Second-Generation Manganese(III) Porphyrins Bearing 3,5-Dichloropyridyl Units: Innovative Homogeneous and Heterogeneous Catalysts for the Epoxidation of Alkenes

 , , and
, , and
Abstract
:
1. Introduction
2. Results and Discussion
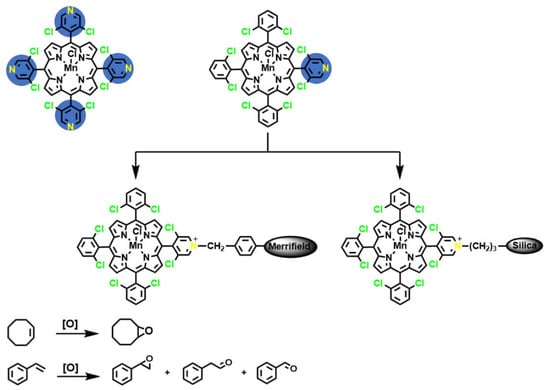
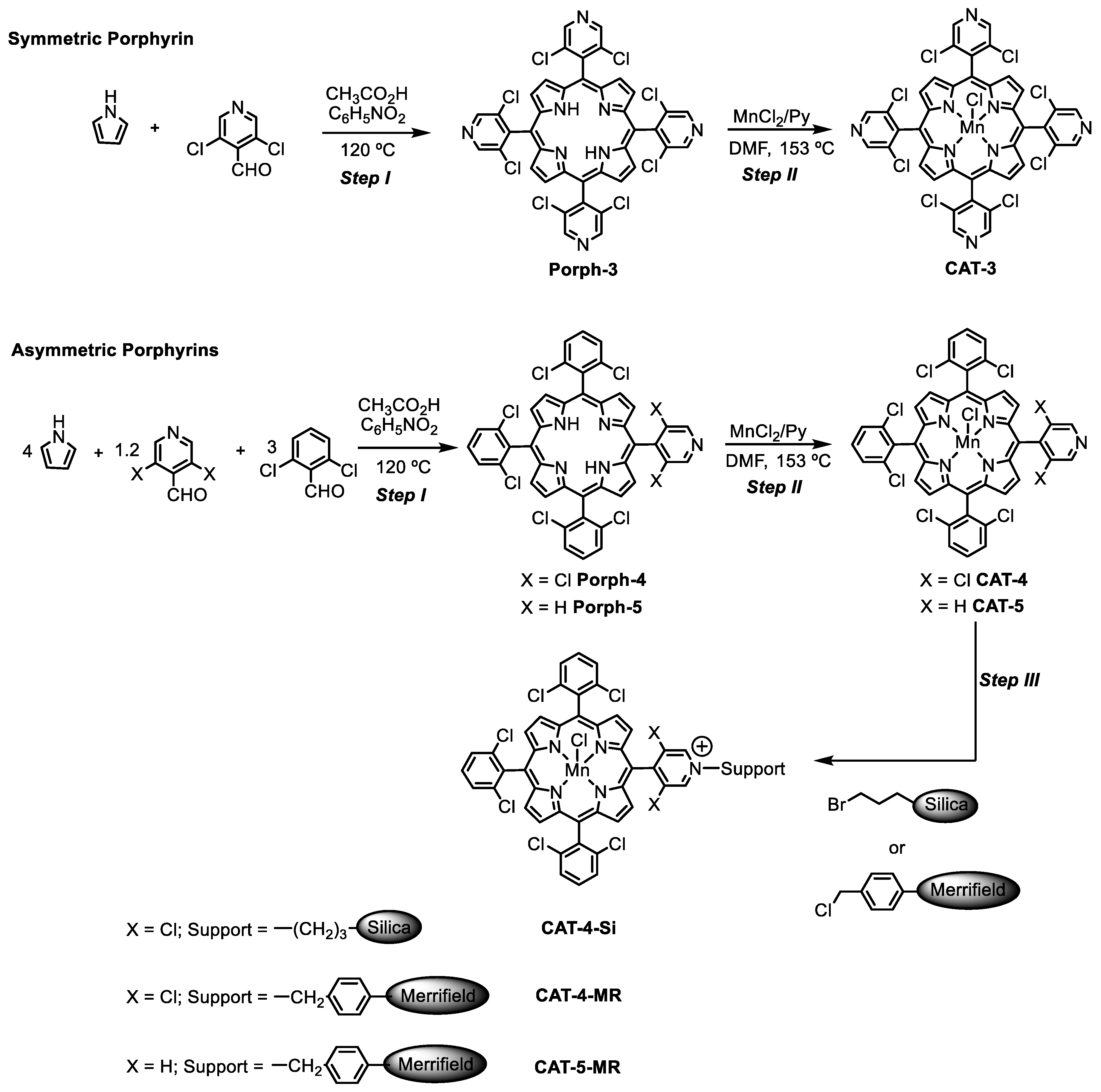
2.1. Synthesis of the Homogeneous and Heterogeneous Metalloporphyrin Catalysts
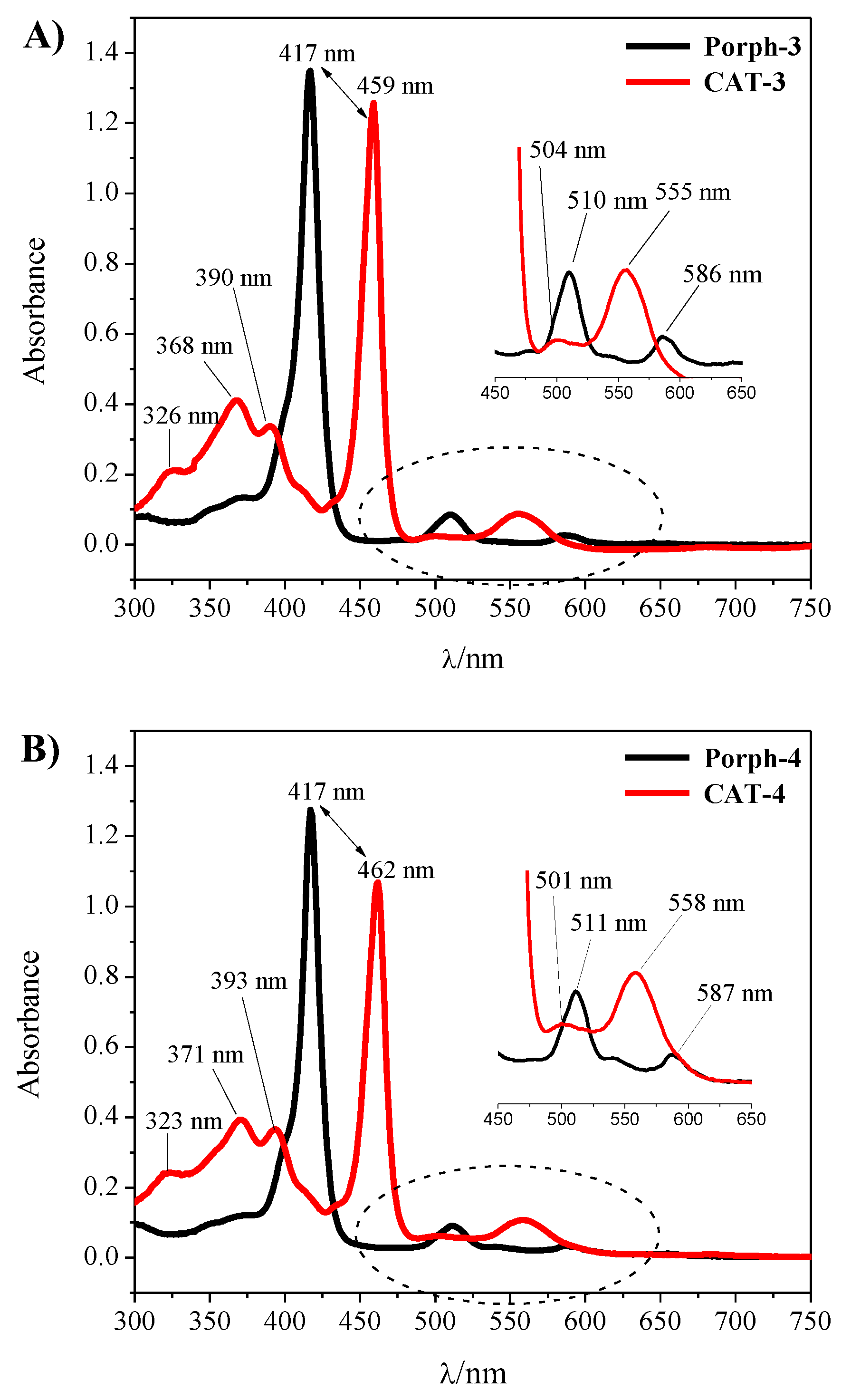
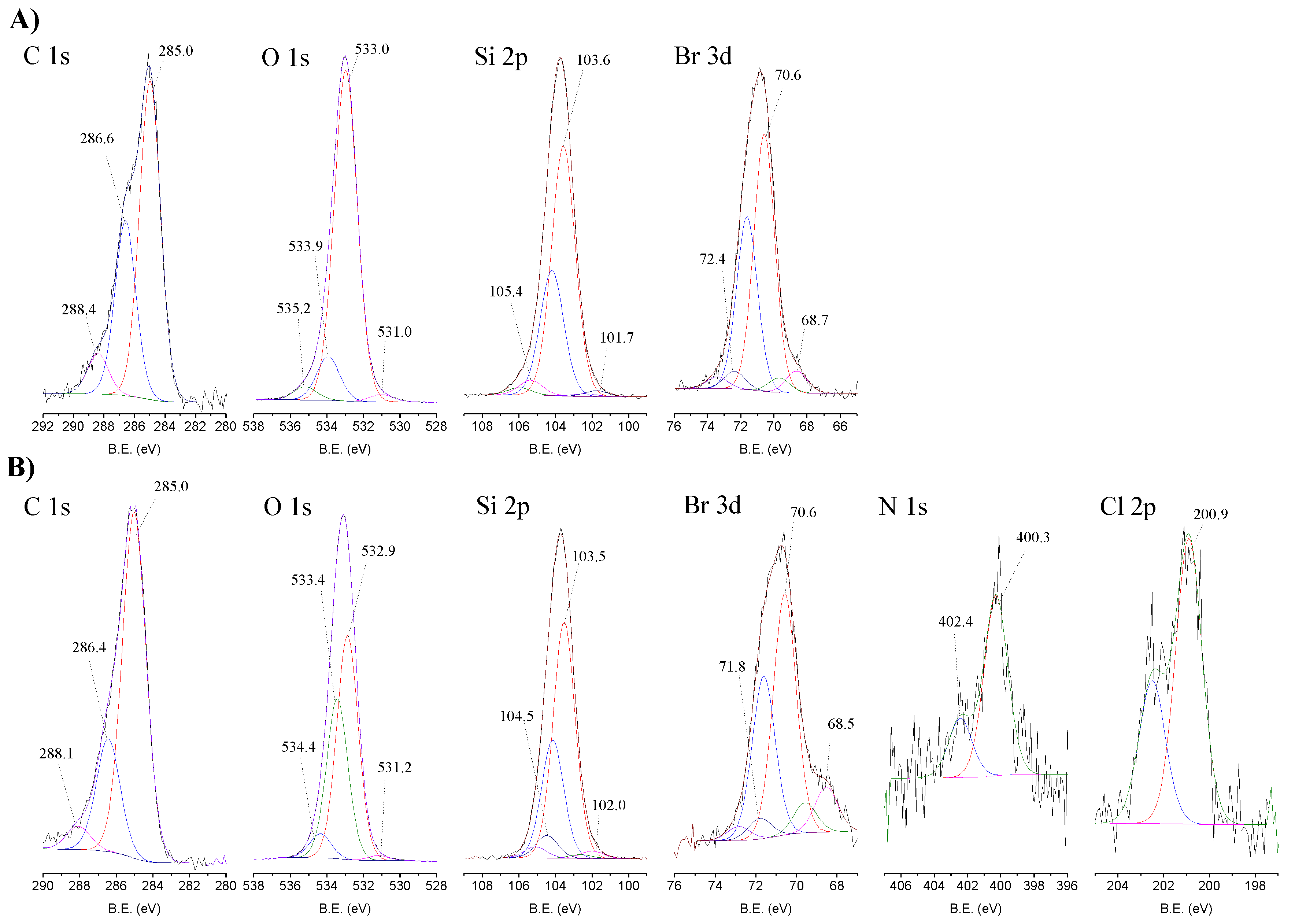
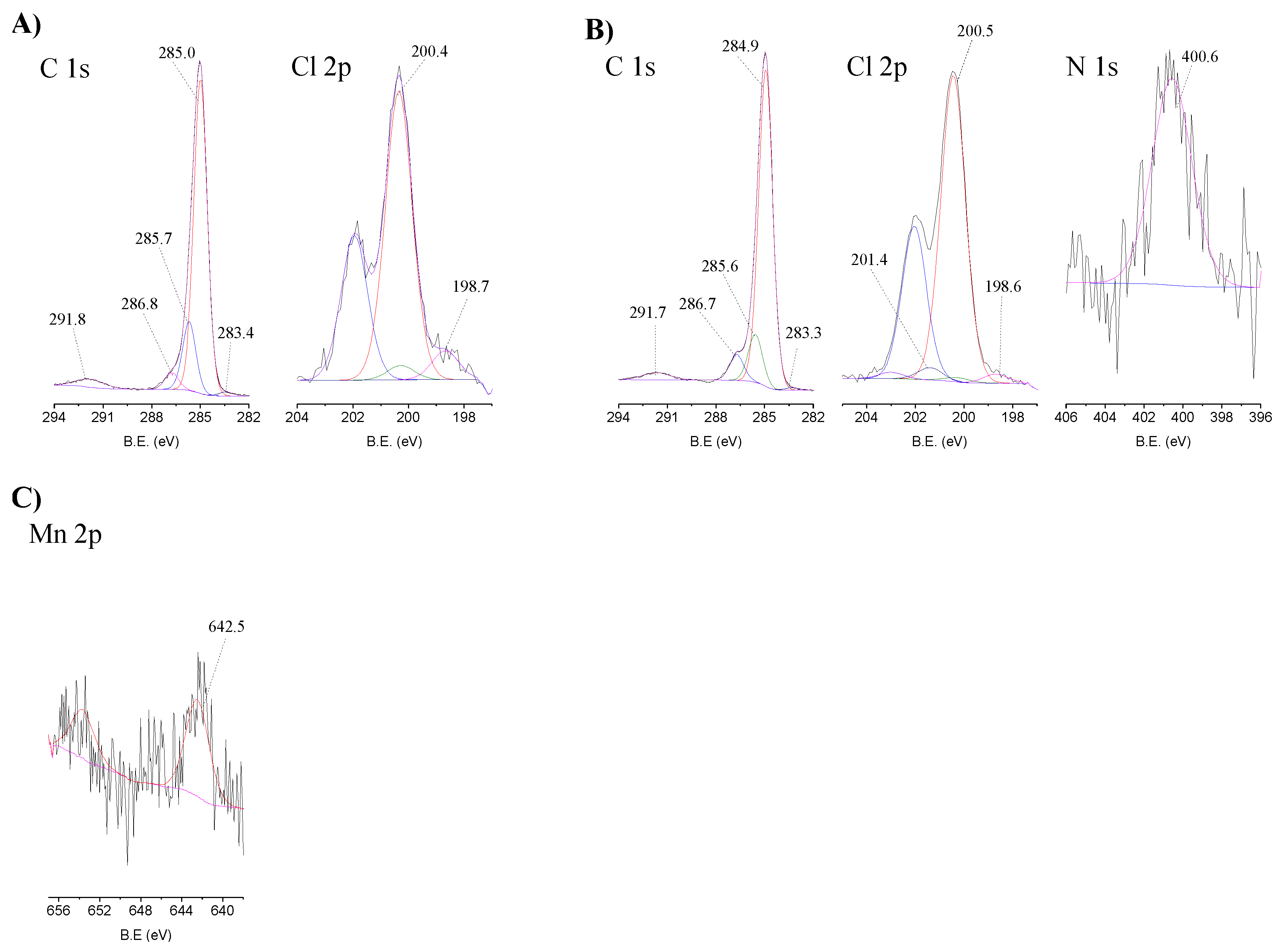
2.2. Structural Characterization
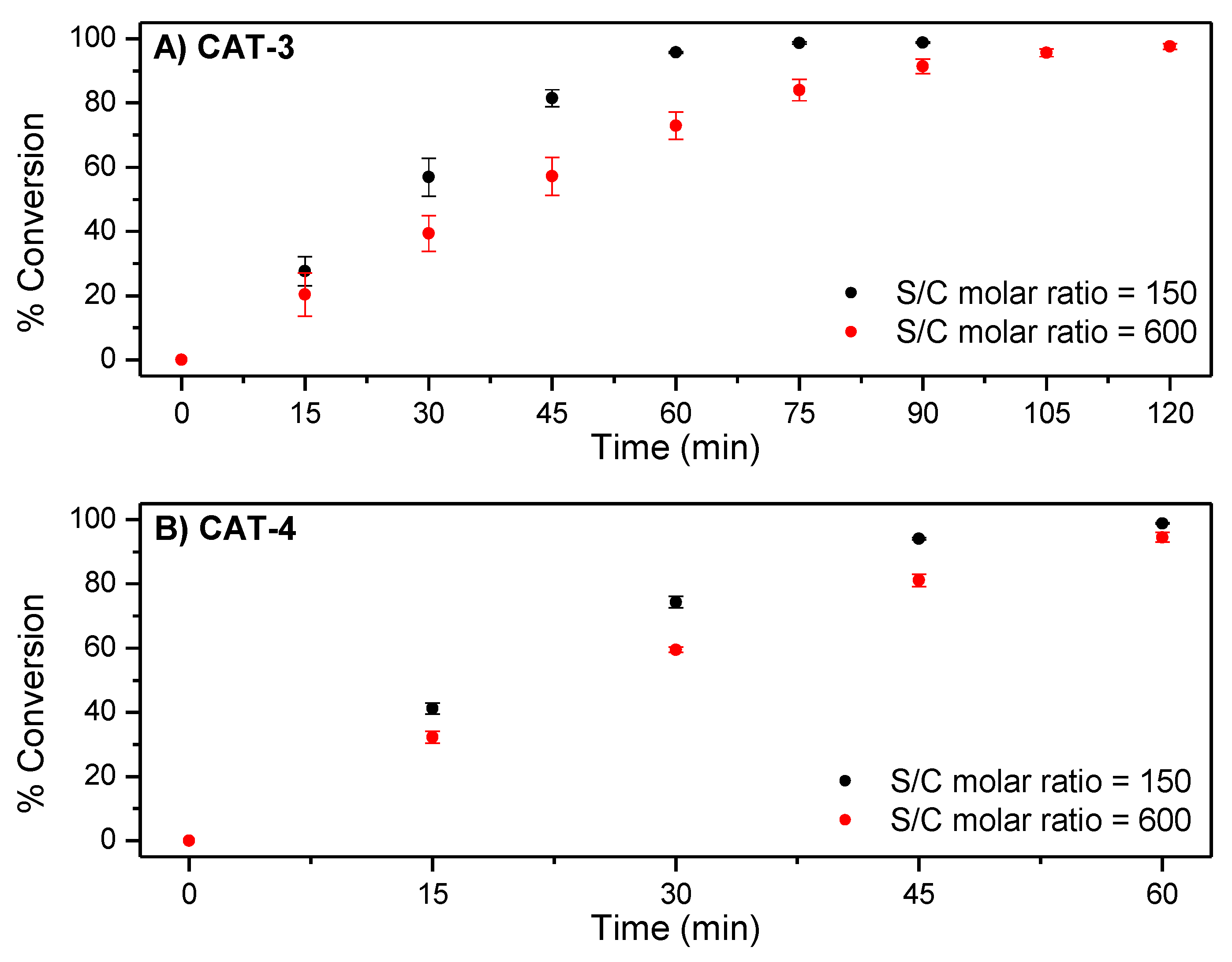
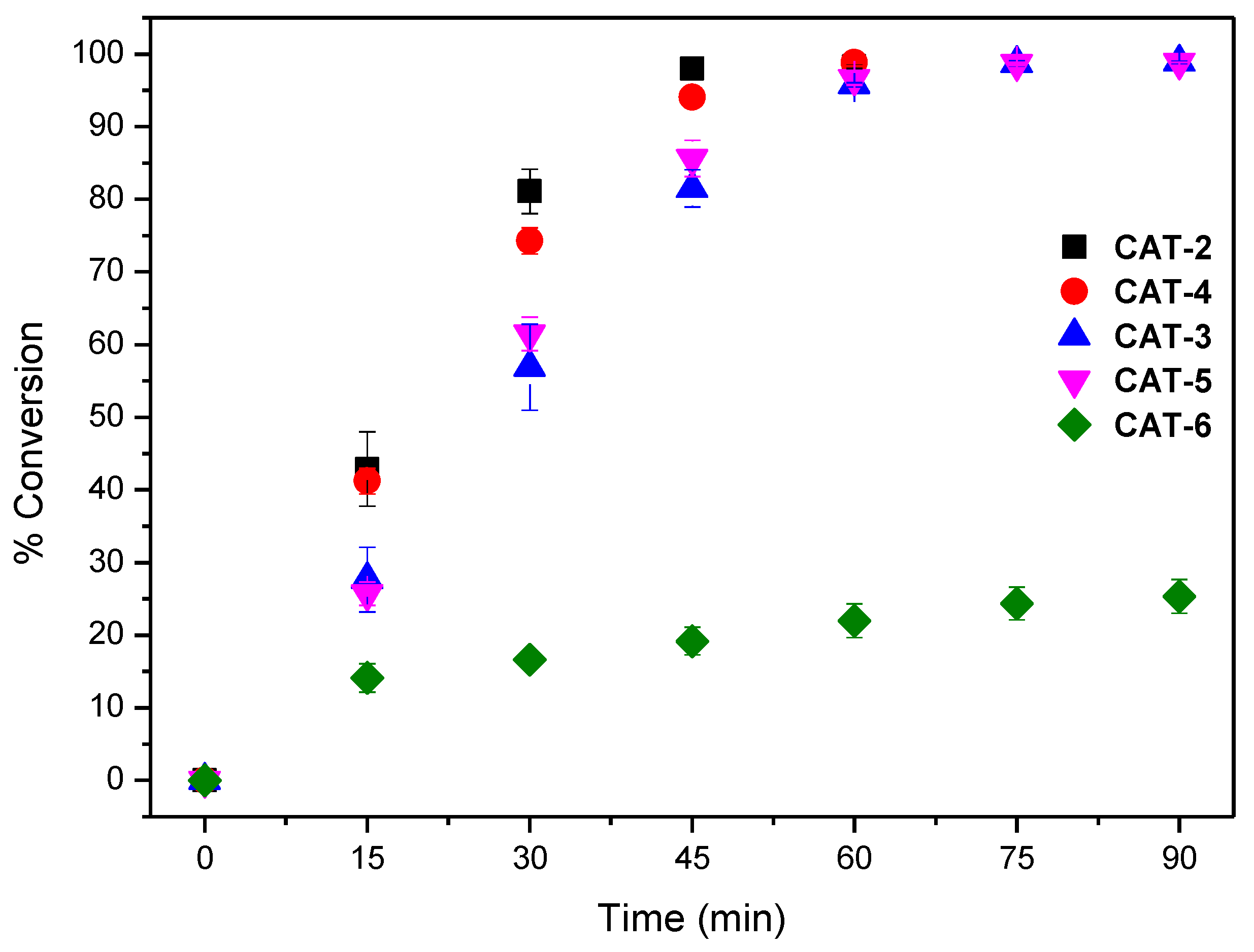
2.3. Oxidation of Cyclooctene Catalysed by CAT-3 and CAT-4 under Homogeneous Conditions
- (i)
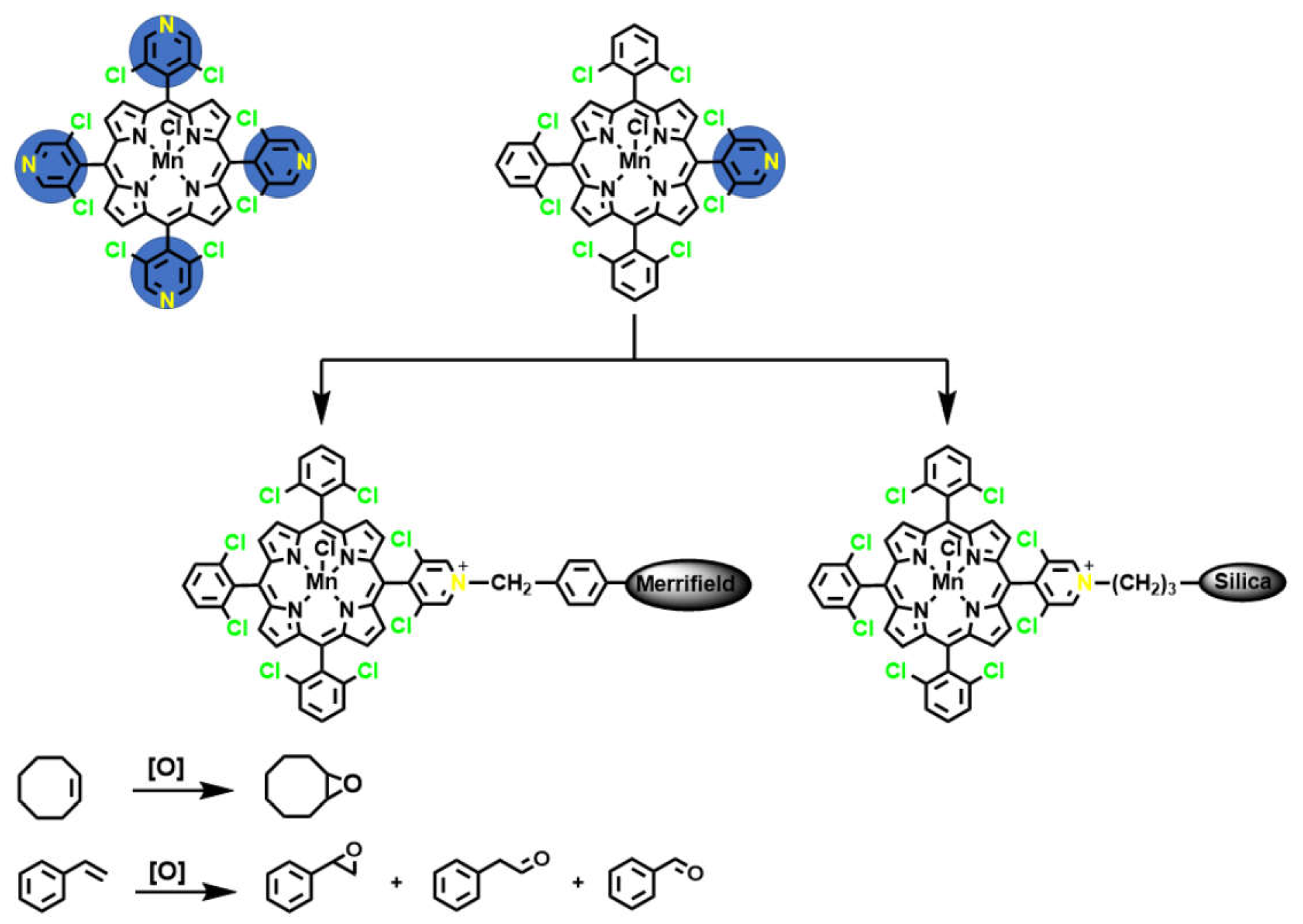
- the chloro[5,10,15,20-tetrakis(2,6-dichlorophenyl)porphyrinate]manganese(III), CAT-2, a highly efficient catalyst having all the meso positions substituted by 2,6-dichlorophenyl units;
- (ii)
- the chloro[5,10,15-tri(2,6-dichlorophenyl)-20-(pyridin-4-yl)porphyrinate]manganese(III), CAT-5, with one meso position substituted by a pyridine unit and the other three meso positions substituted by 2,6-dichlorophenyl groups; and
- (iii)
- the chloro[5,10,15,20-tetra(pyridin-4-yl)porphyrinate]manganese(III), CAT-6, with all the meso positions substituted by pyridine units.
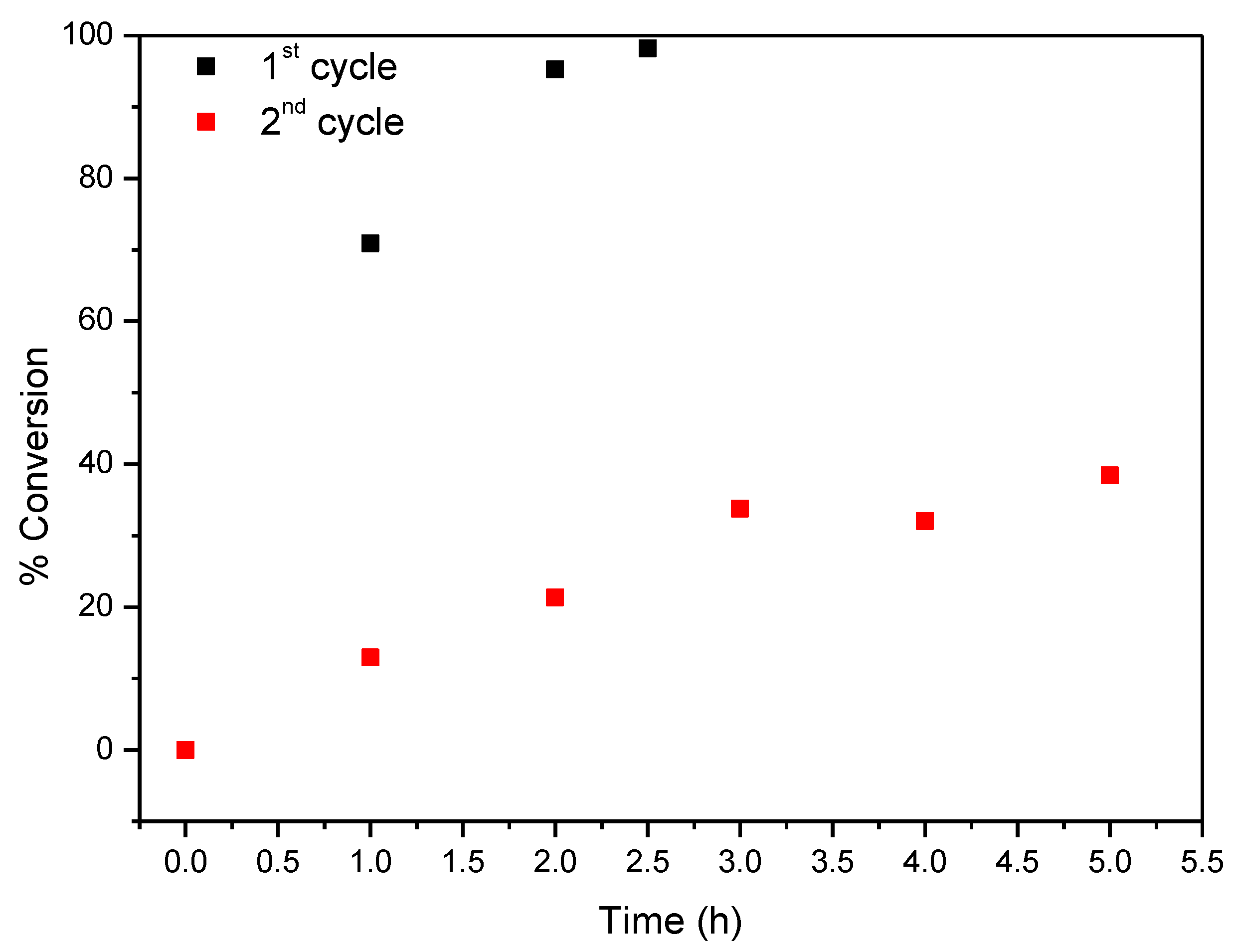
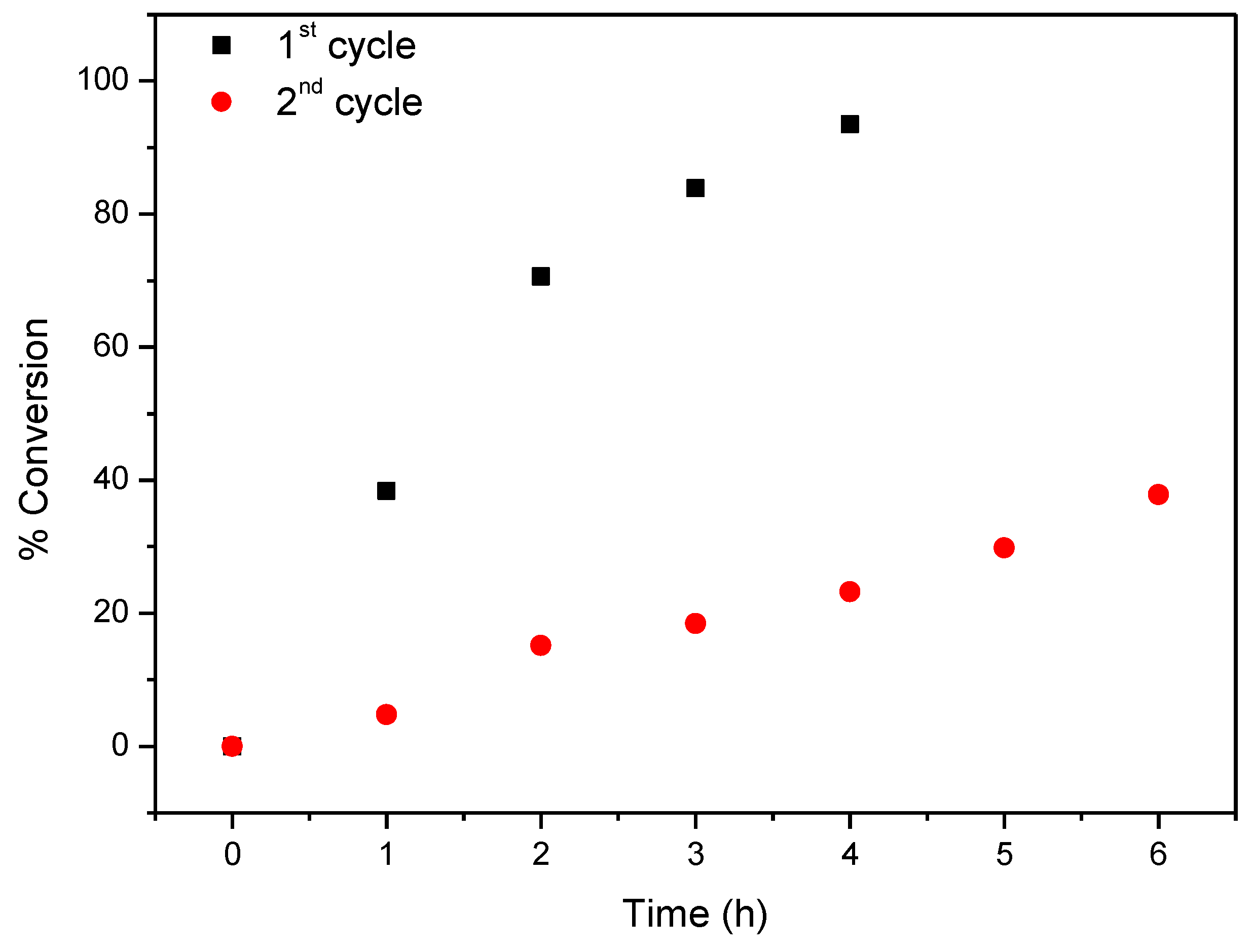
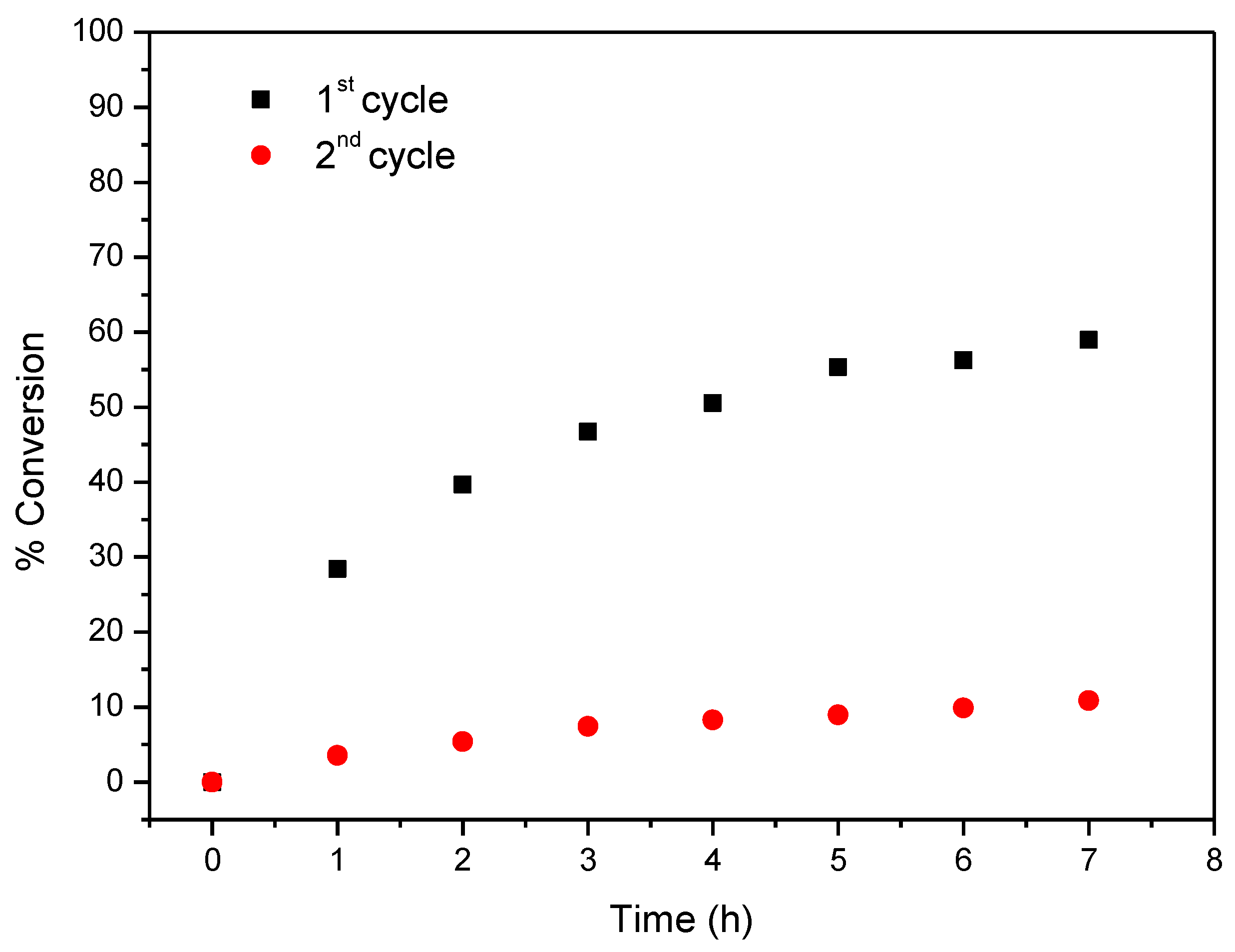
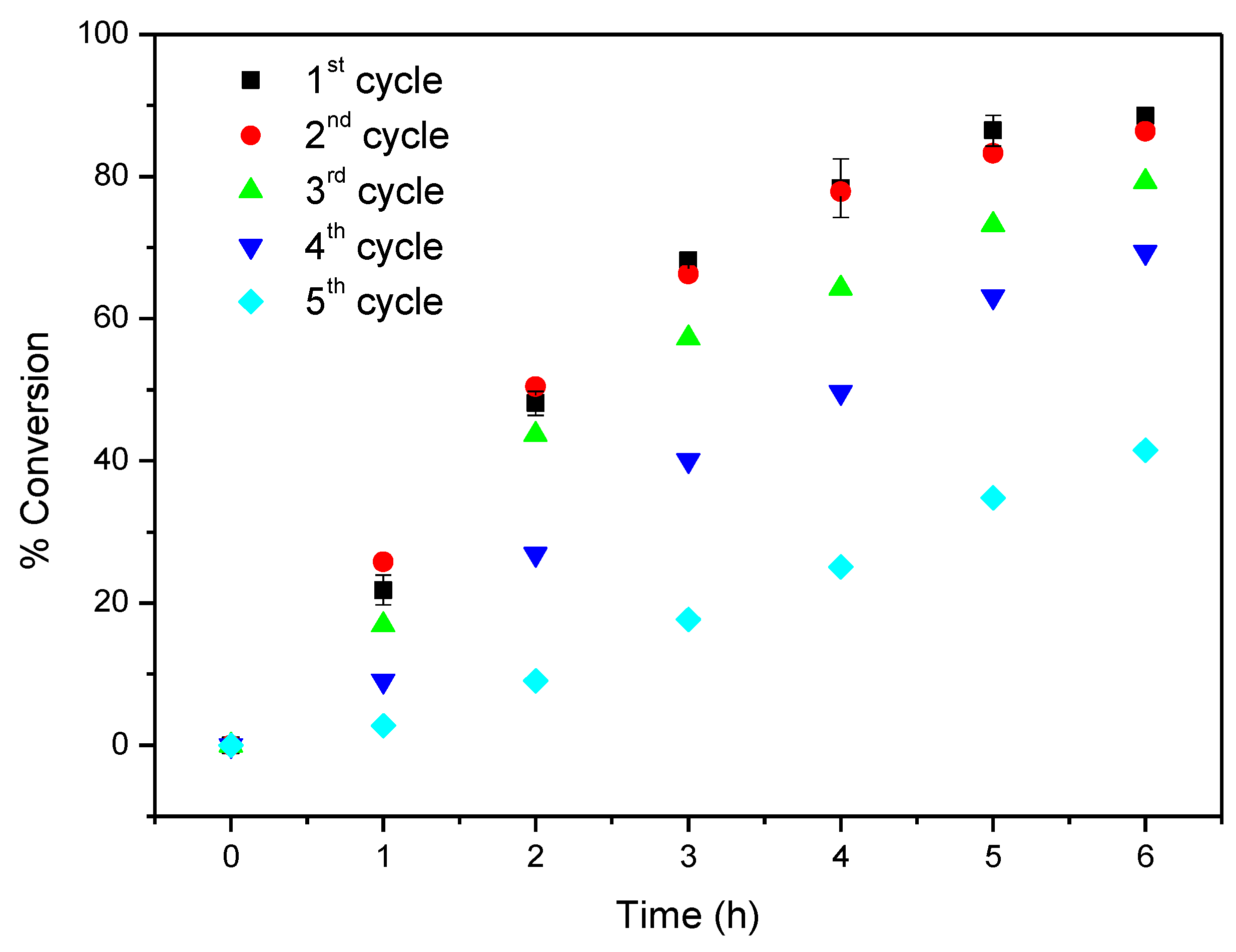
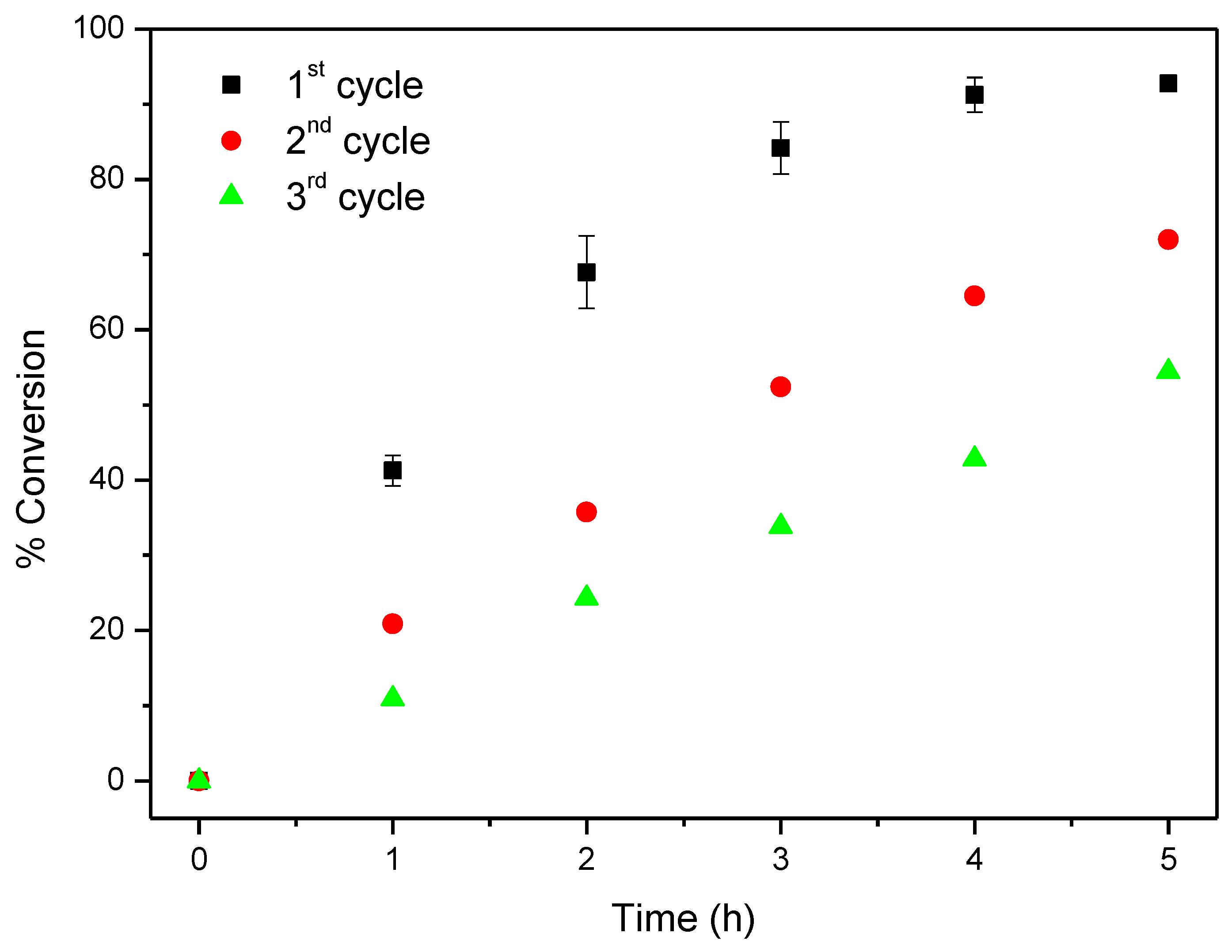
2.4. Oxidation of Cyclooctene under Heterogeneous Catalysis
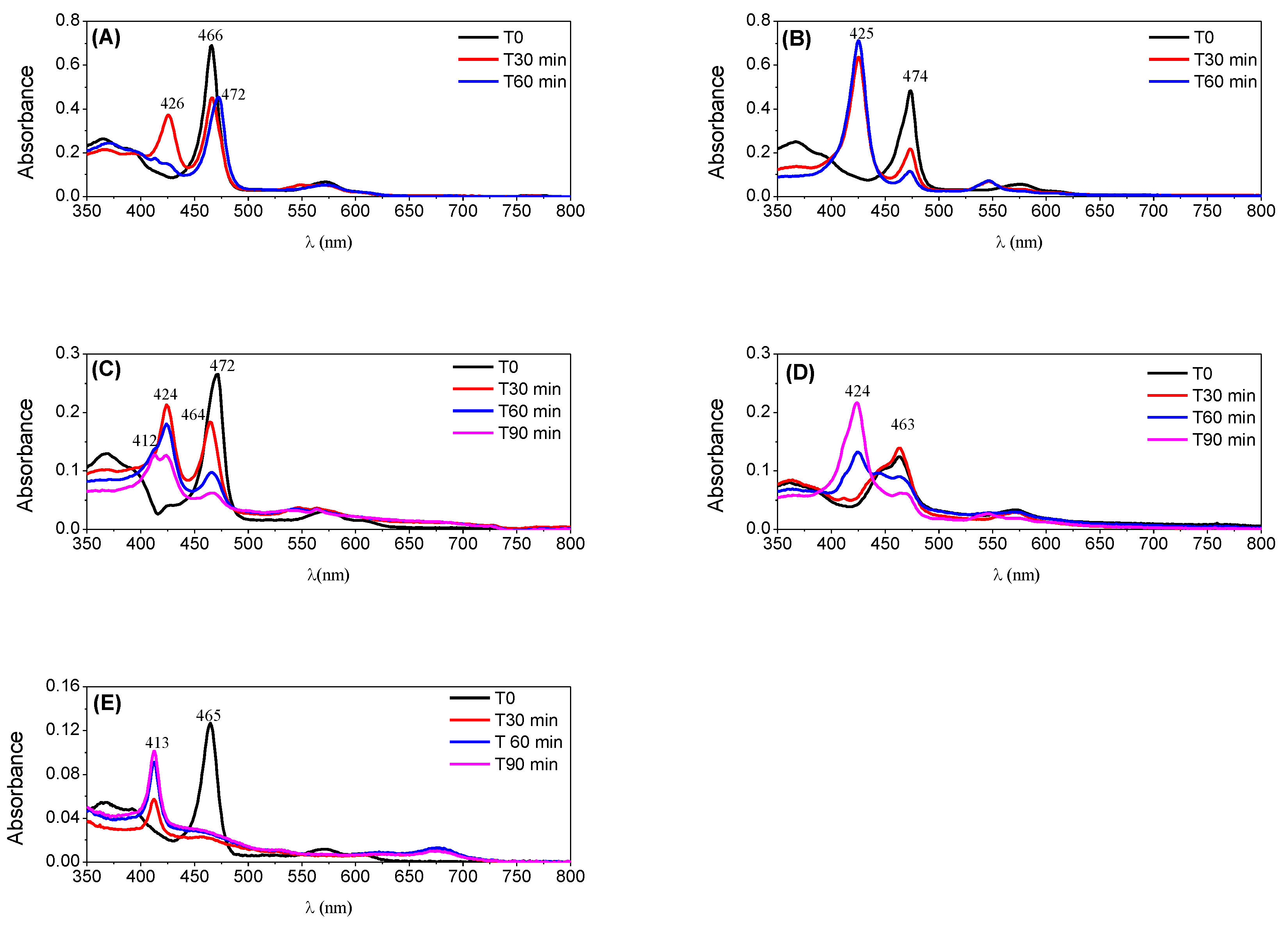
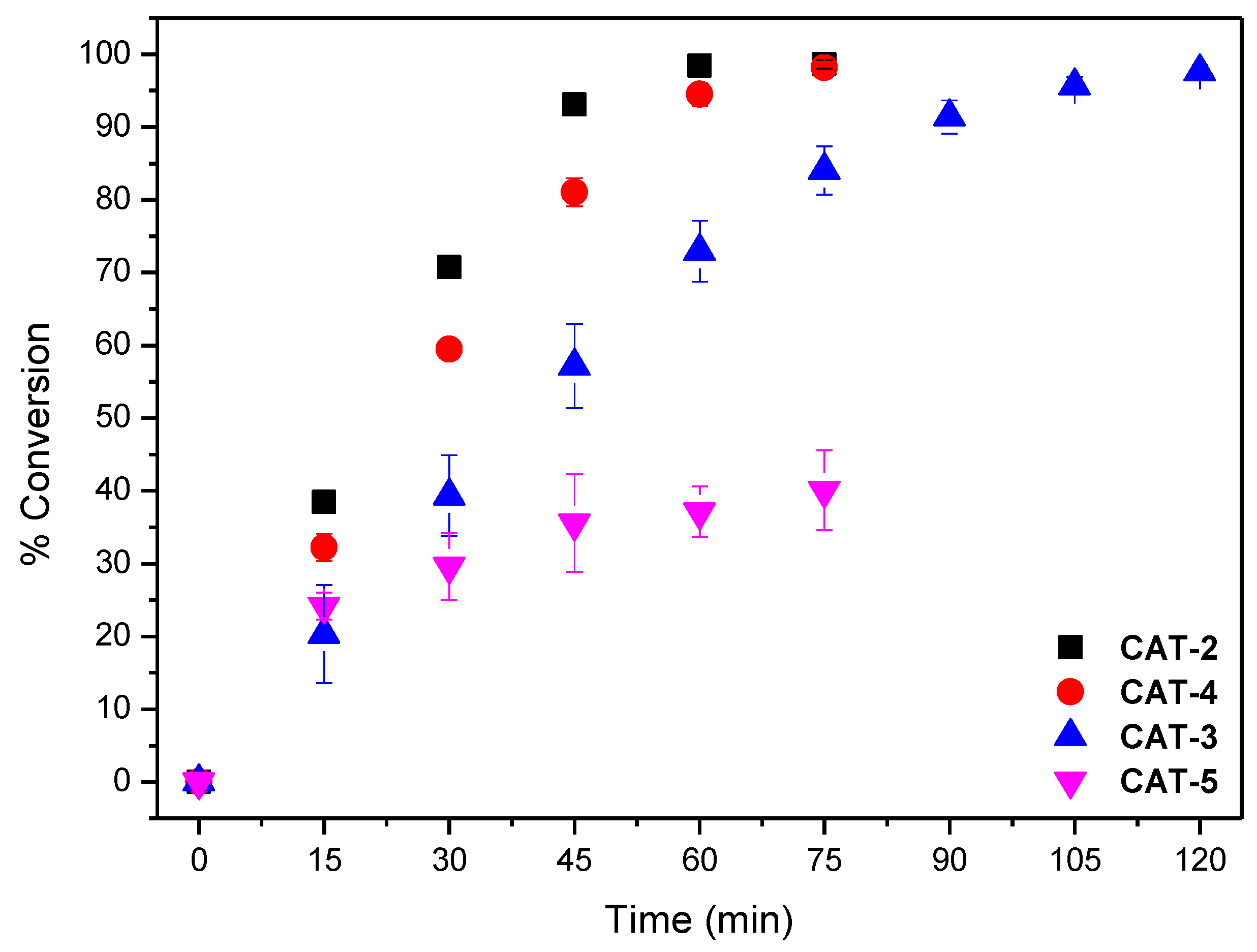
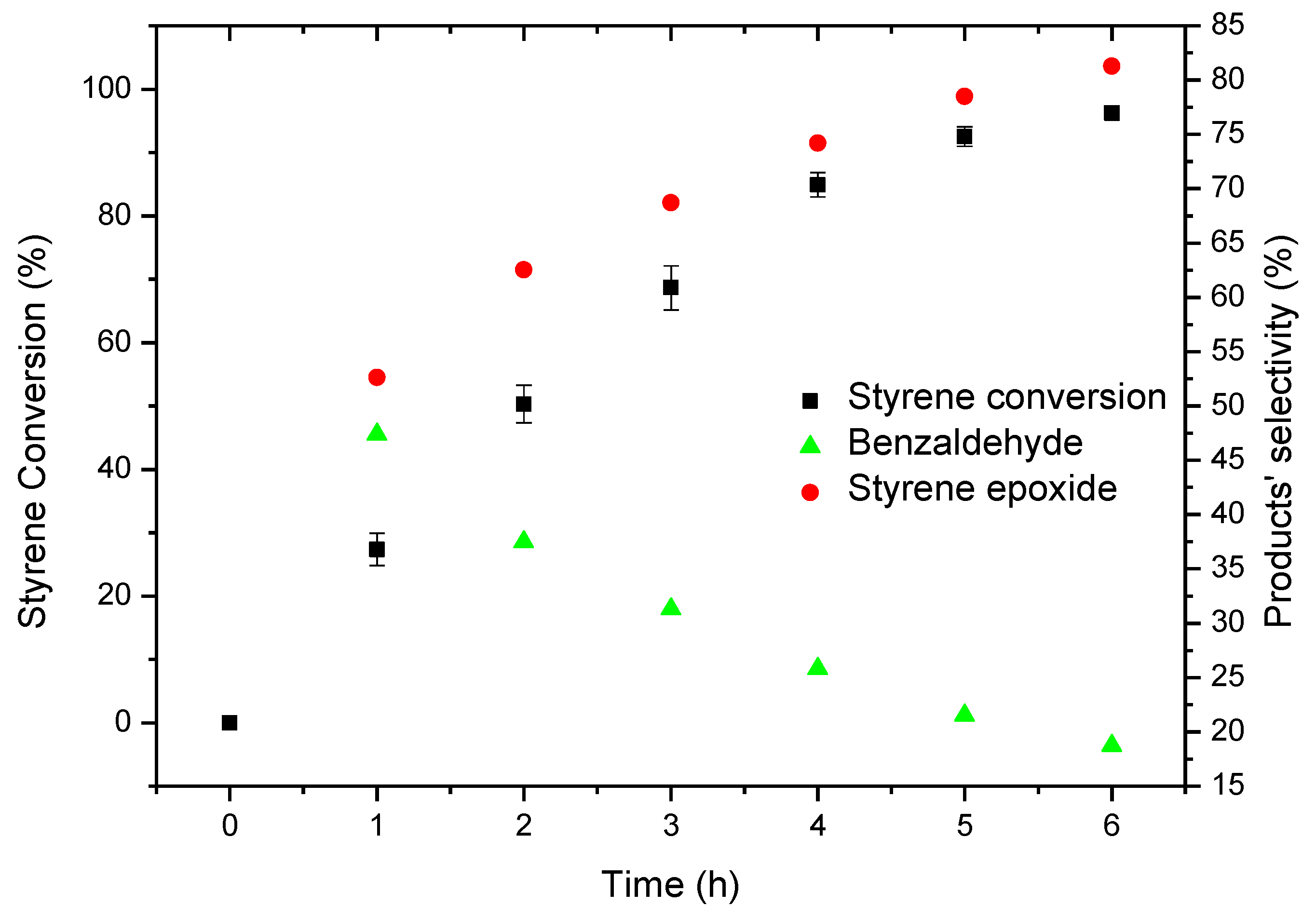
2.5. Oxidation of Styrene under Homogeneous and Heterogeneous Conditions
2.5.1. Homogeneous Oxidation Using CAT-3 or CAT-4 Catalysts
2.5.2. Oxidation of Styrene Catalysed by CAT-4-MR Using TBHP as the Oxidant
3. Materials and Methods
3.1. Reagents and Chemicals
3.2. Equipments
3.3. Synthesis of the Free-Base Porphyrins and Their Manganese(III) Complexes
3.3.1. Porph-3: 5,10,15,20-Tetrakis(3,5-Dichloropyridin-4-yl)Porphyrin
3.3.2. Porph-4: 5,10,15-Tris(2,6-Dichlorophenyl)-20-(3,5-Dichloropyridin-4-yl)Porphyrin
3.3.3. Manganese(III) Porphyrins
CAT-3: Chloro[5,10,15,20-Tetrakis(3,5-Dichloropyridin-4-yl)Porphyrinate]Manganese(III)
CAT-4: Chloro[5,10,15-Tris(2,6-Dichlorophenyl)-20-(3,5-Dichloropyridin-4-yl)Porphyrinate]Manganese(III)
3.4. Immobilization of CAT-4
3.4.1. Classic Heating
3.4.2. Microwave Heating
3.5. Oxidation Reactions
3.5.1. Homogeneous Catalysis
3.5.2. Heterogeneous Catalysis
3.6. Monitoring the Oxidation Reactions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gunter, M.J.; Turner, P. Metalloporphyrins as models for the cytochromes P-450. Coord. Chem. Rev. 1991, 108, 115–161. [Google Scholar] [CrossRef]
- Meunier, B. Metalloporphyrins as versatile catalysts for oxidation reactions and oxidative DNA cleavage. Chem. Rev. 1992, 92, 1411–1456. [Google Scholar] [CrossRef]
- Calvete, M.J.F.; Piñeiro, M.; Dias, L.D.; Pereira, M.M. Hydrogen Peroxide and Metalloporphyrins in Oxidation Catalysis: Old Dogs with Some New Tricks. ChemCatChem 2018, 10, 3615–3635. [Google Scholar] [CrossRef]
- Mansuy, D. A brief history of the contribution of metalloporphyrin models to cytochrome P450 chemistry and oxidation catalysis. C. R. Chim. 2007, 10, 392–413. [Google Scholar] [CrossRef]
- Che, C.; Huang, J. Metalloporphyrin-based oxidation systems: From biomimetic reactions to application in organic synthesis. Chem. Commun. 2009, 3996–4015. [Google Scholar] [CrossRef]
- Simões, M.M.Q.; De Paula, R.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. Metalloporphyrins in the biomimetic oxidative valorization of natural and other organic substrates. J. Porphyr. Phthalocyanines 2009, 13, 589–596. [Google Scholar] [CrossRef]
- Nakagaki, S.; Ferreira, G.K.B.; Ucoski, G.M.; Castro, K.A.D.D.F. Chemical Reactions Catalyzed by Metalloporphyrin-Based Metal-Organic Frameworks. Molecules 2013, 18, 7279–7308. [Google Scholar] [CrossRef]
- Simões, M.M.Q.; Neves, C.M.B.; Pires, S.M.G.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. Mimicking P 450 processes and the use of metalloporphyrins. Pure Appl. Chem. 2013, 85, 1671–1681. [Google Scholar] [CrossRef]
- Barona-Castaño, J.C.; Carmona-Vargas, C.C.; Brocksom, T.J.; De Oliveira, K.T. Porphyrins as Catalysts in Scalable Organic Reactions. Molecules 2016, 21, 310. [Google Scholar] [CrossRef]
- Zucca, P.; Neves, C.M.B.; Simões, M.M.Q.; Neves, M.D.G.P.M.S.; Cocco, G.; Sanjust, E. Immobilized lignin peroxidase-like metalloporphyrins as reusable catalysts in oxidative bleaching of industrial dyes. Molecules 2016, 21, 964. [Google Scholar] [CrossRef]
- Simões, M.M.Q.; Pires, S.M.G.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. Oxidative Transformations of Organic Compounds Mediated by Metalloporphyrins as Catalysts. In Handbook of Porphyrin Science: With Applications to Chemistry, Physics, Materials Science, Engineering, Biology and Medicine; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific Publishing: Singapore, 2016; pp. 197–306. [Google Scholar]
- Groves, J.T.; Nemo, T.E.; Myers, R.S. Hydroxylation and Epoxidation Catalyzed by Iron-Porphine Complexes. Oxygen Transfer from lodosylbenzene. J. Am. Soc. 1979, 101, 1032–1033. [Google Scholar] [CrossRef]
- Traylor, P.S.; Dolphin, D.; Traylor, T.G. Sterically protected hemins with electronegative substituents: Efficient catalysts for hydroxylation and epoxidation. J. Chem. Soc. Chem. Commun. 1984, 279–280. [Google Scholar] [CrossRef]
- Nappa, M.J.; Tolman, C.A. Steric and Electronic Control. Inorg. Chem. 1985, 24, 4711–4719. [Google Scholar] [CrossRef]
- Bartoli, J.F.; Brigaud, O.; Battioni, P.; Mansuy, D. Hydroxylation of Linear Alkanes Catalysed by Iron Porphyrins: Particular Efficacy and Regioselectivity of Perhalogenated Porphyrins. J. Chem. Soc. Chem. Commun. 1991, 440–442. [Google Scholar] [CrossRef]
- Gonsalves, A.M.A.R.; Pereira, M.M.; Serra, A.C.; Johnstone, R.A.W.; Nunes, M.L.P.G. 5,10,15,20-Tetrakisaryl- and 2,3,7,8,12,13,17,18-Octahalogeno-5,10,15,20-tetrakisarylporphyrins and their Metal Complexes as Catalysts in Hypochlorite Epoxidations. J. Chem. Soc. Perkin Trans. 1 1994, 2053–2057. [Google Scholar] [CrossRef]
- Gross, Z.; Simkhovich, L. Hydroxylation of Simple Alkanes by Iodosylbenzene is Catalyzed more Efficiently by Second than by Third Generation Iron(IIl) Porphyrins. Tetrahedron Lett. 1998, 39, 8171–8174. [Google Scholar] [CrossRef]
- Guedes, A.A.; Santos, A.C.M.A.; Assis, M.D. Some factors influencing the selectivity of styrene oxidation by active oxygen donors catalyzed by three generations of ironporphyrins. Kinet. Catal. 2006, 47, 555–563. [Google Scholar] [CrossRef]
- Doro, F.G.; Smith, J.R.L.; Ferreira, A.G.; Assis, M.D. Oxidation of alkanes and alkenes by iodosylbenzene and hydrogen peroxide catalysed by halogenated manganese porphyrins in homogeneous solution and covalently bound to silica. J. Mol. Catal. A Chem. 2000, 164, 97–108. [Google Scholar] [CrossRef]
- Friedermann, G.R.; Halma, M.; Castro, K.A.D.F.; Benedito, F.L.; Doro, F.G.; Drechsel, S.M.; Mangrich, A.S.; das Dores Assis, M.; Nakagaki, S. Intermediate species generated from halogenated manganese porphyrins electrochemically and in homogeneous catalysis of alkane oxidation. Appl. Catal. A Gen. 2006, 308, 172–181. [Google Scholar] [CrossRef]
- Rebelo, S.L.H.; Gonçalves, A.R.; Pereira, M.M.; Simões, M.M.Q.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. Epoxidation reactions with hydrogen peroxide activated by a novel heterogeneous metalloporphyrin catalyst. J. Mol. Catal. A Chem. 2006, 256, 321–323. [Google Scholar] [CrossRef]
- Pires, S.M.G.; De Paula, R.; Simões, M.M.Q.; Neves, M.G.P.M.S.; Santos, I.C.M.S.; Tomé, A.C.; Cavaleiro, J.A.S. A new silica-supported manganese chlorin as a biomimetic oxidation catalyst. Catal. Commun. 2009, 11, 24–28. [Google Scholar] [CrossRef]
- De Paula, R.; Santos, I.C.M.S.; Simões, M.M.Q.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. An immobilized imidazolyl manganese porphyrin for the oxidation of olefins. J. Mol. Catal. A Chem. 2015, 404–405, 156–166. [Google Scholar] [CrossRef]
- Calvete, M.J.F.; Silva, M.; Pereira, M.M.; Burrows, H.D. Inorganic helping organic: Recent advances in catalytic heterogeneous oxidations by immobilised tetrapyrrolic macrocycles in micro and mesoporous supports. RSC Adv. 2013, 3, 22774–22789. [Google Scholar] [CrossRef]
- Nakagaki, S.; Mantovani, K.M.; Machado, G.S.; Castro, K.A.D.D.F.; Wypych, F. Recent Advances in Solid Catalysts Obtained by Metalloporphyrins Immobilization on Layered Anionic Exchangers: A Short Review and Some New Catalytic Results. Molecules 2016, 21, 291. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.F.; Simões, M.M.Q.; Tomé, J.P.C.; Paz, F.A.A. Porphyrin-Based Metal-Organic Frameworks as Heterogeneous Catalysts in Oxidation Reactions. Molecules 2016, 21, 1348. [Google Scholar] [CrossRef] [PubMed]
- Brulé, E.; de Miguel, Y.R. Supported metalloporphyrin catalysts for alkene epoxidation. Org. Biomol. Chem. 2006, 4, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Thellend, A.; Battioni, P.; Mansuy, D. Ammonium acetate as a very simple and efficient cocatalyst for manganese porphyrin-catalysed oxygenation of hydrocarbons by hydrogen peroxide. J. Chem. Soc. Chem. Commun. 1994, 202, 1035–1036. [Google Scholar] [CrossRef]
- Santos, I.C.M.S.; Rebelo, S.L.H.; Balula, M.S.S.; Martins, R.R.L.; Pereira, M.M.M.S.; Simões, M.M.Q.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S.; Cavaleiro, A.M.V. Association of Keggin-type anions with cationic meso-substituted porphyrins: Synthesis, characterization and oxidative catalytic studies. J. Mol. Catal. A Chem. 2005, 231, 35–45. [Google Scholar] [CrossRef]
- Nur, H.; Hamid, H.; Endud, S.; Hamdan, H.; Ramli, Z. Iron-porphyrin encapsulated in poly ( methacrylic acid ) and mesoporous Al-MCM-41 as catalysts in the oxidation of benzene to phenol. Mater. Chem. Phys. 2006, 96, 337–342. [Google Scholar] [CrossRef]
- Gonsalves, A.M.A.R.; Varejão, J.M.T.B.; Pereira, M.M. Some New Aspects Related to the Synthesis of meso-Substituted Porphyrins. J. Heterocycl. Chem. 1991, 28, 635–640. [Google Scholar] [CrossRef]
- Adler, A.D.; Longo, F.R.; Kampas, F.; Kim, J. On the preparation of metalloporphyrins. J. Inorg. Nucl. Chem. 1970, 32, 2443–2445. [Google Scholar] [CrossRef]
- El-Nahhal, I.M.; El-Shetary, B.A.; Mustafa, A.E.-K.B.; El-Ashgar, N.M.; Livage, J.; Chehimi, M.M.; Roberts, A. Structural characterization of immobilized-polysiloxane iminobis(N-diethylenediamineacetamide) ligand system. Solid State Sci. 2003, 5, 1395–1406. [Google Scholar] [CrossRef]
- Lipinska, M.E.; Rebelo, S.L.H.; Freire, C. Iron ( III ) porphyrin anchored onto organosilylated multiwalled carbon nanotubes as an active catalyst for epoxidation reactions under mild conditions. J. Mater. Sci. 2014, 49, 1494–1505. [Google Scholar] [CrossRef]
- Al-Bataineh, S.A.; Britcher, L.G.; Griesser, H.J. XPS characterization of the surface immobilization of antibacterial furanones. Surf. Sci. 2006, 600, 952–962. [Google Scholar] [CrossRef]
- Lavallee, D.K.; Brace, J.; Winograd, N. X-ray Photoelectron Spectra of N-Methyltetraphenylporphyrins: Evidence for a Correlation of Binding Energies with Metal-Nitrogen Bond Distances. Inorg. Chem. 1979, 18, 1776–1780. [Google Scholar] [CrossRef]
- Gassman, P.G.; Ghosh, A.; Almlöf, J. Electronic Effects of Peripheral Substituents in Porphyrins: X-ray Photoelectron Spectroscopy and ab Initio Self-Consistent Field Calculations. J. Am. Chem. Soc. 1992, 114, 9990–10000. [Google Scholar] [CrossRef]
- Goll, J.G.; Moore, K.T.; Ghosh, A.; Therien, M.J. Synthesis, Structure, Electronic Spectroscopy, Photophysics, Electrochemistry, and X-ray Photoelectron Spectroscopy of Highly-Electron-Deficient [5,10,15,20-Tetrakis(perfluoroalkyl)porphinato]zinc(II) Complexes and Their Free Base Derivatives. J. Am. Chem. Soc. 1996, 118, 8344–8354. [Google Scholar] [CrossRef]
- Sarno, D.M.; Jiang, B.; Grosfeld, D.; Afriyie, J.O.; Matienzo, L.J.; Jones, W.E. Self-assembled Molecular Architectures on Surfaces: New Strategies Involving Metal-Organic Copolymers. Langmuir 2000, 16, 6191–6199. [Google Scholar] [CrossRef]
- Jansen, R.J.J.; van Bekkum, H. XPS of Nitrogen-Containing Functional Groups on Activated Carbon. Carbon 1995, 33, 1021–1027. [Google Scholar] [CrossRef]
- Goh, S.H.; Lee, S.Y.; Dai, J.; Tan, K.L. X-ray photoelectron spectroscopic studies of ionic interactions between sulfonated polystyrene and poly(styrene-co-4-vinylpyridine). Polymer 1996, 37, 5305–5308. [Google Scholar] [CrossRef]
- Van der Heide, P. X-Ray Photoelectron Spectroscopy—An Introduction to Principles and Practices; John Wiley & Sons: Hoboken, NJ, USA, 2012; ISBN 9781118062531. [Google Scholar]
- Li, Z.; Xia, C.-G.; Zhang, X.-M. Preparation and catalysis of DMY and MCM-41 encapsulated cationic Mn(III)–porphyrin complex. J. Mol. Catal. A Chem. 2002, 185, 47–56. [Google Scholar] [CrossRef]
- Kaplan, A.; Korin, E.; Bettelheim, A. Structures self-assembled from anionic graphene and cationic manganese porphyrin: Characterization and application in artificial photosynthesis. Eur. J. Inorg. Chem. 2014, 2288–2295. [Google Scholar] [CrossRef]
- Yang, F.; Gao, S.; Xiong, C.; Wang, H.; Chen, J.; Kong, Y. Coordination of manganese porphyrins on amino-functionalized MCM-41 for heterogeneous catalysis of naphthalene hydroxylation. Chin. J. Catal. 2015, 36, 1035–1041. [Google Scholar] [CrossRef]
- Antonangelo, A.R.; Grazia Bezzu, C.; McKeown, N.B.; Nakagaki, S. Highly active manganese porphyrin-based microporous network polymers for selective oxidation reactions. J. Catal. 2019, 369, 133–142. [Google Scholar] [CrossRef]
- Rebelo, S.L.H.; Simões, M.M.Q.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. Oxidation of alkylaromatics with hydrogen peroxide catalysed by manganese(III) porphyrins in the presence of ammonium acetate. J. Mol. Catal. A Chem. 2003, 201, 9–22. [Google Scholar] [CrossRef]
- Rebelo, S.L.H.; Pereira, M.M.; Simões, M.M.Q.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. Mechanistic studies on metalloporphyrin epoxidation reactions with hydrogen peroxide: Evidence for two active oxidative species. J. Catal. 2005, 234, 76–87. [Google Scholar] [CrossRef]
- Groves, J.T.; Kruper, W.J.; Haushalter, R.C. Hydrocarbon Oxidations with Oxometalloporphinates. Isolation and Reactions of a (Porphinato)manganese(V) Complex. J. Am. Chem. Soc. 1980, 102, 6375–6377. [Google Scholar] [CrossRef]
- Groves, J.T.; Stern, M.K. Olefin Epoxidation by Manganese(IV) Porphyrins: Evidence for Two Reaction Pathways. J. Am. Chem. Soc. 1987, 109, 3812–3814. [Google Scholar] [CrossRef]
- Groves, J.T.; Stern, M.K. Synthesis, Characterization, and Reactivity of Oxomanganese(IV) Porphyrin Complexes. J. Am. Chem. Soc. 1988, 110, 8628–8638. [Google Scholar] [CrossRef]
- Groves, J.T.; Lee, J.; Marla, S.S. Detection and Characterization of an Oxomanganese(V) Porphyrin Complex by Rapid-Mixing Stopped-Flow Spectrophotometry. J. Am. Chem. Soc. 1997, 119, 6269–6273. [Google Scholar] [CrossRef]
- Jin, N.; Groves, J.T. Unusual Kinetic Stability of a Ground-State Singlet Oxomanganese(V) Porphyrin. Evidence for a Spin State Crossing Effect. J. Am. Chem. Soc. 1999, 121, 2923–2924. [Google Scholar] [CrossRef]
- Camenzind, M.J.; Hollander, F.J.; Hill, C.L. Syntheses, Ground Electronic State, and Crystal and Molecular Structure of the Monomeric Manganese(IV) Porphyrin Complex Dimethoxy(5,10,15,20-tetraphenylporphinato)manganese(IV). Inorg. Chem. 1982, 21, 4301–4308. [Google Scholar] [CrossRef]
- Zhang, R.; Horner, J.H.; Newcomb, M. Laser flash photolysis generation and kinetic studies of porphyrin-manganese-oxo intermediates. Rate constants for oxidations effected by porphyrin-MnV-oxo species and apparent disproportionation equilibrium constants for porphyrin-MnIV-oxo species. J. Am. Chem. Soc. 2005, 127, 6573–6582. [Google Scholar] [CrossRef] [PubMed]
- La Paglia Fragola, V.; Lupo, F.; Pappalardo, A.; Sfrazzetto, G.T.; Toscano, R.M.; Ballistreri, F.P.; Tomaselli, G.A.; Gulino, A. A surface-confined O=MnV(salen) oxene catalyst and high turnover values in asymmetric epoxidation of unfunctionalized olefins. J. Mater. Chem. 2012, 22, 20561–20565. [Google Scholar] [CrossRef]
- Kang, Y.; Wang, F.; Reinhard, F.G.C.; Xia, C.; de Visser, S.P.; Wang, Y. Can Manganese(III)-Iodosylarene Act as an Oxidant Alongside High-Valent Manganese(V)-Oxo Complexes? Chem. Sel. 2018, 3, 3208–3213. [Google Scholar] [CrossRef]
- Ballistreri, F.P.; Gangemi, C.M.A.; Pappalardo, A.; Tomaselli, G.A.; Toscano, R.M.; Sfrazzetto, G.T. (Salen)Mn(III) catalyzed asymmetric epoxidation reactions by hydrogen peroxide in water: A green protocol. Int. J. Mol. Sci. 2016, 17, 1112. [Google Scholar] [CrossRef] [Green Version]
- Cuiping, B.; Wensheng, X.; Dexin, F.; Mo, X.; Dong, G.; Zhongxue, G.; Yanshui, Z. Efficient decolorization of Malachite Green in the Fenton reaction catalyzed by [Fe(III)-salen]Cl complex. Chem. Eng. J. 2013, 215–216, 227–234. [Google Scholar]
- Liu, J.-Y.; Li, X.-F.; Li, Y.-Z.; Chang, W.-B.; Huang, A.-J. Oxidation of styrene by various oxidants with different kinds of metalloporphyrins. J. Mol. Catal. A Chem. 2002, 187, 163–167. [Google Scholar] [CrossRef]
- Xie, M.-H.; Yang, X.-L.; Wu, C.-D. A metalloporphyrin functionalized metal–organic framework for selective oxidization of styrene. Chem. Commun. 2011, 47, 5521. [Google Scholar] [CrossRef]
- De Paula, R.; Simões, M.M.Q.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. Homogeneous olefin epoxidation catalysed by an imidazolium-based manganese porphyrin. Catal. Commun. 2008, 10, 57–60. [Google Scholar] [CrossRef]
- De Visser, S.P.; Kumar, D.; Shaik, S. How do aldehyde side products occur during alkene epoxidation by cytochrome P450? Theory reveals a state-specific multi-state scenario where the high-spin component leads to all side products. J. Inorg. Biochem. 2004, 98, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; De Visser, S.P.; Shaik, S. Multistate Reactivity in Styrene Epoxidation by Compound I of Cytochrome P450: Mechanisms of Products and Side Products Formation. Chem. Eur. J. 2005, 11, 2825–2835. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, H.; Lu, Y.; Cai, Y.; Liu, X. Mild oxidation of styrene and its derivatives catalyzed by ionic manganese porphyrin embedded in a similar structured ionic liquid. Green Chem. 2007, 9, 1114–1119. [Google Scholar] [CrossRef]
- Fontecave, M.; Mansuy, D. Alkene Epoxidation by lodosylbenzene Catalysed by Porphyrin and Non-porphyrin Iron Complexes: The Importance of the Porphyrin Ligand in Cytochrome P-450 and Heme Model Reactions. J. Chem. Soc. Chem. Commun. 1984, 879–881. [Google Scholar] [CrossRef]
- Gilmartin, C.; Smith, J.R.L. Alkene Epoxidation by lodosylbenzene Catalysed. J. Chem. Soc. Perkin Trans. 2 1995, 243–251. [Google Scholar] [CrossRef]
- Groves, J.T.; Myers, R.S. Catalytic Asymmetric Epoxidations with Chiral Iron Porphyrins. J. Am. Chem. Soc. 1983, 105, 5791–5796. [Google Scholar] [CrossRef]
- Mansuy, D.; Leclaire, J.; Fontecave, M.; Dansette, P. Regioselectivity of olefin oxidation by iodosobenzene catalyzed by metalloporphyrins: Control by the catalyst. Tetrahedron 1984, 40, 2847–2857. [Google Scholar] [CrossRef]
- Collman, J.P.; Kodadek, T.; Brauman, J.I. Oxygenation of Styrene by Cytochrome P-450 Model Systems: A Mechanistic Study. J. Am. Chem. Soc. 1986, 108, 2588–2594. [Google Scholar] [CrossRef]
- De Paula, R.; Simões, M.M.Q.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. Oxidation of styrene and of some derivatives with H2O2 catalyzed by novel imidazolium-containing manganese porphyrins: A mechanistic and thermodynamic interpretation. J. Mol. Catal. A Chem. 2011, 345, 1–11. [Google Scholar] [CrossRef]
- Gonsalves, A.M.A.R.; Serra, A.C. Observations on the origin of phenylacetaldehyde in styrene epoxidation and the mechanism of oxidations catalysed by manganese complexes of porphyrins. J. Chem. Soc. Perkin Trans. 2 2002, 715–719. [Google Scholar] [CrossRef]
- Arasasingham, R.D.; He, G.-X.; Bruice, T.C. Mechanism of Manganese Porphyrin-Catalyzed Oxidation of Alkenes. Role of Manganese( IV)-Oxo Species. J. Am. Chem. Soc. 1993, 115, 7985–7991. [Google Scholar] [CrossRef]
- Gonsalves, A.M.A.R.; Serra, A.C. On the mechanism of carboxylic acid co-catalyst assisted metalloporphyrin oxidations. J. Mol. Catal. A Chem. 2001, 168, 25–32. [Google Scholar] [CrossRef] [Green Version]
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
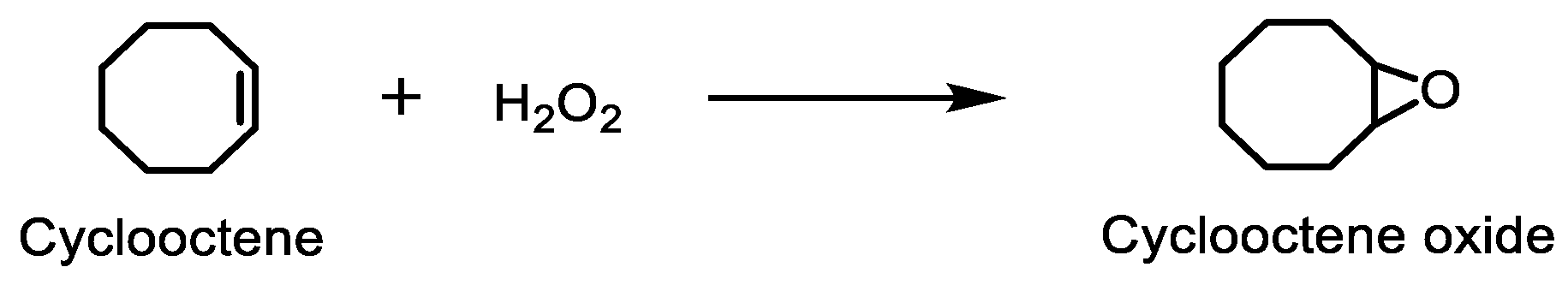{kind=link}
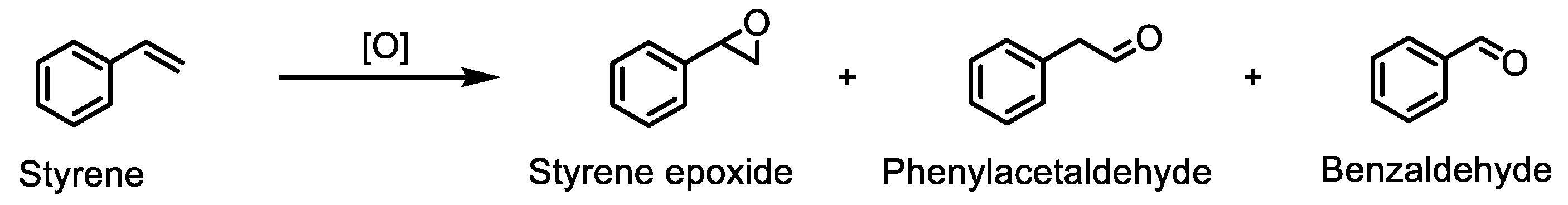{kind=link}
| Entry | Material | Atomic (%) | ||||||
|---|---|---|---|---|---|---|---|---|
| C 1s | N 1s | O 1s | Si 2p | Cl 2p | Br 3d | Mn 2p3 | ||
| 1 | Si | 15.4 | - | 59.1 | 24.0 | - | 1.4 | - |
| 2 | CAT-4-Si | 17.0 | 0.5 | 56.4 | 25.0 | 0.4 | 0.7 | n.d. |
| 3 | MR | 93.4 | - | 5.6 | - | 1.1 | - | - |
| 4 | CAT-4-MR | 88.9 | 0.7 | 7.0 | - | 3.3 | - | 0.06 |
| 5 | CAT-5-MR | 86.3 | 0.7 | 9.5 | - | 3.4 | - | 0.1 |
| Catalyst | S/C Molar Ratio | H2O2 (eq) | Conversion (%) | Time (min) |
|---|---|---|---|---|
| CAT-2 | 150 | 2 | 98.3 | 60 |
| 600 | 2.5 | 98.6 | 75 | |
| CAT-3 | 150 | 3 | 98.8 | 90 |
| 600 | 4 | 97.6 | 120 | |
| CAT-4 | 150 | 2 | 98.8 | 60 |
| 600 | 2.5 | 98.2 | 75 | |
| CAT-5 | 150 | 2 | 98.8 | 90 |
| 600 | 2.5 | 40.1 | 75 | |
| CAT-6 | 150 | 3 | 25.4 | 90 |
| Catalyst | Oxidant | Cycle | Oxidant (eq.) | Time (h) | Conversion (%) |
|---|---|---|---|---|---|
| CAT-4-Si 2 | H2O2 5 | 1st | 4.5 | 2.5 | 98.2 |
| 2nd | 10 | 5 | 38.4 | ||
| TBHP 7 | 1st | 8 | 4 | 93.5 | |
| 2nd | 12 | 6 | 37.8 | ||
| CAT-4-MR 3 | H2O2 5 | 1st | 14 (15) | 7 (24) | 58.9 (65.6) |
| 2nd | 10.8 (16.7) | ||||
| H2O2 6 | 1st | 12 | 6 (24) | 46.6 (74.3) | |
| 2nd | 6 (72) | 10.9 (33.4) | |||
| TBHP 7 | 1st | 12 | 6 | 88.6 | |
| 2nd | 86.3 | ||||
| 3rd | 79.2 | ||||
| 4th | 69.4 | ||||
| 5th | 41.5 | ||||
| Without | - | 12 | 6 | 9.8 | |
| CAT-5-MR 4 | TBHP 7 | 1st | 10 | 5 | 92.8 |
| 2nd | 72.0 | ||||
| 3rd | 54.4 | ||||
| Without | - | 10 | 5 | 10.3 | |
| MR | TBHP 7 | - | 12 | 6 | 6.3 |
| Sample | Atomic (%) | ||||||
|---|---|---|---|---|---|---|---|
| C 1s | N 1s | O 1s | Si 2p | Cl 2p | Br 3d | Mn 2p | |
| CAT-4-MR | 88.9 | 0.7 | 7.0 | - | 3.3 | - | 0.06 |
| CAT-4-MR-H2O2 | 91.7 | 0.5 | 4.5 | - | 3.2 | - | 0.05 |
| CAT-4-MR-TBHP | 81.0 | 3.7 | 14.5 | - | 0.9 | - | n.d. |
| CAT-4-Si | 17.0 | 0.5 | 56.4 | 25.0 | 0.4 | 0.7 | n.d. |
| CAT-4-Si-H2O2 | 16.6 | 0.4 | 56.7 | 24.7 | 0.1 | 1.4 | 0.07 |
| CAT-4-Si-TBHP | 16.7 | 0.8 | 58.2 | 22.9 | 0.2 | 1.1 | n.d. |
| Catalyst | Time (min) | H2O2 (eq) | Conversion (%) | Selectivity (%) | ||
|---|---|---|---|---|---|---|
| Benzaldehyde | Phenylacetaldehyde | Styrene Epoxide | ||||
| CAT-2 | 75 | 2.5 | 100 | 0.6 | 36.7 | 62.6 |
| CAT-3 | 150 | 5 | 97.6 | 2.1 | 30.3 | 67.6 |
| CAT-4 | 105 | 3.5 | 99.0 | 1.7 | 32.6 | 65.6 |
| None | 150 | 5 | - | - | - | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neves, C.M.B.; Rebelo, S.L.H.; Faustino, M.A.F.; Neves, M.G.P.M.S.; Simões, M.M.Q. Second-Generation Manganese(III) Porphyrins Bearing 3,5-Dichloropyridyl Units: Innovative Homogeneous and Heterogeneous Catalysts for the Epoxidation of Alkenes. Catalysts 2019, 9, 967. https://0-doi-org.brum.beds.ac.uk/10.3390/catal9110967
Neves CMB, Rebelo SLH, Faustino MAF, Neves MGPMS, Simões MMQ. Second-Generation Manganese(III) Porphyrins Bearing 3,5-Dichloropyridyl Units: Innovative Homogeneous and Heterogeneous Catalysts for the Epoxidation of Alkenes. Catalysts. 2019; 9(11):967. https://0-doi-org.brum.beds.ac.uk/10.3390/catal9110967
Chicago/Turabian StyleNeves, Cláudia M. B., Susana L. H. Rebelo, M. Amparo F. Faustino, M. Graça P. M. S. Neves, and Mário M. Q. Simões. 2019. "Second-Generation Manganese(III) Porphyrins Bearing 3,5-Dichloropyridyl Units: Innovative Homogeneous and Heterogeneous Catalysts for the Epoxidation of Alkenes" Catalysts 9, no. 11: 967. https://0-doi-org.brum.beds.ac.uk/10.3390/catal9110967