | |
| 4 | Chirazyme L-2, C-3 | 72 | 52 | 57 | 53 | 6 | |
| 5 | CAL-B Sigma L4777 | 72 | 8 | 6 | 71 | 6 | |
| 6 | Novozym 435-STREM | 72 | 46 | 55 | 65 | 8 | |
| 7 | CAL-B-Immobead 150 | 72 | 16 | 14 | 76 | 8 | |
| 8 |  | Novozym 435 | 24 | 65 | 87 | 46 | 7 |
| 9 | Lipozyme 435 | 24 | 44 | 52 | 65 | 8 | |
| 10 | Chirazyme L-2, C-2 | 24 | 61 | 62 | 39 | 4 | |
| 11 | Chirazyme L-2, C-3 | 24 | 46 | 55 | 66 | 8 | |
| 12 | CAL-B Sigma L4777 | 24 | 52 | 57 | 52 | 5 | |
| 13 | Novozym 435-STREM | 24 | 68 | 91 | 42 | 7 | |
| 14 | CAL-B-Immobead 150 | 24 | 55 | 76 | 63 | 10 |
3. Materials and Methods
3.1. General Procedure for the Synthesis of Racemic NSAIDs’ Esters rac-1a–b, rac-2a–b, rac-3a–b, rac-4a–b, rac-5a–b
3.2. General Procedure for Enzyme Screening for EKR of rac-1 and rac-2 through Enantioselective Esterification with Trimethyl Orthoacetate
3.3. General Procedure for Alkoxy Group Donor Screening for EKR of rac-1 and rac-2 through Enantioselective Esterification
3.4. General Procedure for Organic Solvent Screening for EKR of rac-1 and rac-2 through Enantioselective Esterification with Trialkyl Orthoacetates
3.5. General Procedure for EKR of rac-3–5 through Enantioselective Esterification with Triethyl Orthoacetate
3.6. General Procedure for Preparative Scale EKR of Racemic NSAIDs rac-1–4 through Enantioselective Esterification with Trialkyl Orthoacetates
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mayer, J.M. Stereoselective metabolism of anti-inflammatory 2-arylpropionates. Acta Pharm. Nord. 1990, 2, 197–216. [Google Scholar] [PubMed]
- Evans, A.M. Pharmacodynamics and pharmacokinetics of the profens: Enantioselectivity, clinical implications, and special reference to (S)-(+)-ibuprofen. J. Clin. Pharmacol. 1996, 36, 7S–15S. [Google Scholar] [PubMed]
- Davies, N.M.; Anderson, K.E. Clinical pharmacokinetics of naproxen. Clin. Pharmacokinet. 1997, 32, 268–293. [Google Scholar] [CrossRef]
- Suri, A.; Grundy, B.L.; Derendorf, H. Pharmacokinetics and pharmacodynamics of enantiomers of ibuprofen and flurbiprofen after oral administration. Int. J. Clin. Pharmacol. Therap. 1997, 35, 1–8. [Google Scholar]
- Davies, N.M. Clinical pharmacokinetics of ibuprofen. The first 30 years. Clin. Pharmacokinet. 1998, 34, 101–154. [Google Scholar] [CrossRef]
- Hao, H.; Wang, G.; Sun, J. Enantioselective pharmacokinetics of ibuprofen and involved mechanisms. Drug Metabol. Rev. 2005, 37, 215–234. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.S.; Bresloff, P.; Mason, C.G. Pharmacological differences between the optical isomers of ibuprofen: Evidence for metabolic inversion of the (–)-isomer. J. Pharm. Pharmacol. 1976, 28, 256–257. [Google Scholar] [CrossRef]
- Hutt, A.J.; Caldwell, J. The metabolic chiral inversion of 2-arylpropionic acids–a novel route with pharmacological consequences. J. Pharm. Pharmacol. 1983, 35, 693–704. [Google Scholar] [CrossRef]
- Neupert, W.; Brugger, R.; Euchenhofer, C.; Brune, K.; Geisslinger, G. Effects of ibuprofen enantiomers and its coenzyme a thioesters on human prostaglandin endoperoxide synthases. Br. J. Pharmacol. 1997, 122, 487–492. [Google Scholar] [CrossRef] [Green Version]
- Tegeder, I.; Williams, K.; Geisslinger, G. Metabolic chiral inversion of 2-arylpropionic acids. In Stereochemical Aspects of Drug Action and Disposition; Eichelbaum, M., Testa, B., Somogyi, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2003; Volume 153, pp. 341–354. [Google Scholar]
- Rudy, A.C.; Liu, Y.; Brater, C.; Hall, S.D. Stereoselective pharmacokinetics and inversion of (r)- ketoprofen in healthy volunteers. J. Clin. Pharmacol. 1998, 38, 3S–10S. [Google Scholar] [CrossRef]
- Martin, J.E.; Le Leu, R.K.; Hu, Y.; Young, G.P. R-flurbiprofen suppresses distal nonmucin-producing colorectal tumors in azoxymethane-treated rats, without suppressing eicosanoid production. Am. J. Phys. Gastrointest. Liver Phys. 2010, 298, G860–G864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, B.W.; Jamali, F. Enantiomeric interaction of flurbiprofen in the rat. J. Pharm. Sci. 1989, 78, 632–634. [Google Scholar] [CrossRef]
- Geisslinger, G.; Schaible, H.G. New insights into the site and mode of antinociceptive action of flurbiprofen enantiomers. J. Clin. Pharmacol. 1996, 36, 513–520. [Google Scholar] [CrossRef]
- Barbanoj, M.J.; Antonijoan, R.M.; Gich, I. Clinical pharmacokinetics of dexketoprofen. Clin. Pharmacokinet. 2001, 40, 245–262. [Google Scholar] [CrossRef] [PubMed]
- Hancu, G.; Modroiu, A. Chiral switch: Between therapeutical benefit and marketing strategy. Pharmaceuticals 2022, 15, 240. [Google Scholar] [CrossRef] [PubMed]
- Calcaterra, A.; D’Acquarica, I. The market of chiral drugs: Chiral switches versus de novo enantiomerically pure compounds. J. Pharm. Biomed. Anal. 2018, 147, 323–340. [Google Scholar] [CrossRef] [PubMed]
- Agranat, I.; Caner, H.; Caldwell, J. Putting chirality to work: The strategy of chiral switches. Nat. Rev. Drug Discov. 2002, 1, 753–768. [Google Scholar] [CrossRef] [PubMed]
- Foster, R.T.; Jamali, F.; Russell, A.S.; Alballa, S.R. Pharmacokinetics of ketoprofen enantiomers in healthy subjects following single and multiple doses. J. Pharm. Sci. 1988, 77, 70–73. [Google Scholar] [CrossRef]
- Gu, Q.-M.; Chen, C.-S.; Sih, C.J. A facile enzymatic resolution process for the preparation of (+)-2-(6-hethoxy-2-naphthyl)propionic acid (Naproxen). Tetrahedron Lett. 1986, 27, 1763–1766. [Google Scholar] [CrossRef]
- Lalonde, J.J.; Govardhan, C.; Khalaf, N.; Martinez, A.G.; Visuri, K.; Margolin, A.L. Cross-Linked Crystals of Candida rugosa Lipase: Highly Efficient Catalysts for the Resolution of Chiral Esters. J. Am. Chem. Soc. 1995, 117, 6845–6852. [Google Scholar] [CrossRef]
- Gill, I.; Pastor, E.; Ballesteros, A. Lipase−Silicone Biocomposites: Efficient and Versatile Immobilized Biocatalysts. J. Am. Chem. Soc. 1999, 121, 9487–9496. [Google Scholar] [CrossRef]
- Cernia, E.; Delfini, M.; Di Cocco, E.; Palocci, C.; Soro, S. Investigation of lipase-catalysed hydrolysis of naproxen methyl ester: Use of NMR spectroscopy methods to study substrate–enzyme interaction. Bioorg. Chem. 2002, 30, 276–284. [Google Scholar] [CrossRef]
- Long, W.S.; Kamaruddin, A.; Bhatia, S. Chiral resolution of racemic ibuprofen ester in an enzymatic membrane reactor. J. Memb. Sci. 2005, 247, 185–200. [Google Scholar] [CrossRef]
- Giorno, L.; Damore, E.; Drioli, E.; Cassano, R.; Picci, N. Influence of OR ester group length on the catalytic activity and enantioselectivity of free lipase and immobilized in membrane used for the kinetic resolution of naproxen esters. J. Catal. 2007, 247, 194–200. [Google Scholar] [CrossRef]
- Gilani, S.L.; Najafpour, G.D.; Heydarzadeh, H.D.; Moghadamnia, A. Enantioselective synthesis of (S)-naproxen using immobilized lipase on chitosan beads. Chirality 2017, 29, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Basak, A.; Nag, A.; Bhattacharya, G.; Mandal, S.; Nag, S. Chemoenzymatic synthesis of antiinflammatory drugs in enantiomerically pure form. Tetrahedron Asymmetry 2000, 11, 2403–2407. [Google Scholar] [CrossRef]
- Gérard, D.; Guéroult, M.; Casas-Godoy, L.; Condoret, J.-S.; André, I.; Marty, A.; Duquesne, S. Efficient resolution of profen ethyl ester racemates by engineered Yarrowia lipolytica Lip2p lipase. Tetrahedron Asymmetry 2017, 28, 433–441. [Google Scholar] [CrossRef] [Green Version]
- Smeets, J.W.H.; Kieboom, A.P.G. Enzymatic enantioselective ester hydrolysis by carboxylesterase NP. Rec. Trav. Chim. Pays-Bas 2010, 111, 490–495. [Google Scholar] [CrossRef]
- Kumar, I.; Manju, K.; Jolly, R.S. A new biocatalyst for the preparation of enantiomerically pure 2-arylpropanoic acids. Tetrahedron Asymmetry 2001, 12, 1431–1434. [Google Scholar] [CrossRef]
- Koul, S.; Parshad, R.; Taneja, S.C.; Qazi, G.N. Enzymatic resolution of naproxen. Tetrahedron Asymmetry 2003, 14, 2459–2465. [Google Scholar] [CrossRef]
- Wang, P.-Y.; Wu, C.-H.; Ciou, J.-F.; Wu, A.-C.; Tsai, S.-W. Kinetic resolution of (R,S)-pyrazolides containing substituents in the leaving pyrazole for increased lipase enantioselectivity. J. Mol. Catal. B Enzym. 2010, 66, 113–119. [Google Scholar] [CrossRef]
- Wang, P.-Y.; Chen, Y.-J.; Wu, A.-C.; Lin, Y.-S.; Kao, M.-F.; Chen, J.-R.; Ciou, J.-F.; Tsai, S.-W. (R,S)-Azolides as Novel Substrates for Lipase-Catalyzed Hydrolytic Resolution in Organic Solvents. Adv. Synth. Catal. 2009, 351, 2333–2341. [Google Scholar] [CrossRef]
- Toledo, M.V.; José, C.; Suster, C.R.L.; Collins, S.E.; Portela, R.; Bañares, M.A.; Briand, L.E. Catalytic and molecular insights of the esterification of ibuprofen and ketoprofen with glycerol. Mol. Catal. 2021, 513, 111811. [Google Scholar] [CrossRef]
- Ghanem, A. Direct enantioselective HPLC monitoring of lipase-catalyzed kinetic resolution of flurbiprofen. Chirality 2010, 22, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Shang, W.; Zhang, X.; Yang, X.; Zhang, S. High pressure CO2-controlled reactors: Enzymatic chiral resolution in emulsions. RSC Adv. 2014, 4, 24083–24088. [Google Scholar] [CrossRef]
- Mohammadi, M.; Gandomkar, S.; Habibi, Z.; Yousefi, M. One pot three-component reaction for covalent immobilization of enzymes: Application of immobilized lipases for kinetic resolution of rac-ibuprofen. RSC Adv. 2016, 6, 52838–52849. [Google Scholar] [CrossRef]
- Pérez-Venegas, M.; Rodríguez-Treviño, A.M.; Juaristi, E. Dual Mechanoenzymatic Kinetic Resolution of (±)-Ketorolac. ChemCatChem 2020, 12, 1782–1788. [Google Scholar] [CrossRef]
- Contesini, F.J.; de Oliveira Carvalho, P. Esterification of (R,S)-Ibuprofen by native and commercial lipases in a two-phase system containing ionic liquids. Tetrahedron Asymmetry 2006, 17, 2069–2073. [Google Scholar] [CrossRef]
- Kato, D.-I.; Tatsumi, T.; Bansho, A.; Teruya, K.; Yoshida, H.; Takeo, M.; Negoro, S. Enantiodifferentiation of ketoprofen by Japanese firefly luciferase from Luciola Lateralis. J. Mol. Catal. B Enzym. 2011, 69, 140–146. [Google Scholar] [CrossRef]
- Chavez-Flores, D.; Salvador, J.M. Facile conversion of racemic ibuprofen to (S)-ibuprofen. Tetrahedron Asymmetry 2012, 23, 237–239. [Google Scholar] [CrossRef]
- Terao, Y.; Ijima, Y.; Kakidani, H.; Ohta, H. Enzymatic synthesis of (R)-flurbiprofen. Bull. Chem. Soc. Jap. 2003, 76, 2395–2397. [Google Scholar] [CrossRef]
- Enoki, J.; Linhorst, M.; Busch, F.; Baraibar, Á.G.; Miyamoto, K.; Kourist, R.; Mügge, C. Preparation of optically pure flurbiprofen via an integrated chemo-enzymatic synthesis pathway. Mol. Catal. 2019, 467, 135–142. [Google Scholar] [CrossRef]
- Tassano, E.; Faber, K.; Hall, M. Biocatalytic parallel interconnected dynamic asymmetric disproportionation of alpha-substituted aldehydes: Atom-efficient access to enantiopure (S)-profens and profenols. Adv. Synth. Catal. 2018, 360, 2742–2751. [Google Scholar] [CrossRef] [Green Version]
- Galletti, P.; Emer, E.; Gucciardo, G.; Quintavalla, A.; Pori, M.; Giacomini, D. Chemoenzymatic synthesis of (2S)-2-arylpropanols through a dynamic kinetic resolution of 2-arylpropanals with alcohol dehydrogenases. Org. Biomol. Chem. 2010, 8, 4117–4123. [Google Scholar] [CrossRef] [PubMed]
- Friest, J.A.; Maezato, Y.; Broussy, S.; Blum, P.; Berkowitz, D.B. Use of a robust dehydrogenase from an archael hyperthermophile in asymmetric catalysis-dynamic reductive kinetic resolution entry into (S)-profens. J. Am. Chem. Soc. 2010, 132, 5930–5931. [Google Scholar] [CrossRef] [PubMed]
- Galletti, P.; Pori, M.; Funiciello, F.; Soldati, R.; Ballardini, A.; Giacomini, D. Laccase-mediator system for alcohol oxidation to carbonyls or carboxylic acids: Toward a sustainable synthesis of profens. ChemSusChem 2014, 7, 2684–2689. [Google Scholar] [CrossRef]
- Lotti, M.; Pleiss, J.; Valero, F.; Ferrer, P. Enzymatic Production of Biodiesel: Strategies to Overcome Methanol Inactivation. Biotechnol. J. 2018, 13, e1700155. [Google Scholar] [CrossRef] [PubMed]
- Lotti, M.; Pleiss, J.; Valero, F.; Ferrer, P. Effects of methanol on lipases: Molecular, kinetic and process issues in the production of biodiesel. Biotechnol. J. 2015, 10, 22–30. [Google Scholar] [CrossRef]
- Kulschewski, T.; Sasso, F.; Secundo, F.; Lotti, M.; Pleiss, J. Molecular mechanism of deactivation of C. antarctica lipase B by methanol. J. Biotechnol. 2013, 168, 462–469. [Google Scholar] [CrossRef]
- José, C.; Bonetto, R.D.; Gambaro, L.A.; Torres, M.d.P.G.; Foresti, M.L.; Ferreira, M.L.; Briand, L.E. Investigation of the causes of deactivation–degradation of the commercial biocatalyst Novozym® 435 in ethanol and ethanol–aqueous media. J. Mol. Catal. B Enzym. 2011, 71, 95–107. [Google Scholar] [CrossRef]
- José, C.; Briand, L.E. Deactivation of Novozym® 435 during the esterification of ibuprofen with ethanol: Evidences of the detrimental effect of the alcohol. React. Kinet. Mech. Catal. 2009, 99, 17–22. [Google Scholar] [CrossRef]
- Mangiagalli, M.; Ami, D.; de Divitiis, M.; Brocca, S.; Catelani, T.; Natalello, A.; Lotti, M. Short-chain alcohols inactivate an immobilized industrial lipase through two different mechanisms. Biotechnol. J. 2022, 2100712, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Stergiou, P.Y.; Foukis, A.; Filippou, M.; Koukouritaki, M.; Parapouli, M.; Theodorou, L.G.; Hatziloukas, E.; Afendra, A.; Pandey, A.; Papamichael, E.M. Advances in lipase-catalyzed esterification reactions. Biotechnol. Adv. 2013, 31, 1846–1859. [Google Scholar] [CrossRef] [PubMed]
- Chaibakhsh, N.; Abdul Rahman, M.B.; Basri, M.; Salleh, A.B.; Rahman, R.N.Z.R.A. Effect of alcohol chain length on the optimum conditions for lipase-catalyzed synthesis of adipate esters. Biocatal. Biotrans. 2009, 27, 303–308. [Google Scholar] [CrossRef]
- Shimada, Y.; Watanabe, Y.; Samukawa, T.; Sugihara, A.; Noda, H.; Fukuda, H.; Tominaga, Y. Conversion of vegetable oil to biodiesel using immobilized Candida antarctica lipase. J. Am. Oil Chem. Soc. 1999, 76, 789–793. [Google Scholar] [CrossRef]
- Wang, L.; Du, W.; Liu, D.; Li, L.; Dai, N. Lipase-catalyzed biodiesel production from soybean oil deodorizer distillate with absorbent present in tert-butanol system. J. Mol. Catal. B Enzym. 2006, 43, 29–32. [Google Scholar] [CrossRef]
- Panico, A.M.; Cardile, V.; Vittorio, F.; Ronsisvalle, G.; Scoto, G.M.; Parenti, C.; Gentile, B.; Morrone, R.; Nicolosi, G. Different in vitro activity of flurbiprofen and its enantiomers on human articular cartilage. Il Farm. 2003, 58, 1339–1344. [Google Scholar] [CrossRef]
- Morrone, R.; Piattelli, M.; Nicolosi, G. Resolution of Racemic Acids by Irreversible Lipase-Catalyzed Esterification in Organic Solvents. Eur. J. Org. Chem. 2001, 2001, 1441–1443. [Google Scholar] [CrossRef]
- Brodzka, A.; Koszelewski, D.; Zysk, M.; Ostaszewski, R. The mechanistic promiscuity of the enzymatic esterification of chiral carboxylic acids. Catal. Commun. 2018, 106, 82–86. [Google Scholar] [CrossRef]
- Koszelewski, D.; Brodzka, A.; Żądło, A.; Paprocki, D.; Trzepizur, D.; Zysk, M.; Ostaszewski, R. Dynamic Kinetic Resolution of 3-Aryl-4-pentenoic Acids. ACS Catal. 2016, 6, 3287–3292. [Google Scholar] [CrossRef]
- Brodzka, A.; Koszelewski, D.; Cwiklak, M.; Ostaszewski, R. Studies on the chemoenzymatic synthesis of 3-phenyl-GABA and 4-phenyl-pyrrolid-2-one: The influence of donor of the alkoxy group on enantioselective esterification. Tetrahedron Asymmetry 2013, 24, 427–433. [Google Scholar] [CrossRef]
- Brodzka, A.; Koszelewski, D.; Ostaszewski, R. The studies on chemoenzymatic synthesis of Femoxetine. J. Mol. Catal. B Enzym. 2012, 82, 96–101. [Google Scholar] [CrossRef]
- Koszelewski, D.; Zysk, M.; Brodzka, A.; Zadlo, A.; Paprocki, D.; Ostaszewski, R. Evaluation of a new protocol for enzymatic dynamic kinetic resolution of 3-hydroxy-3-(aryl)propanoic acids. Org. Biomol. Chem. 2015, 13, 11014–11020. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.S.; Fujimoto, Y.; Girdaukas, G.; Sih, C.J. Quantitative analyses of biochemical kinetic resolutions of enantiomers. J. Am. Chem. Soc. 2002, 104, 7294–7299. [Google Scholar] [CrossRef]
- Faber, K. (Ed.) Biotransformations in Organic Chemistry: A Textbook, 7th ed.; Springer: New York, NY, USA, 2018. [Google Scholar]
- Klibanov, A.M. Improving enzymes by using them in organic solvents. Nature 2001, 409, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Laane, C.; Boeren, S.; Vos, K.; Veeger, C. Rules for optimization of biocatalysis in organic solvents. Biotechnol. Bioeng. 1987, 30, 81–87. [Google Scholar] [CrossRef]
- Chua, L.S.; Sarmidi, M.R. Effect of solvent and initial water content on (R,S)-1-phenylethanol resolution. Enzym. Microb. Technol. 2006, 38, 551–556. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, Q.; Zhang, Z.; Ma, J.; Feng, Y. Solvent effects on the enantioselectivity of the thermophilic lipase QLM in the resolution of (R,S)-2-octanol and (R,S)-2-pentanol. J. Mol. Catal. B Enzym. 2009, 56, 146–150. [Google Scholar] [CrossRef]
- Ke, T.; Wescott, C.R.; Klibanov, A.M. Prediction of the Solvent Dependence of Enzymatic Prochiral Selectivity by Means of Structure-Based Thermodynamic Calculations. J. Am. Chem. Soc. 1996, 118, 3366–3374. [Google Scholar] [CrossRef]
- Wescott, C.R.; Noritomi, H.; Klibanov, A.M. Rational Control of Enzymatic Enantioselectivity through Solvation Thermodynamics. J. Am. Chem. Soc. 1996, 118, 10365–10370. [Google Scholar] [CrossRef]
- Savile, C.K.; Kazlauskas, R.J. How substrate solvation contributes to the enantioselectivity of subtilisin toward secondary alcohols. J. Am. Chem. Soc. 2005, 127, 12228–12229. [Google Scholar] [CrossRef] [PubMed]
- Carrea, G.; Riva, S. Properties and Synthetic Applications of Enzymes in Organic Solvents. Angew. Chem. Int. Ed. 2000, 39, 2226–2254. [Google Scholar] [CrossRef]
- Duan, Z.Q.; Du, W.; Liu, D.H. The solvent influence on the positional selectivity of Novozym 435 during 1,3-diolein synthesis by esterification. Biores. Technol. 2010, 101, 2568–2571. [Google Scholar] [CrossRef]
- Danelius, E.; Andersson, H.; Jarvoll, P.; Lood, K.; Grafenstein, J.; Erdelyi, M. Halogen Bonding: A Powerful Tool for Modulation of Peptide Conformation. Biochemistry 2017, 56, 3265–3272. [Google Scholar] [CrossRef] [PubMed]
- Sirimulla, S.; Bailey, J.B.; Vegesna, R.; Narayan, M. Halogen interactions in protein-ligand complexes: Implications of halogen bonding for rational drug design. J. Chem. Inf. Model. 2013, 53, 2781–2791. [Google Scholar] [CrossRef]
- Borozan, S.Z.; Stojanovic, S.D. Halogen bonding in complexes of proteins and non-natural amino acids. Comput. Biol. Chem. 2013, 47, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Mizuguchi, E.; Itanami, M.; Achiwa, K. Enzymatic resolution of new anti-inflammatory drug etodolac. Heterocycles 1997, 46, 149–152. [Google Scholar]
- Ghanem, A.; Aboul-Enein, M.N.; El-Azzouny, A.; El-Behairy, M.F. Lipase-mediated enantioselective kinetic resolution of racemic acidic drugs in non-standard organic solvents: Direct chiral liquid chromatography monitoring and accurate determination of the enantiomeric excesses. J. Chromatogr. A 2010, 1217, 1063–1074. [Google Scholar] [CrossRef]
- Ren, W.; Chang, W.; Wang, Y.; Li, J.; Shi, Y. Pd-Catalyzed Regiodivergent Hydroesterification of Aryl Olefins with Phenyl Formate. Org. Lett. 2015, 17, 3544–3547. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, J.; Huang, Z.; Liao, H.; Peng, S.; Lehmann, J.; Zhang, Y. NO-donating tacrine derivatives as potential butyrylcholinesterase inhibitors with vasorelaxation activity. Bioorg. Med. Chem. Lett. 2013, 23, 3162–3165. [Google Scholar] [CrossRef]
- Cikla, P.; Ozsavci, D.; Bingol-Ozakpinar, O.; Sener, A.; Cevik, O.; Ozbas-Turan, S.; Akbuga, J.; Sahin, F.; Kucukguzel, S.G. Synthesis, cytotoxicity, and pro-apoptosis activity of etodolac hydrazide derivatives as anticancer agents. Archiv. Pharm. 2013, 346, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Rana, N.K.; Singh, V.K. Enantioselective enolate protonation in sulfa-michael addition to alpha-substituted n-acryloyloxazolidin-2-ones with bifunctional organocatalyst. Org. Lett. 2011, 13, 6520–6523. [Google Scholar] [CrossRef]
- Miyamoto, K.; Ohta, H. Enzyme-mediated asymmetric decarboxylation of disubstituted malonic acids. J. Am. Chem. Soc. 2002, 112, 4077–4078. [Google Scholar] [CrossRef]
- Chikusa, Y.; Fujimoto, T.; Ikunaka, M.; Inoue, T.; Kamiyama, S.; Maruo, K.; Matsumoto, J.; Matsuyama, K.; Moriwaki, M.; Nohira, H.; et al. (s)-3-methyl-2-phenylbutylamine, a versatile agent to resolve chiral, racemic carboxylic acids1. Org. Proc. Res. Dev. 2002, 6, 291–296. [Google Scholar] [CrossRef]



| Entry | Substrate a | Alkoxy Donor | Time (h) | Conv. (%) b | ees (%) c | eep (%) c | Ed |
|---|---|---|---|---|---|---|---|
| 1 |  | Trimethyl orthoacetate (Alk-D1) | 72 | 46 | 55 | 65 | 8 |
| 2 | Triethyl orthoacetate (Alk-D2) | 72 | 17 | 13 | 62 | 5 | |
| 3 | Trimethyl orthobenzoate (Alk-D3) | 72 | 68 | 63 | 29 | 3 | |
| 4 | Triethyl orthobenzoate (Alk-D4) | 72 | 29 | 25 | 62 | 5 | |
| 5 |  | Trimethyl orthoacetate (Alk-D1) | 7 | 42 | 44 | 60 | 6 |
| 6 | Triethyl orthoacetate (Alk-D2) | 24 | 41 | 50 | 71 | 10 | |
| 7 | Trimethyl orthobenzoate (Alk-D3) | 36 | 38 | 27 | 44 | 3 | |
| 8 | Triethyl orthobenzoate (Alk-D4) | 7 | 38 | 27 | 45 | 3 |

| Entry | Substrate a | Solvent (log p) b | Time (h) | Conv. (%) c | ees (%) d | eep (%) d | Ee |
|---|---|---|---|---|---|---|---|
| 1 |  | 1,4-Dioxane (−0.31) | 72 | 0 | N.D. f | N.D. f | N.D. f |
| 2 | CH3CN (0.17) | 72 | 0 | N.D. f | N.D. f | N.D. f | |
| 3 | Acetone (0.20) | 72 | 0 | N.D. f | N.D. f | N.D. f | |
| 4 | THF (0.40) | 72 | 0 | N.D. f | N.D. f | N.D. f | |
| 5 | CH2Cl2 (1.01) | 72 | 27 | 3 | 8 | 1 | |
| 6 | t-Amyl alcohol (1.09) | 72 | 0 | N.D. f | N.D. f | N.D. f | |
| 7 | CHCl3 (1.67) | 72 | 0 | N.D. f | N.D. f | N.D. f | |
| 8 | Cyclohexane (2.50) | 72 | 72 | 26 | 10 | 2 | |
| 9 | PhCH3 (2.52) | 72 | 46 | 55 | 65 | 8 | |
| 10 | n-Hexane (3.00) | 72 | 64 | 16 | 9 | 1 | |
| 11 | Xylene (3.01) | 72 | 62 | 61 | 38 | 4 | |
| 12 | Isooctane (3.75) | 72 | 89 | 23 | 3 | 1 | |
| 13 |  | 1,4-Dioxane (−0.31) | 7 | 0 | N.D. f | N.D. f | N.D. f |
| 14 | CH3CN (0.17) | 7 | 0 | N.D. f | N.D. f | N.D. f | |
| 15 | Acetone (0.20) | 7 | 0 | N.D. f | N.D. f | N.D. f | |
| 16 | THF (0.40) | 7 | 0 | N.D. f | N.D. f | N.D. f | |
| 17 | CH2Cl2 (1.01) | 7 | 17 | 2 | 10 | 1 | |
| 18 | t-Amyl alcohol (1.09) | 7 | 0 | N.D. f | N.D. f | N.D. f | |
| 19 | CHCl3 (1.67) | 7 | 0 | N.D. f | N.D. f | N.D. f | |
| 20 | Cyclohexane (2.50) | 7 | 47 | 54 | 62 | 7 | |
| 21 | PhCH3 (2.52) | 7 | 19 | 17 | 75 | 8 | |
| 22 | n-Hexane (3.00) | 7 | 59 | 74 | 52 | 7 | |
| 23 | Xylene (3.01) | 7 | 37 | 29 | 49 | 4 | |
| 24 | Isooctane (3.75) | 7 | 57 | 63 | 48 | 5 |

| Entry | Substrate a | Solvent | Conv. (%) b | ees (%) c | eep (%) c | Ed |
|---|---|---|---|---|---|---|
| 1 |  | n-Hexane | 77 | 69 | 21 | 3 |
| 2 | PhCH3 | 16 | 4 | 21 | 2 | |
| 3 |  | n-Hexane | 66 | 97 | 49 | 11 |
| 4 | PhCH3 | 53 | 81 | 71 | 14 | |
| 5 |  | n-Hexane | 0 | N.D. e | N.D. e | N.D. e |
| 6 | PhCH3 | 0 | N.D. e | N.D. e | N.D. e |

| Entry | Substrate a | Alkoxy Donor | Solvent | Time (h) | Conv. (%) b | ees (%) c/ Yield (%) d | eep (%) c/ Yield (%) d | Ee |
|---|---|---|---|---|---|---|---|---|
| 1 |  | Alk-D1 | PhCH3 | 72 | 66 | 57/42 | 29/49 | 3 |
| 2 |  | Alk-D2 | n-Hexane | 7 | 46 | 56/45 | 67/32 | 9 |
| 3 |  | Alk-D2 | n-Hexane | 48 | 73 | 69/23 | 25/45 | 3 |
| 4 |  | Alk-D2 | PhCH3 | 48 | 66 | 97/28 | 51/52 | 12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zdun, B.; Cieśla, P.; Kutner, J.; Borowiecki, P. Expanding Access to Optically Active Non-Steroidal Anti-Inflammatory Drugs via Lipase-Catalyzed KR of Racemic Acids Using Trialkyl Orthoesters as Irreversible Alkoxy Group Donors. Catalysts 2022, 12, 546. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12050546
Zdun B, Cieśla P, Kutner J, Borowiecki P. Expanding Access to Optically Active Non-Steroidal Anti-Inflammatory Drugs via Lipase-Catalyzed KR of Racemic Acids Using Trialkyl Orthoesters as Irreversible Alkoxy Group Donors. Catalysts. 2022; 12(5):546. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12050546
Chicago/Turabian StyleZdun, Beata, Piotr Cieśla, Jan Kutner, and Paweł Borowiecki. 2022. "Expanding Access to Optically Active Non-Steroidal Anti-Inflammatory Drugs via Lipase-Catalyzed KR of Racemic Acids Using Trialkyl Orthoesters as Irreversible Alkoxy Group Donors" Catalysts 12, no. 5: 546. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12050546