Biomimetic Oxidation of Benzofurans with Hydrogen Peroxide Catalyzed by Mn(III) Porphyrins

 , , and
, , and
Abstract
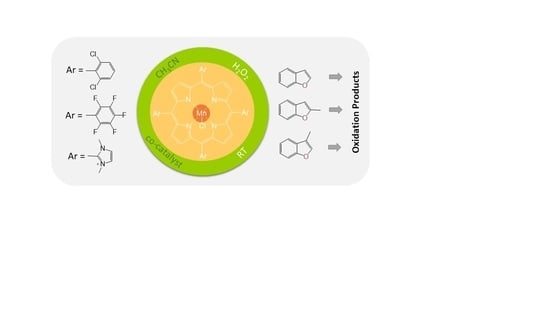
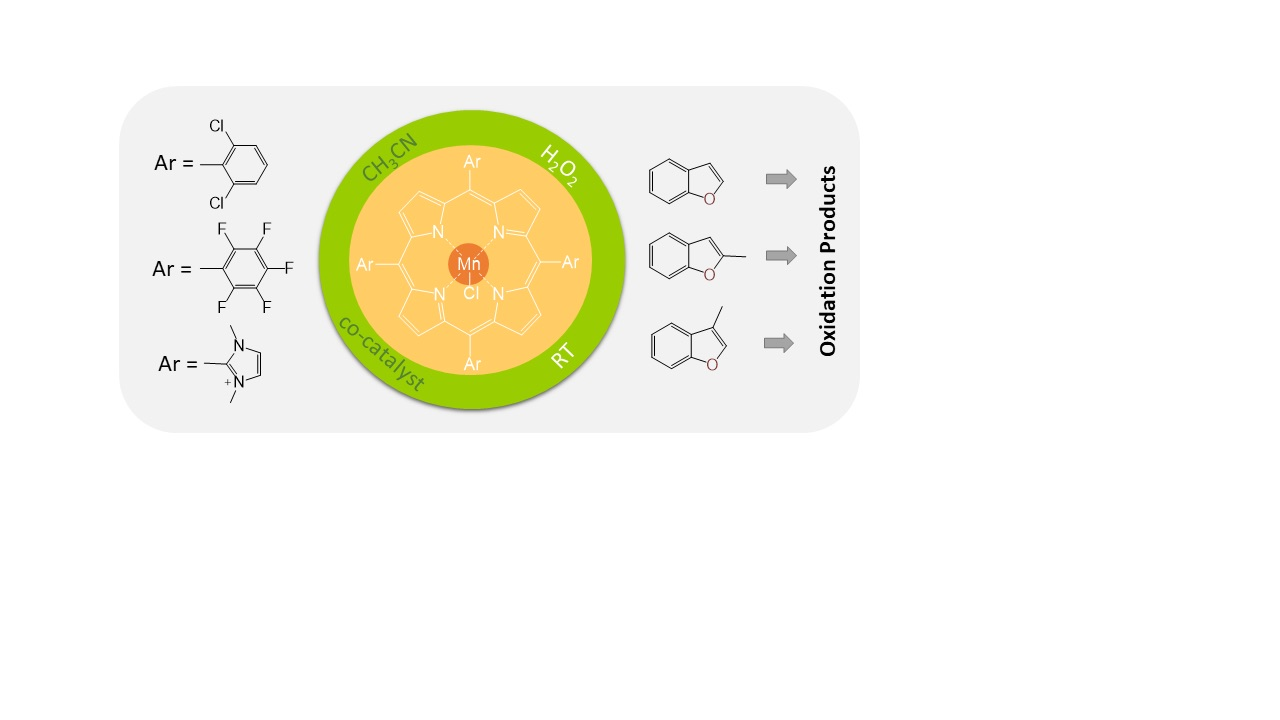
:
1. Introduction
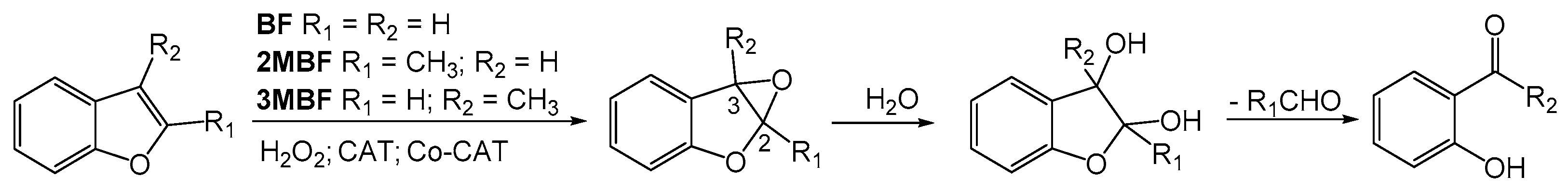
2. Results and Discussion
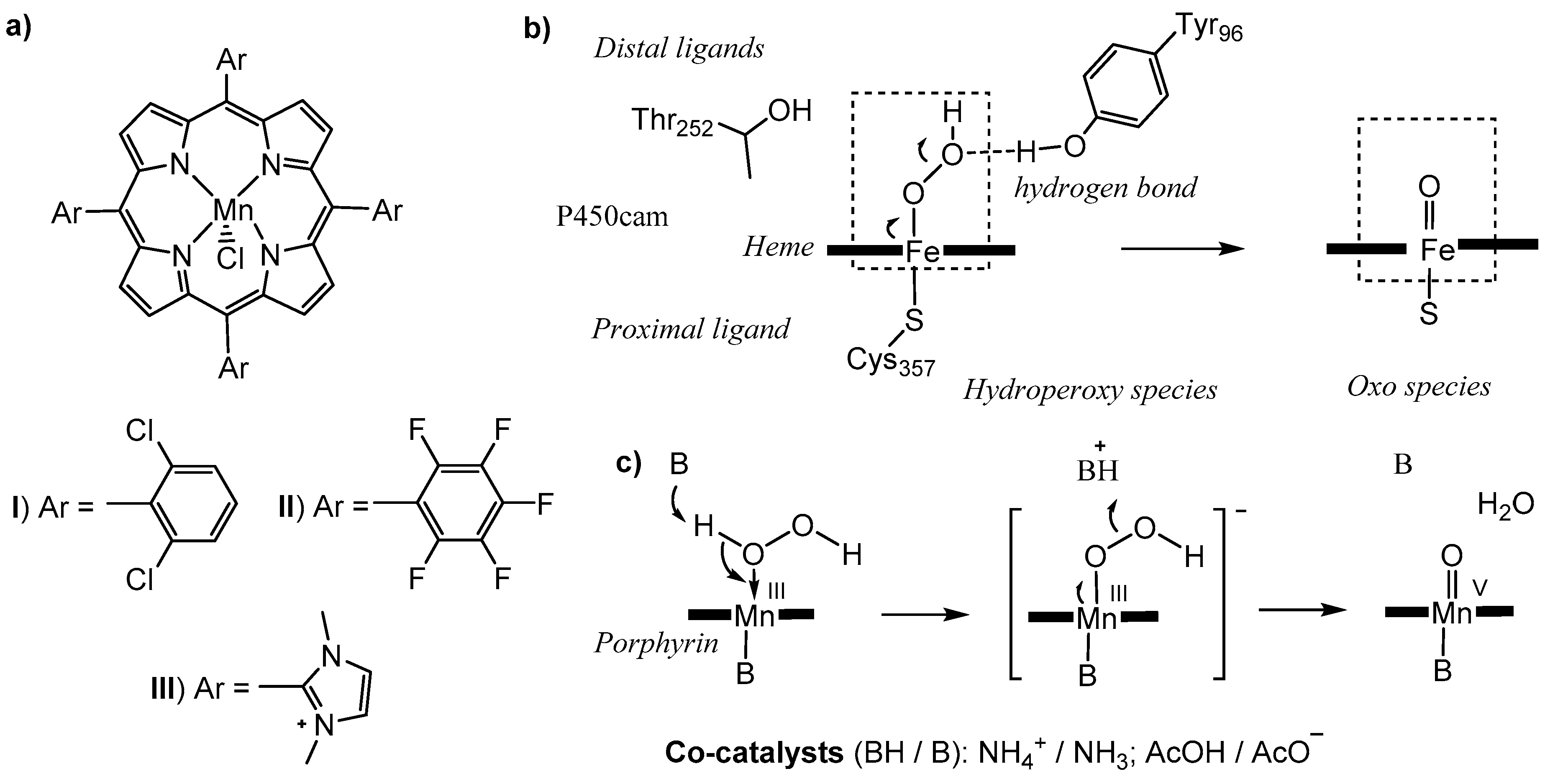
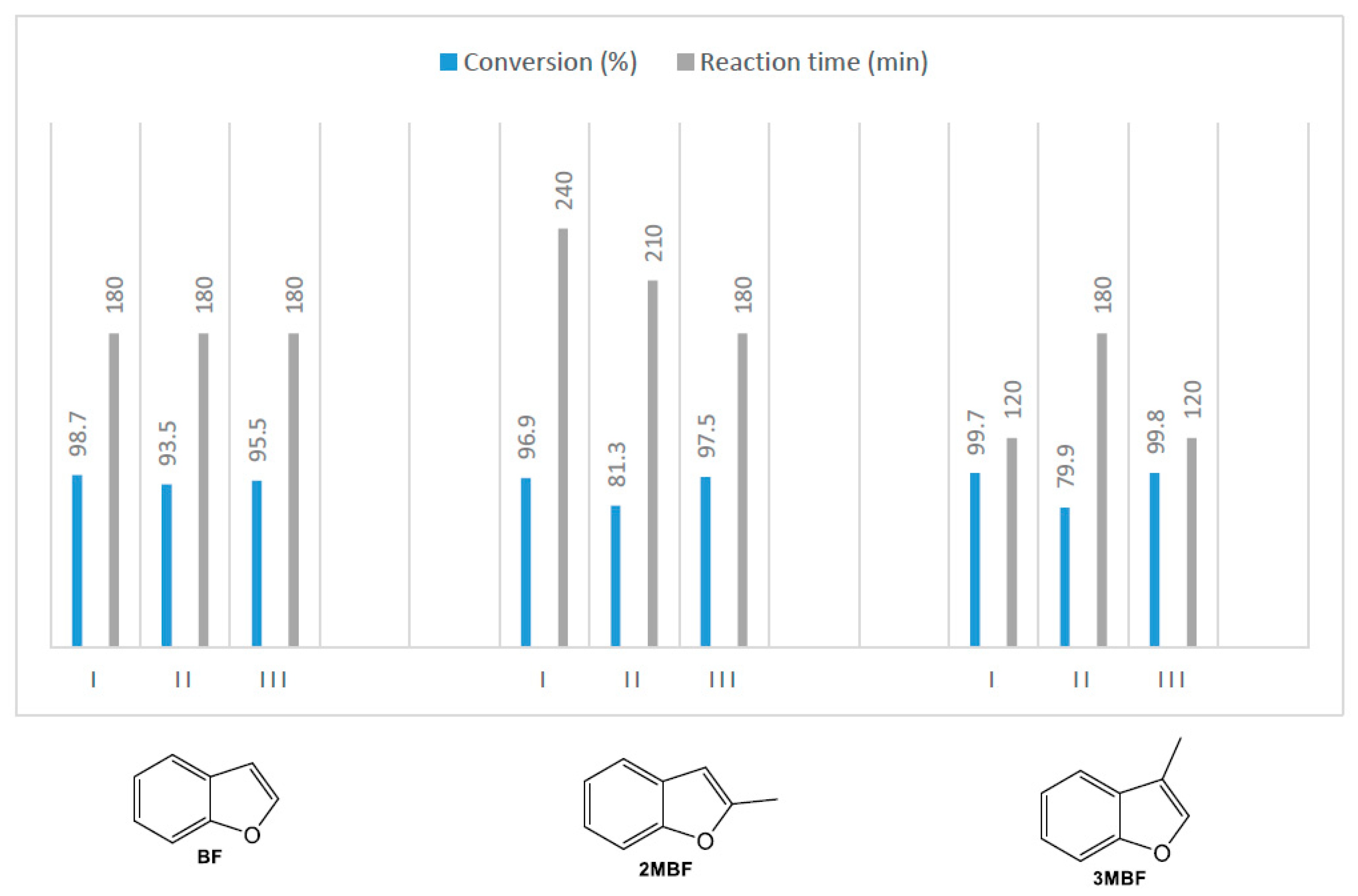
2.1. Comparisons of Metalloporphyrin Catalyst Performance
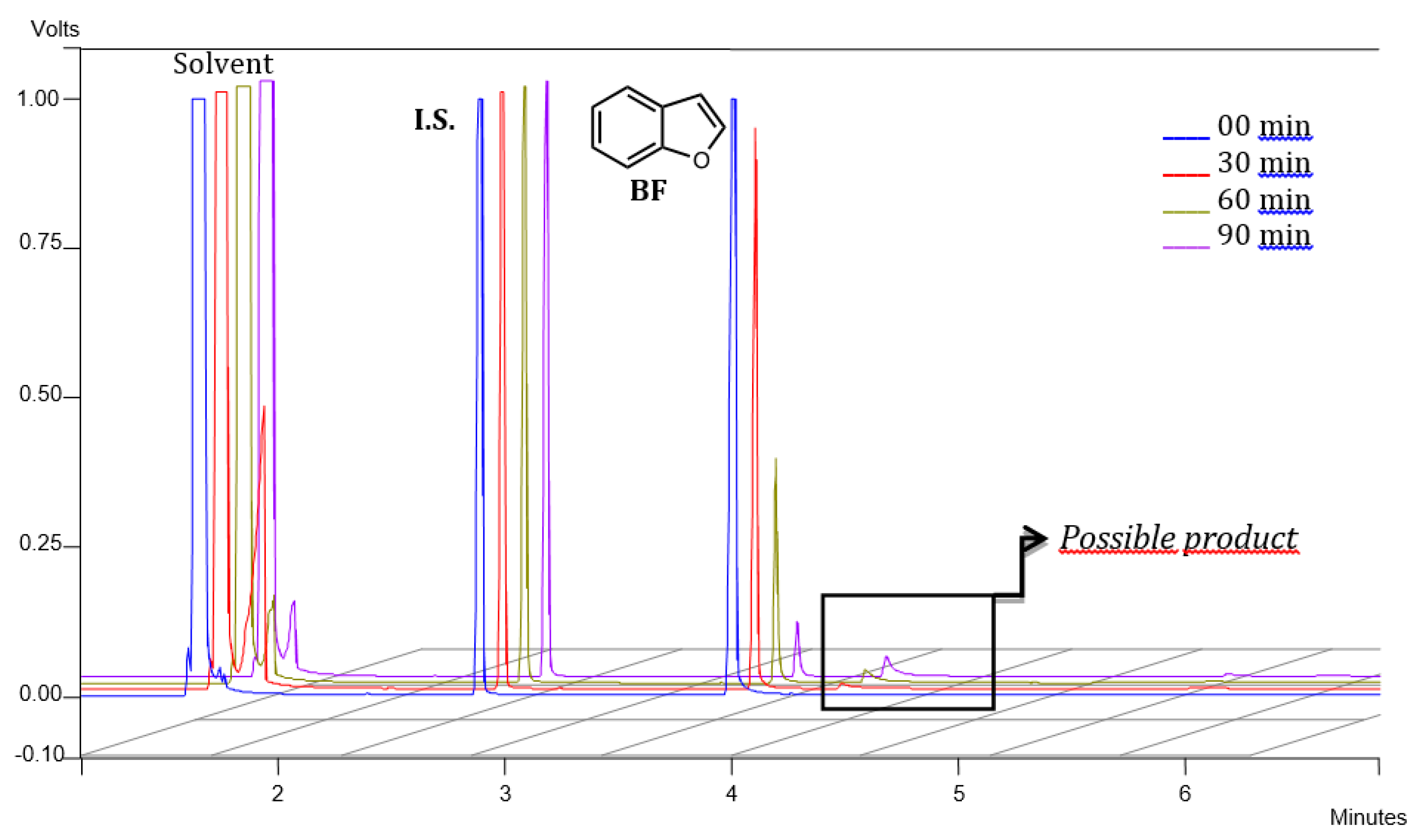
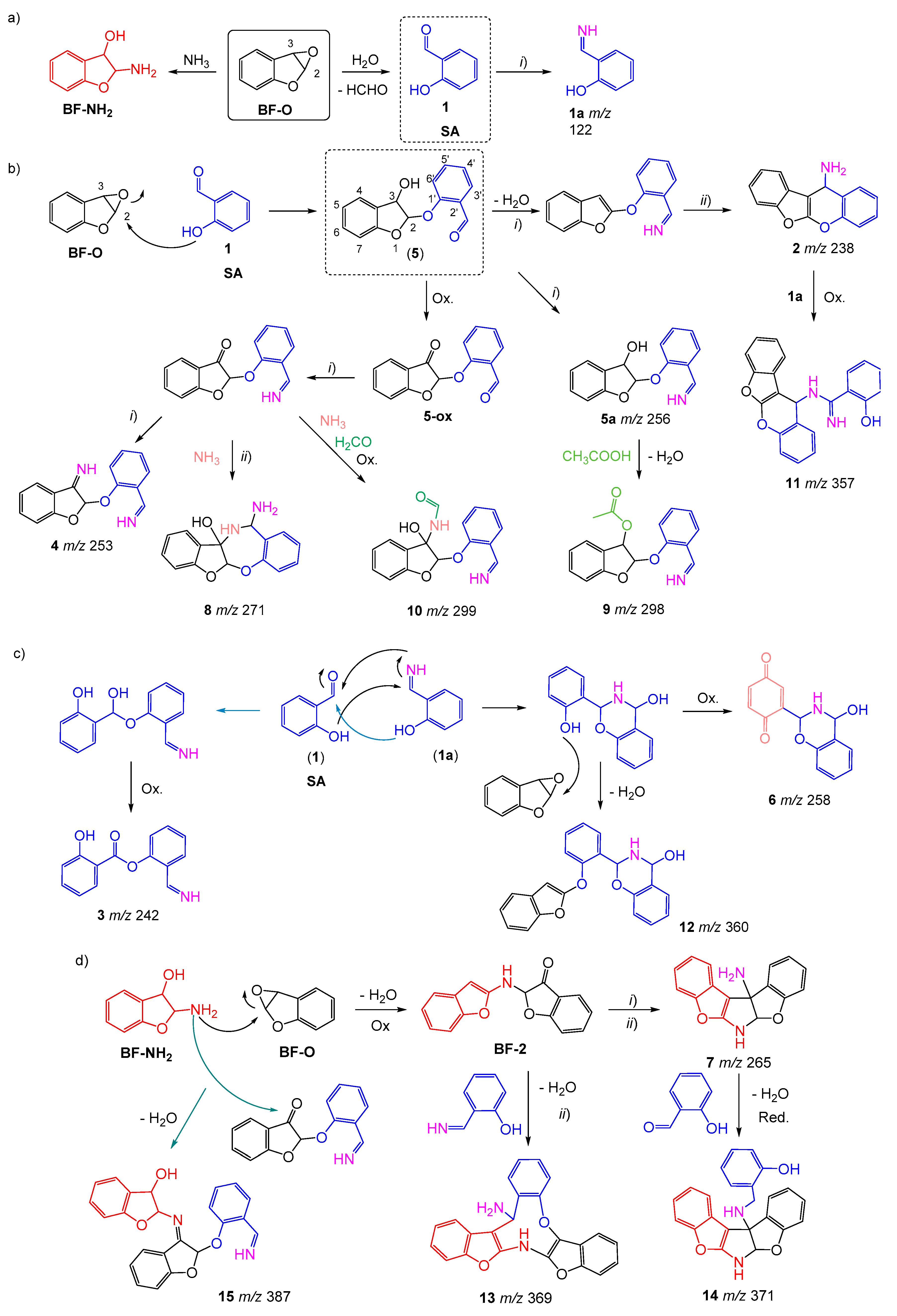
2.2. Benzofuran (BF) Oxidation Reactions
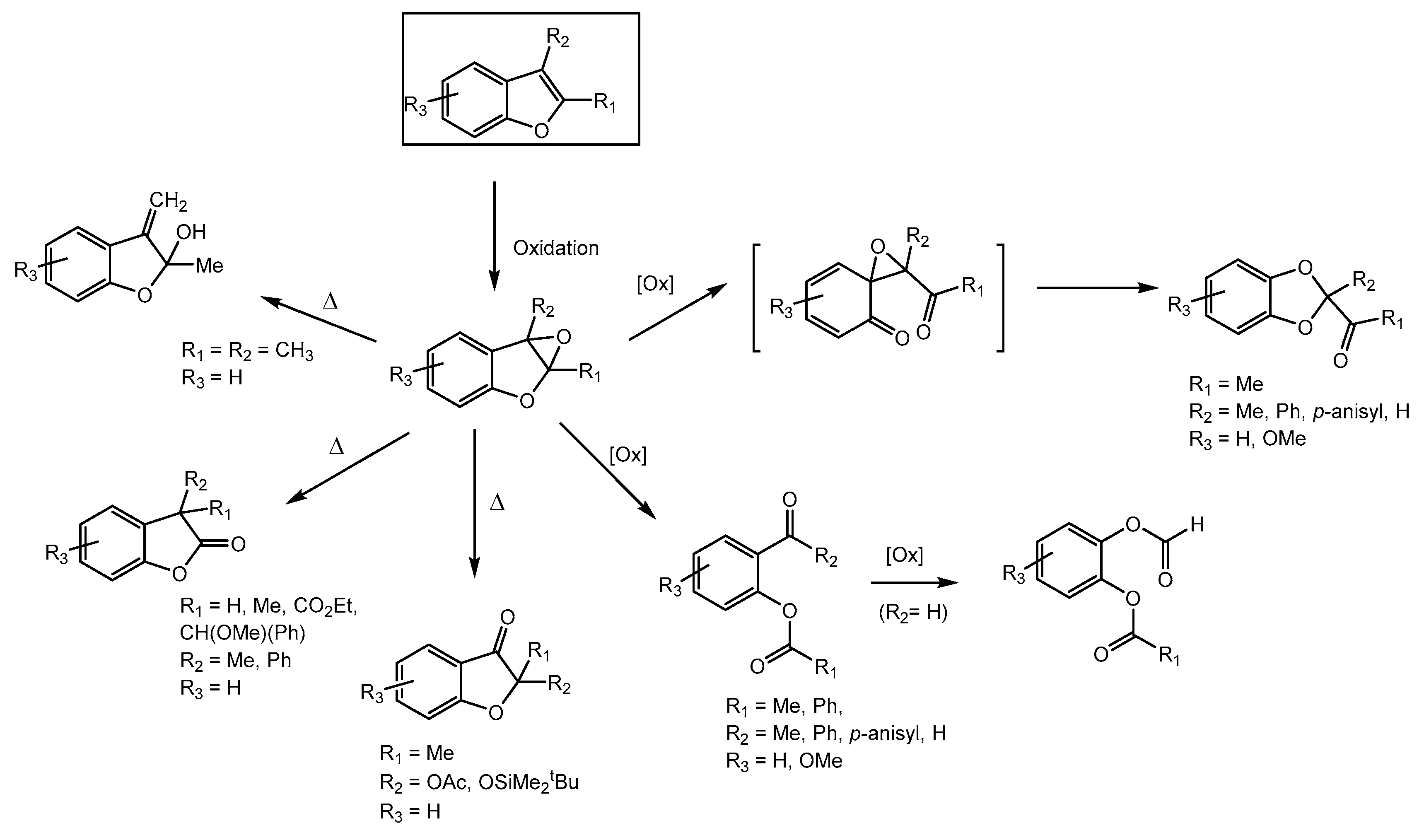
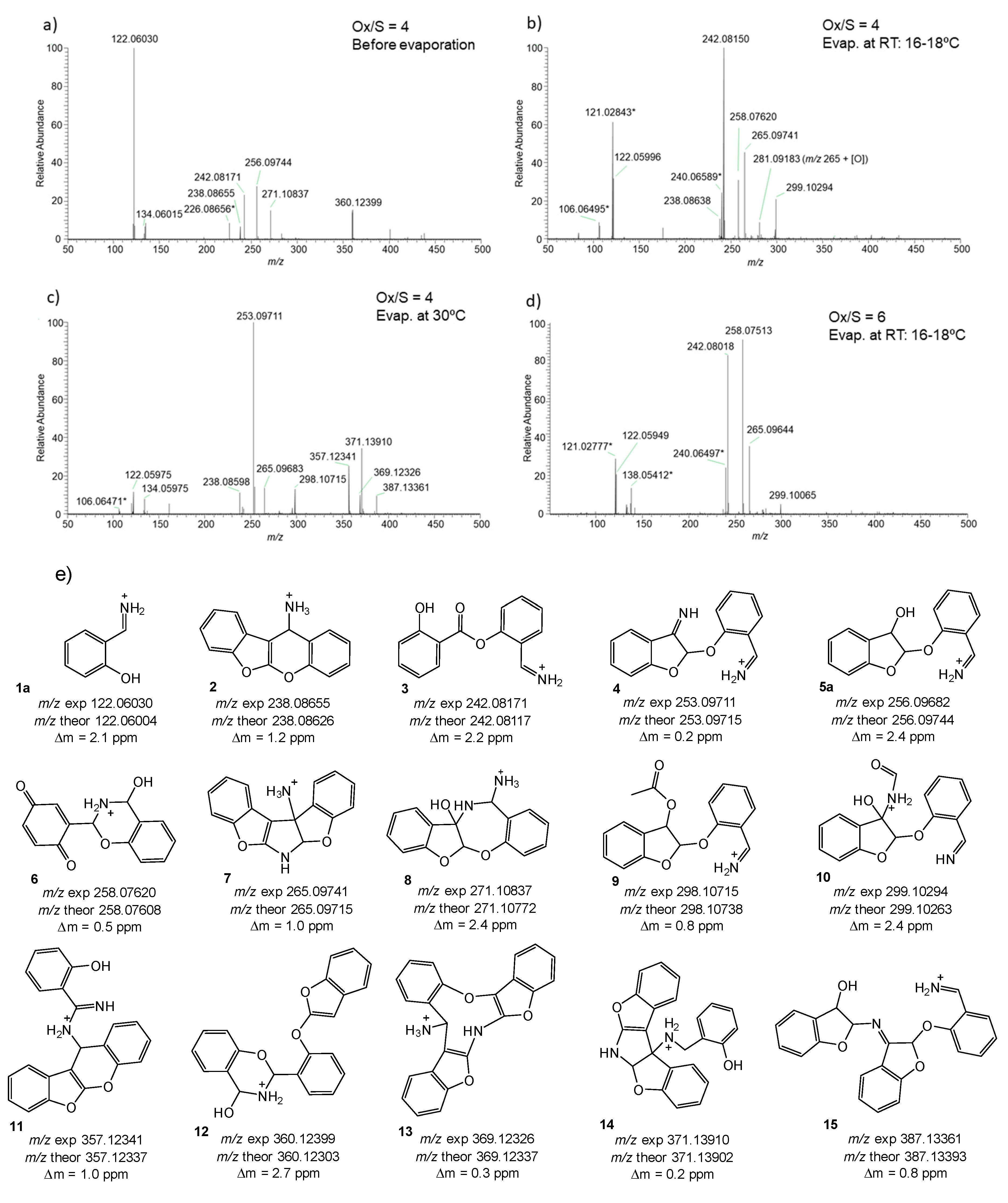
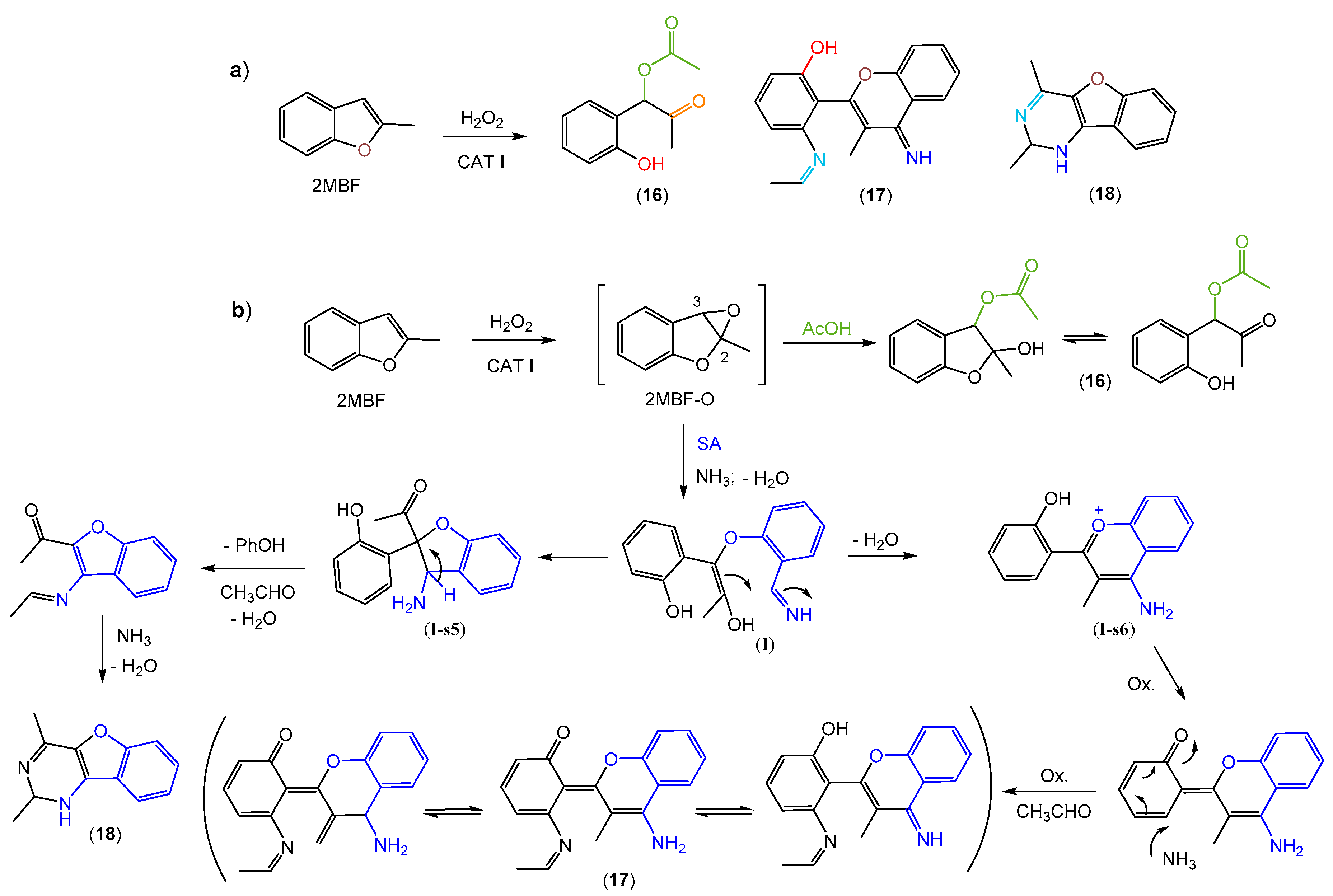
2.3. Oxidation Products of 2-Methylbenzofuran (2MBF)
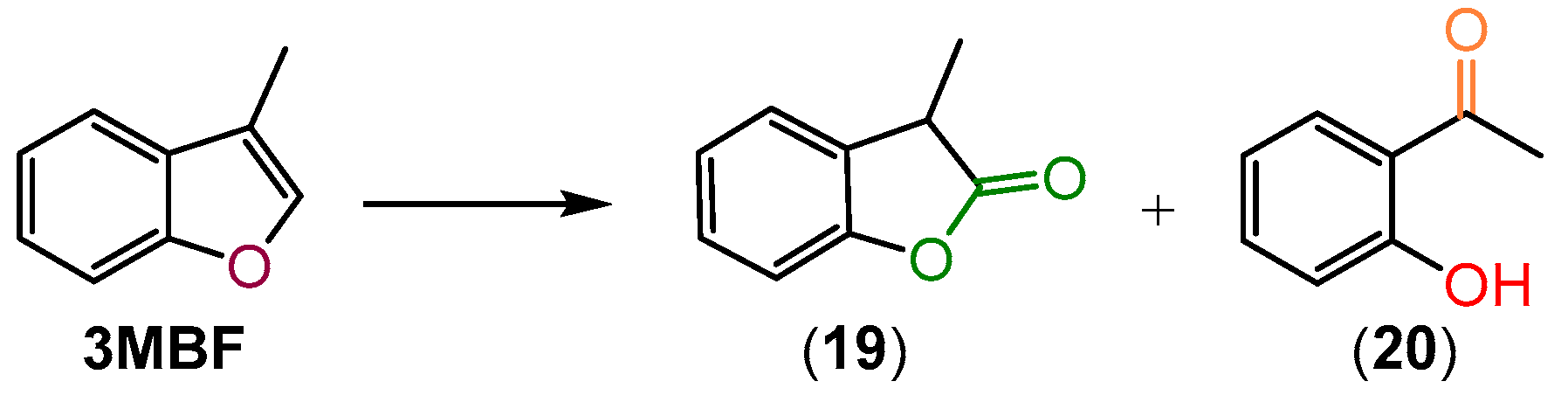
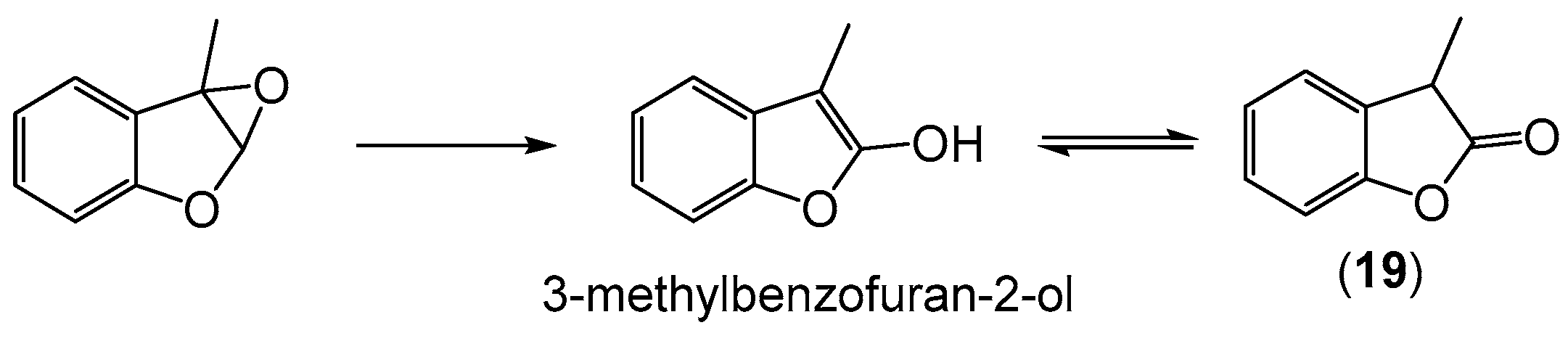
2.4. Oxidation of 3-Methylbenzofuran (3MBF)
3. Materials and Methods
3.1. Reagents and Instrumentation
3.2. Catalytic Experiments
3.3. Characterization Data of Isolated Fractions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Chen, J.; Jiang, S.; Wang, J.; Renukuntla, J.; Sirimulla, S.; Chen, J. A comprehensive review of cytochrome P450 2E1 for xenobiotic metabolism. Drug Metab. Rev. 2019, 51, 178–195. [Google Scholar] [CrossRef] [PubMed]
- Olsen, L.; Oostenbrink, C.; Jørgensen, F.S. Prediction of cytochrome P450 mediated metabolism. Adv. Drug Metab. Rev. 2015, 86, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Foroozesh, M.; Sridhar, J.; Goyal, N.; Liu, J. Coumarins and P450s, Studies Reported to-Date. Molecules 2019, 24, 1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guengerich, F.P. Cytochrome P450 and chemical toxicology. Chem. Res. Toxicol. 2008, 21, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Simões, M.M.Q.; Pires, S.M.G.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. Oxidative transformations of organic compounds mediated by metalloporphyrins as catalysts. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guillard, R., Eds.; World Scientific: Singapore, 2016; Volume 44, p. 197. [Google Scholar]
- Zhao, M.; Ou, S.; Wu, C.-D. Porous metal−organic frameworks for heterogeneous biomimetic catalysis. Acc. Chem. Res. 2014, 47, 1199–1207. [Google Scholar] [CrossRef]
- Bernadou, J.; Meunier, B. Biomimetic chemical catalysts in the oxidative activation of drugs. Adv. Synth. Catal. 2004, 346, 171–184. [Google Scholar] [CrossRef]
- Lage, A.L.A.; Meireles, A.M.; Marciano, A.C.; Ribeiro, J.M.; Souza-Fagundes, E.M.; Martins, D.C.S. Ciprofloxacin degradation by first-, second-, and third-generation manganese porphyrins. J. Hazard. Mater. 2018, 360, 445–451. [Google Scholar] [CrossRef]
- Rebelo, S.L.H.; Pereira, M.M.; Monsanto, P.V.; Burrows, H.D. Catalytic oxidative degradation of s-triazine and phenoxyalkanoic acid based herbicides with metalloporphyrins and hydrogen peroxide: Identification of two distinct reaction schemes. J. Mol. Catal. A Chem. 2009, 297, 35–43. [Google Scholar] [CrossRef]
- Rebelo, S.L.; Linhares, M.; Simões, M.M.; Silva, A.M.; Neves, M.G.P.; Cavaleiro, J.A.; Freire, C. Indigo dye production by enzymatic mimicking based on an iron (III) porphyrin. J. Catal. 2014, 315, 33–40. [Google Scholar] [CrossRef]
- Linhares, M.; Rebelo, S.L.H.; Simões, M.M.; Silva, A.M.; Neves, M.G.P.; Cavaleiro, J.A.; Freire, C. Biomimetic oxidation of indole by Mn (III) porphyrins. Appl. Catal. A Gen. 2014, 470, 427–433. [Google Scholar] [CrossRef]
- Pires, S.M.; Simões, M.M.; Santos, I.C.; Rebelo, S.L.; Paz, F.A.A.; Neves, M.G.P.; Cavaleiro, J.A. Oxidation of organosulfur compounds using an iron (III) porphyrin complex: An environmentally safe and efficient approach. Appl. Catal. B: Environ. 2014, 160–161, 80–88. [Google Scholar] [CrossRef]
- Santos, E.H.; Carvalho, C.; Terzi, C.M.; Nakagaki, S. Recent advances in catalyzed sequential reactions and the potential use of tetrapyrrolic macrocycles as catalysts. Molecules 2018, 23, 2796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Silva, G.; Pires, S.M.; Silva, V.L.; Simões, M.M.; Neves, M.G.P.; Rebelo, S.L.; Silva, A.M.S.; Cavaleiro, J.A.S. A green and sustainable method for the oxidation of 1, 3-dihydrobenzo[c]thiophenes to sulfones using metalloporphyrin complexes. Catal. Commun. 2014, 56, 68–71. [Google Scholar] [CrossRef]
- Berijani, K.; Farokhi, A.; Hosseini-Monfared, H.; Janiak, C. Enhanced enantioselective oxidation of olefins catalyzed by Mnporphyrin immobilized on graphene oxide. Tetrahedron 2018, 74, 2202–2210. [Google Scholar] [CrossRef]
- Neves, C.M.B.; Tomé, J.P.C.; Hou, Z.; Dehaen, W.; Hoogenboom, R.; Neves, M.G.P.; Simões, M.M.Q. Oxidation of monoterpenes catalysed by a water-soluble MnIIIPEG-porphyrin in a biphasic medium. ChemCatChem 2018, 10, 2804–2809. [Google Scholar] [CrossRef]
- Maljutenko, K.; Borovkov, V.; Kananovich, D.; Järving, I.; Lopp, M. Aerobic cascade oxidation of substituted cyclopentane-1, 2-diones using metalloporphyrin catalysts. Tetrahedron 2018, 74, 661–664. [Google Scholar] [CrossRef]
- Connelly, J.C.; Connor, S.C.; Monte, S.; Bailey, N.J.C.; Borgeaud, N.; Holmes, E.; Troke, J.; Nicholson, J.K.; Gavaghan, C.L. Application of directly coupled high performance liquid chromatography-NMR-mass spectometry and 1H NMR spectroscopic studies to the investigation of 2,3-benzofuran metabolism in sprague-dawley rats. Drug Metab. Dispos. 2002, 30, 1357–1363. [Google Scholar] [CrossRef] [Green Version]
- Linhares, M.; Rebelo, S.L.H.; Biernacki, K.; Magalhães, A.L.; Freire, C. Biomimetic One-Pot Route to Acridine Epoxides. J. Org. Chem. 2015, 80, 281–289. [Google Scholar] [CrossRef]
- Nevagi, R.J.; Dighe, S.N.; Dighe, S.N. Biological and medicinal significance of benzofuran. Eur. J. Med. Chem. 2015, 97, 561–581. [Google Scholar] [CrossRef]
- Khanam, H. Shamsuzzaman, Bioactive benzofuran derivatives: A review. Eur. J. Med. Chem. 2015, 97, 483–505. [Google Scholar] [CrossRef]
- Hiremathad, A.; Patil, M.R.; Chethana, K.R.; Chand, K.; Santos, M.A.; Keri, R.S. Benzofuran: An emerging scaffold for antimicrobial agents. RSC Adv. 2015, 5, 96809–96828. [Google Scholar] [CrossRef]
- Dawood, K.M. Benzofuran derivatives: A patent review. Expert Opin. Ther. Pat. 2013, 23, 1133–1156. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Zhu, H.; Zhang, H.; Lang, Q.; Tang, L.; Huang, Q.; Yu, L. In vitro and in vivo characterization of a benzofuran derivative, a potential anticancer agent, as a novel Aurora B kinase inhibitor. Eur. J. Med. Chem. 2015, 89, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.-J.; Zhang, X.-W.; Yang, L.; Li, W.; Li, J.-H.; Wang, J.-X.; Chen, J. Synthesis and evaluation of xanthine oxidase inhibitory and antioxidant activities of 2-arylbenzo[b]furan derivatives based on salvianolic acid C. Eur. J. Med. Chem. 2016, 124, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Zha, X.; Lamba, D.; Zhang, L.; Lou, Y.; Xu, C.; Kang, D.; Chen, L.; Xu, Y.; Zhang, L.; De Simone, A.; et al. Novel Tacrine-benzofuran hybrids as potent multitarget-directed ligands for the treatment of Alzheimer’s disease: Design, synthesis, biological evaluation, and X-ray crystallography. J. Med. Chem. 2016, 59, 114–131. [Google Scholar] [CrossRef]
- Sauter, M.; Adam, W. Oxyfunctionalization of benzofurans by singlet oxygen, dioxiranes, and peracids: Chemical model studies for the DNA-damaging activity of benzofuran dioxetanes (oxidation) and epoxides (alkylation). Acc. Chem. Res. 1995, 28, 289–298. [Google Scholar] [CrossRef]
- Rebelo, S.L.H.; Pires, S.M.G.; Simões, M.M.Q.; Medforth, C.J.; Cavaleiro, J.A.S.; Neves, M.G.P. A green and versatile route to highly functionalized benzofuran derivatives using biomimetic oxygenation. ChemistrySelect 2018, 3, 1392–1403. [Google Scholar] [CrossRef]
- Rebelo, S.L.H.; Moniz, T.; Medforth, C.J.; Castro, B.; Rangel, M. EPR spin trapping studies of H2O2 activation in metaloporphyrin catalyzed oxygenation reactions: Insights on the biomimetic mechanism. Mol. Catal. 2019, 475, 110500. [Google Scholar] [CrossRef]
- Costa, P.; Linhares, M.; Rebelo, S.L.H.; Neves, M.G.P.; Freire, C. Direct access to polycyclic peripheral diepoxy-mesoquinone derivatives from acene catalytic oxidation. RSC Adv. 2013, 3, 5350–5353. [Google Scholar] [CrossRef]
- Rebelo, S.L.H.; Simões, M.M.Q.; Neves, M.G.P.; Cavaleiro, J.A.S. Oxidation of alkylaromatics with hydrogen peroxide catalysed by manganese(III) porphyrins in the presence of ammonium acetate. J. Mol. Catal. 2003, 201, 9–22. [Google Scholar] [CrossRef]
- Rebelo, S.L.H.; Silva, A.M.N.; Medforth, C.J.; Freire, C. Iron (III) Fluorinated Porphyrins: Greener Chemistry from Synthesis to Oxidative Catalysis Reactions. Molecules 2016, 21, 481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, J.; Silva, A.M.N.; Rebelo, S.L.H.; Cunha-Silva, L.; Rangel, M.; Castro, B.; Leite, A.; Silva, A.M.G. Synthesis and coordination studies of 5-(4’-carboxyphenyl)-10,15,20-tris(pentafluorophenyl)porphyrin and its pyrrolidine-fused chlorin derivative. New J. Chem. 2018, 42, 8169–8179. [Google Scholar] [CrossRef]
- Rocha, M.; Rebelo, S.L.H.; Freire, C. Enantioselective arene epoxidation under mild conditions by Jacobsen catalyst. Appl. Catal. A Gen. 2013, 460–461, 116–123. [Google Scholar] [CrossRef]
- De Paula, R.; Simões, M.M.Q.; Neves, M.G.P.; Cavaleiro, J.A.S. Homogeneous olefin epoxidation catalysed by an imidazolium-based manganese porphyrin. Catal. Commun. 2008, 10, 57–60. [Google Scholar] [CrossRef]
- Takahashi, M.; Micalizio, G.C. Regio- and stereoselective cross-coupling of substituted olefins and imines. J. Am. Chem. Soc. 2007, 129, 7514–7516. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Loading (mol%) | Conversion (%) | Time (min) | Selectivity (η) a | |
|---|---|---|---|---|---|
| 19 | 20 | ||||
| I | 0.3 | 99.7 | 120 | 91.9 (91.6) | 8.1 (8.1) |
| I | 0.7 | 99.9 | 90 | 88.3 (88.2) | 11.7 (11.7) |
| II | 0.3 | 79.9 | 240 | 86.8 (69.4) | 13.2 (10.5) |
| II | 0.7 | 84.3 | 180 | 87.0 (73.3) | 13.0 (11.0) |
| III | 0.3 | 99.8 | 120 | 98.8 (98.6) | 1.1 (1.1) |
| III | 0.7 | 99.9 | 90 | 98.4 (98.3) | 1.6 (1.6) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rebelo, S.L.H.; Pires, S.M.G.; Simões, M.M.Q.; de Castro, B.; Neves, M.G.P.M.S.; Medforth, C.J. Biomimetic Oxidation of Benzofurans with Hydrogen Peroxide Catalyzed by Mn(III) Porphyrins. Catalysts 2020, 10, 62. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10010062
Rebelo SLH, Pires SMG, Simões MMQ, de Castro B, Neves MGPMS, Medforth CJ. Biomimetic Oxidation of Benzofurans with Hydrogen Peroxide Catalyzed by Mn(III) Porphyrins. Catalysts. 2020; 10(1):62. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10010062
Chicago/Turabian StyleRebelo, Susana L. H., Sónia M. G. Pires, Mário M. Q. Simões, Baltazar de Castro, M. Graça P. M. S. Neves, and Craig J. Medforth. 2020. "Biomimetic Oxidation of Benzofurans with Hydrogen Peroxide Catalyzed by Mn(III) Porphyrins" Catalysts 10, no. 1: 62. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10010062