Crystal Structures and Electrical Resistivity of Three Exotic TMTSF Salts with I
3
−
: Determination of Valence by DFT and MP2 Calculations



Abstract
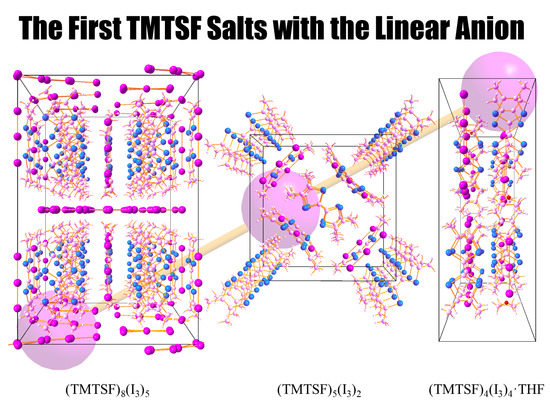
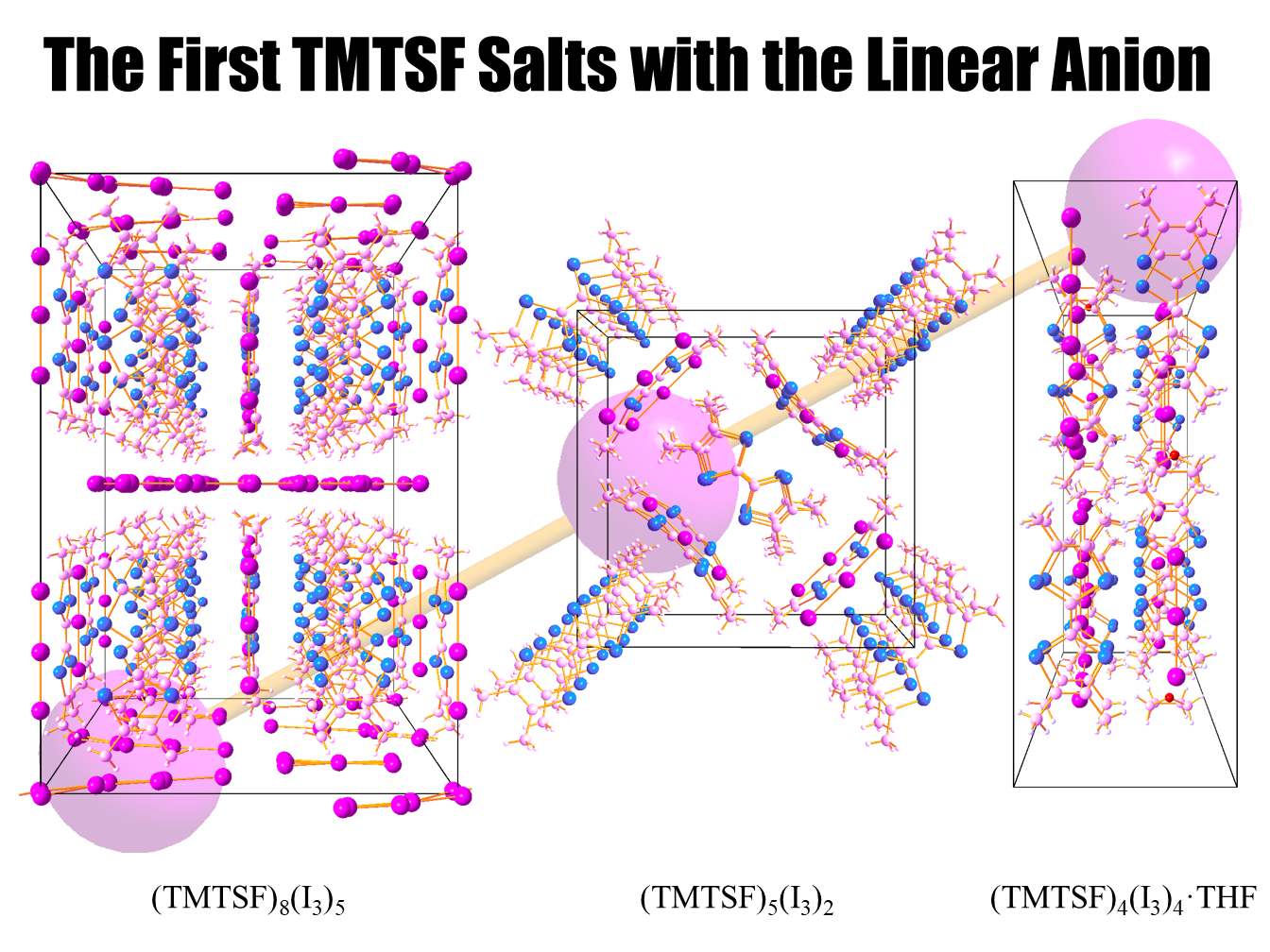
:
1. Introduction
2. Results
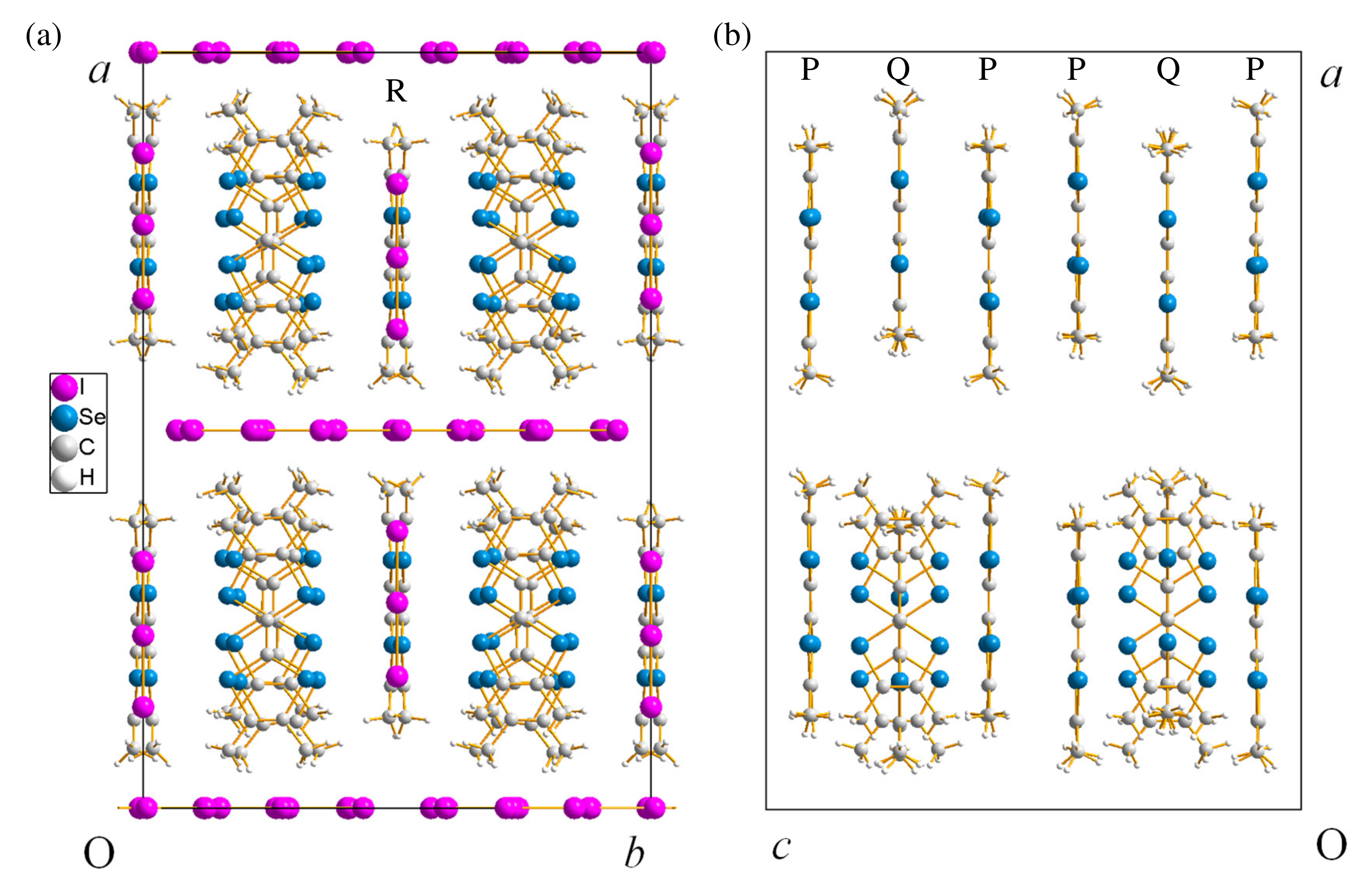
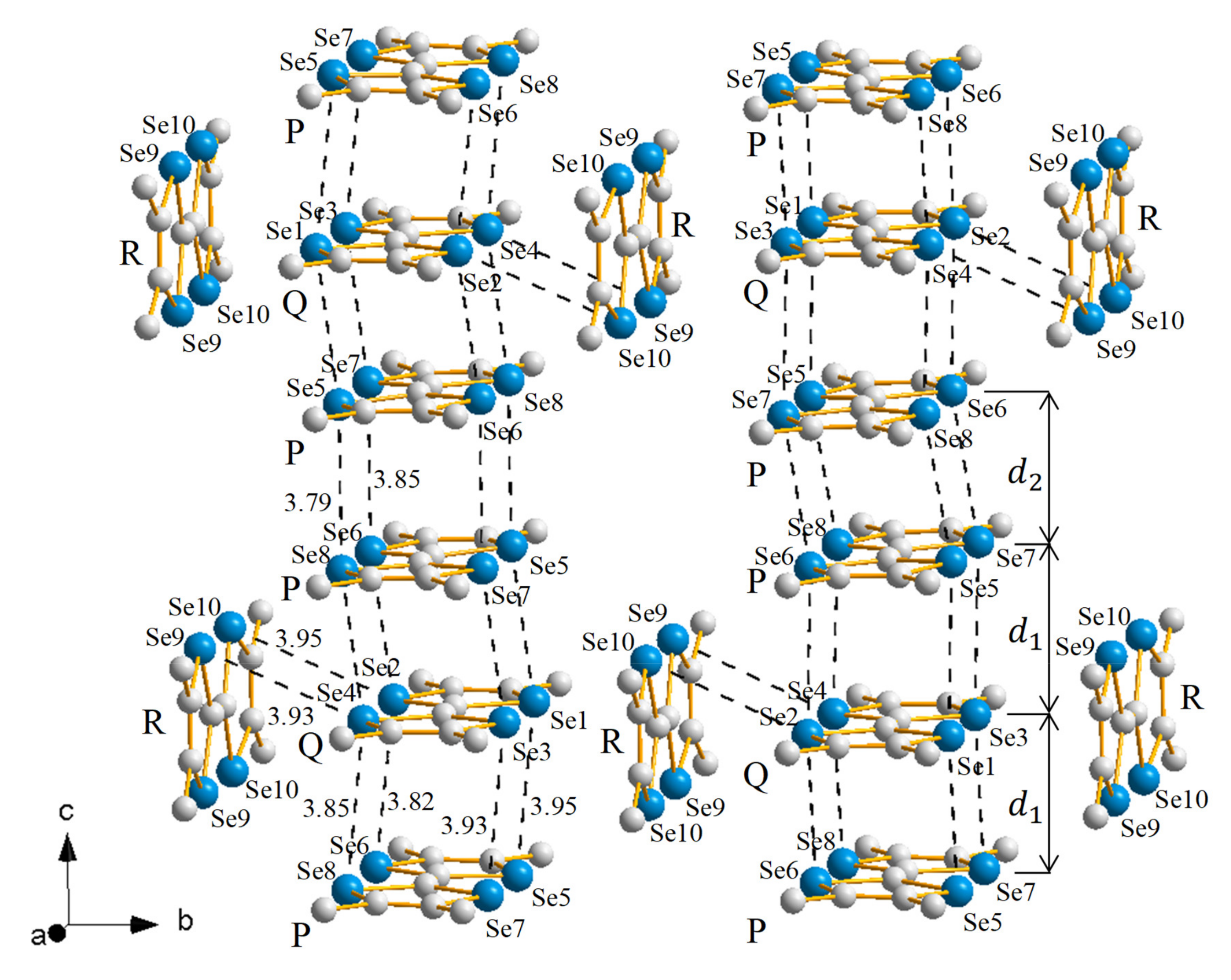
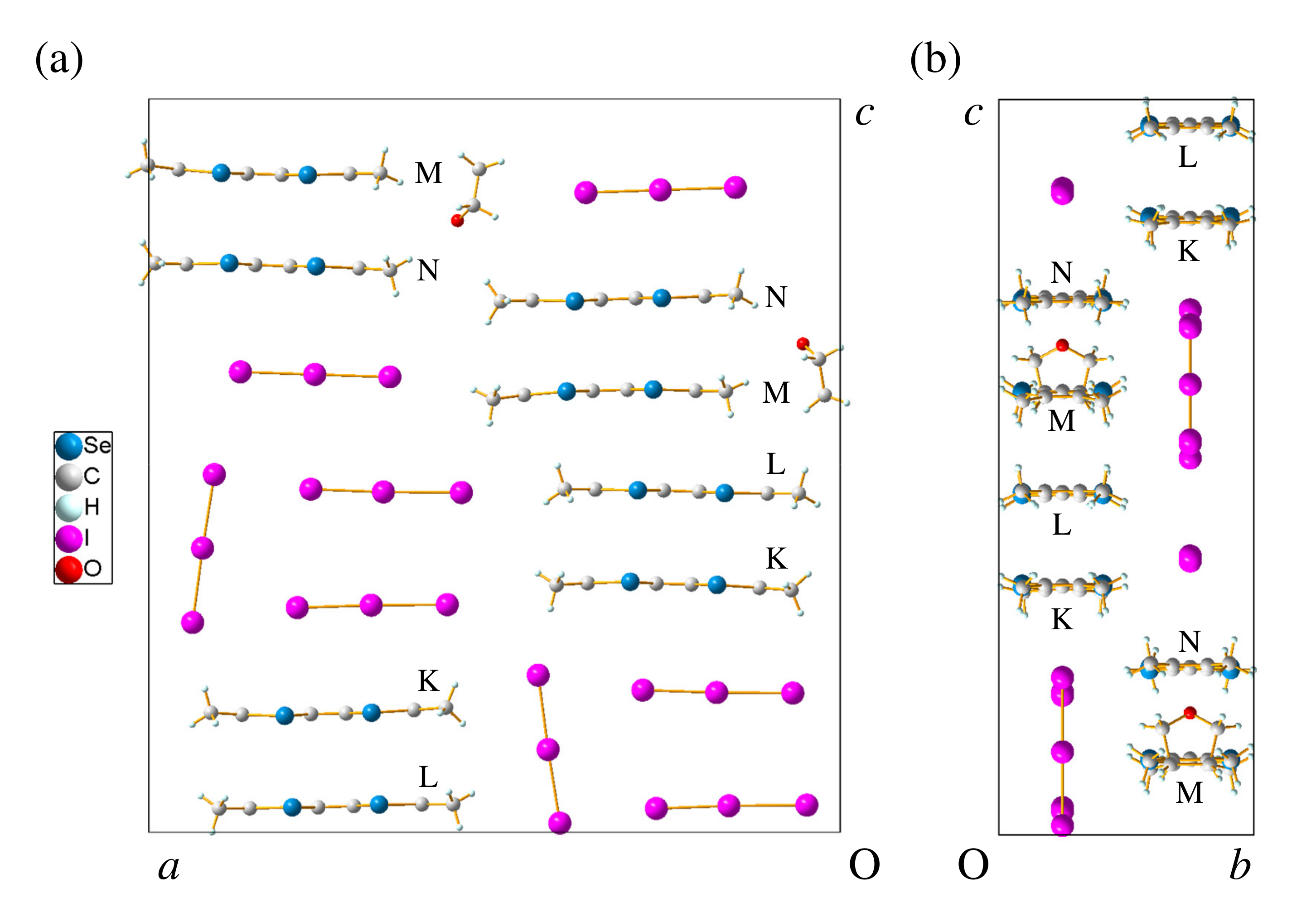
2.1. (TMTSF)(I)
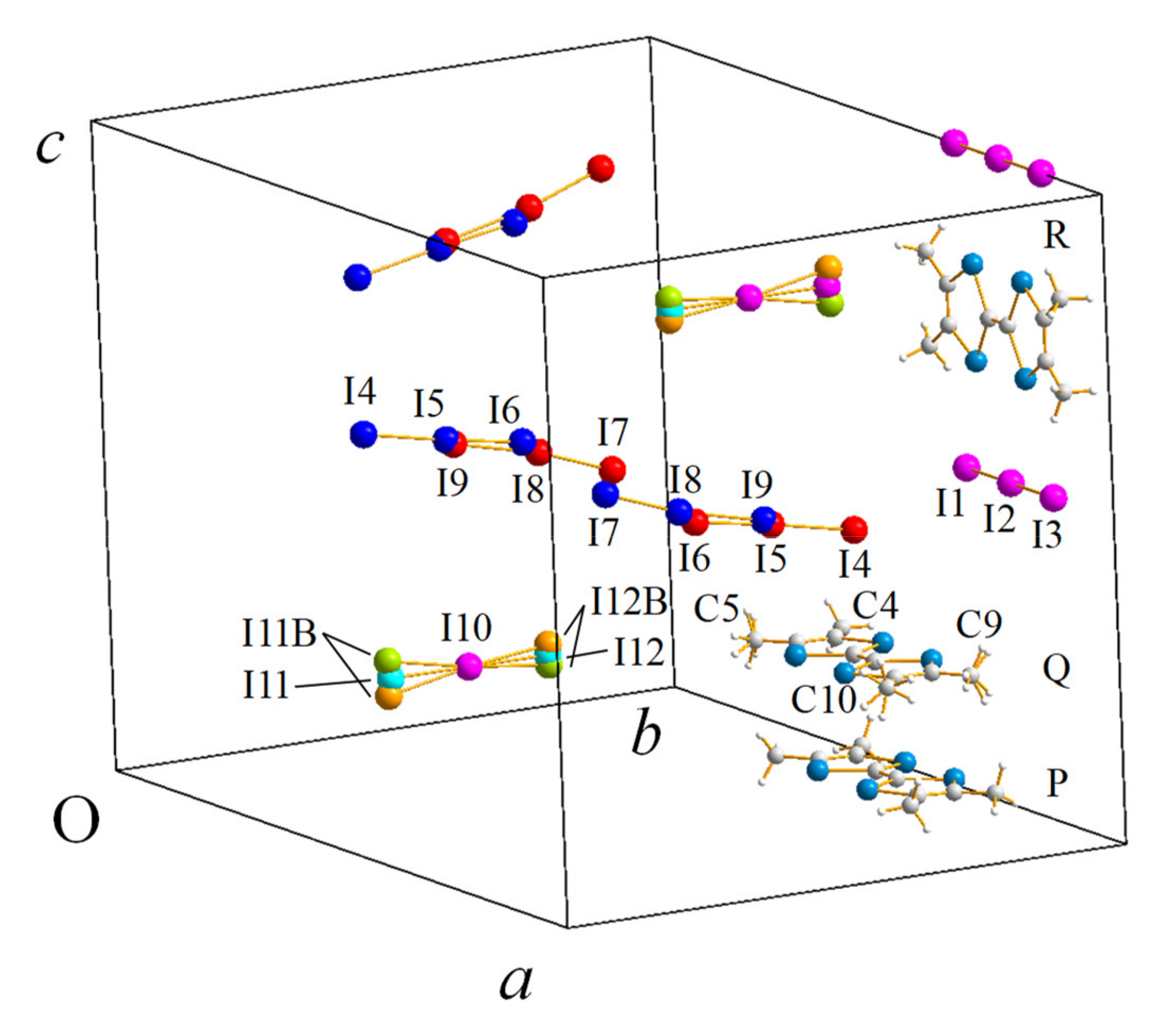
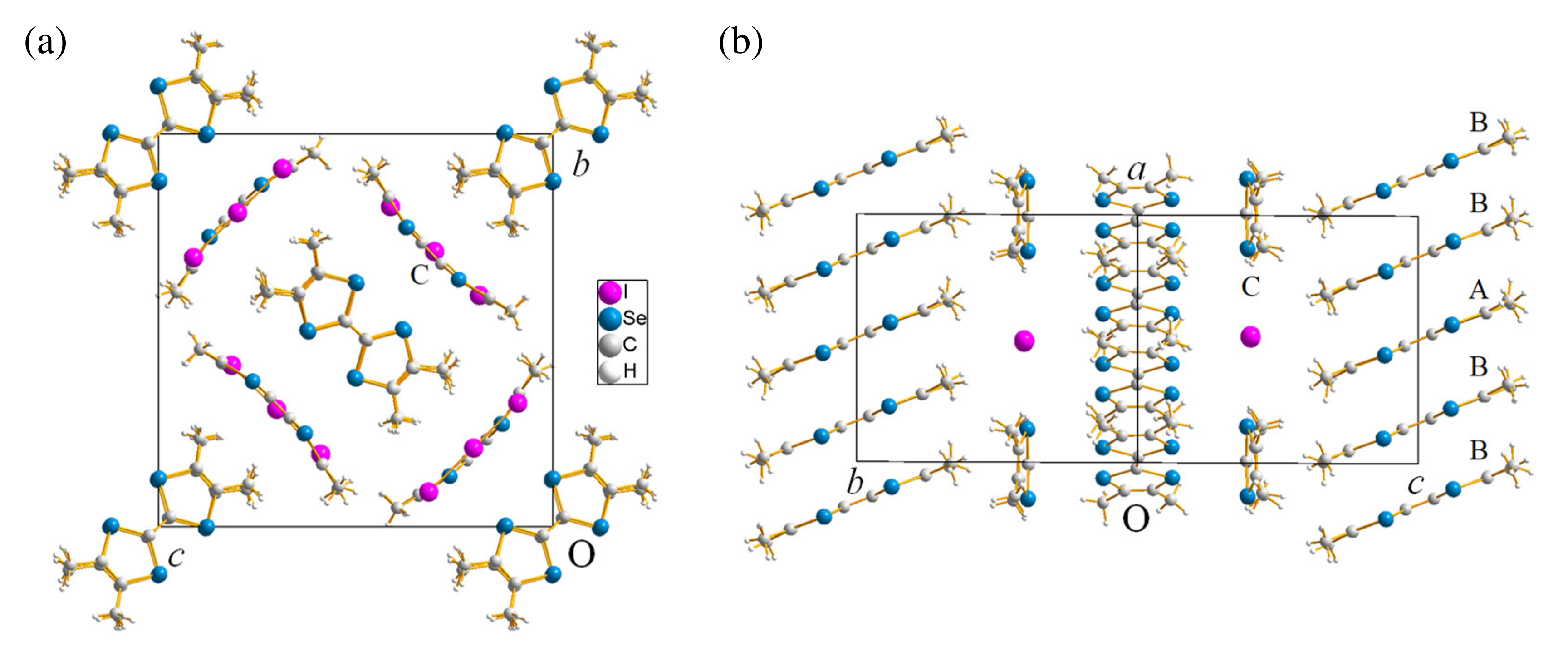
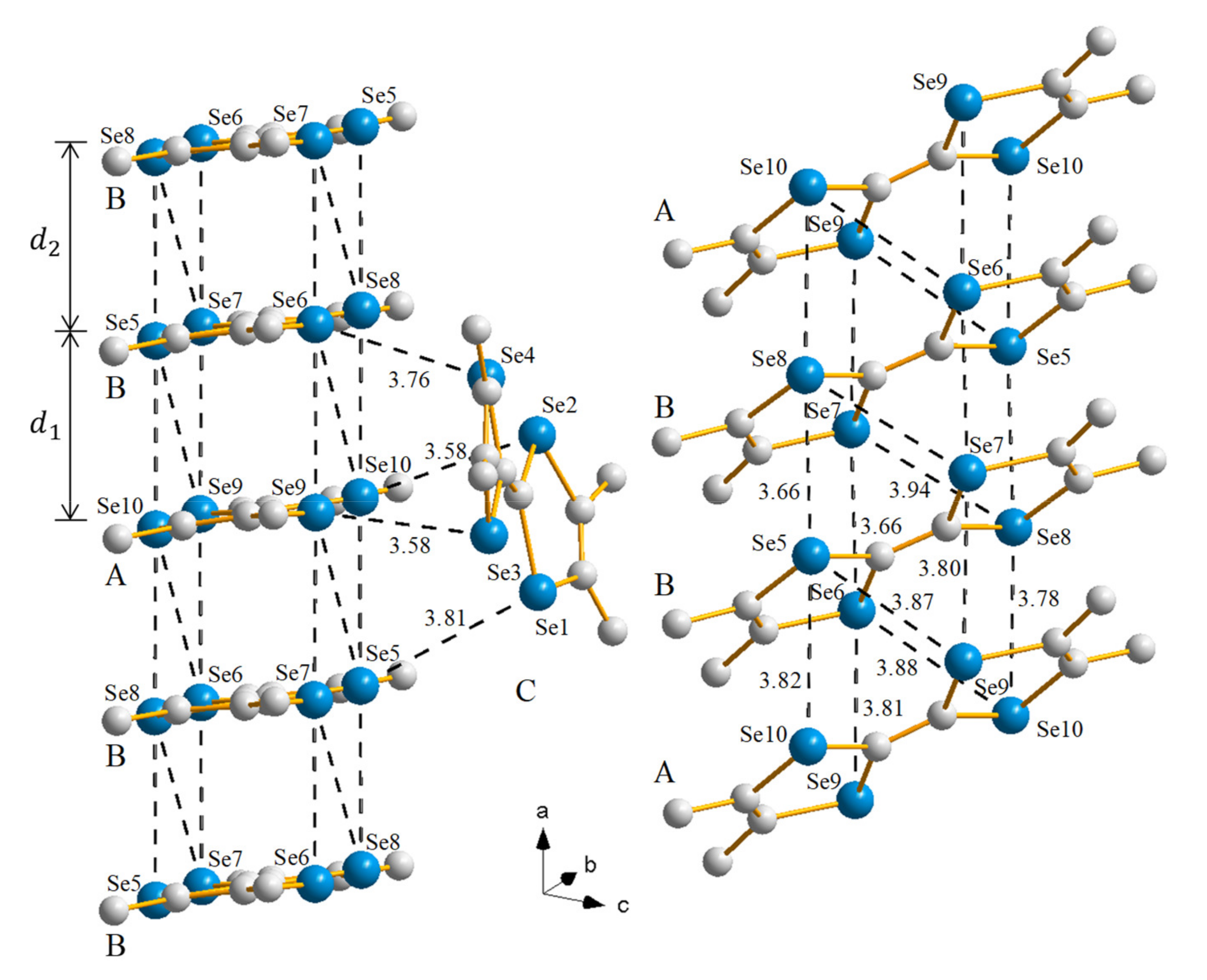
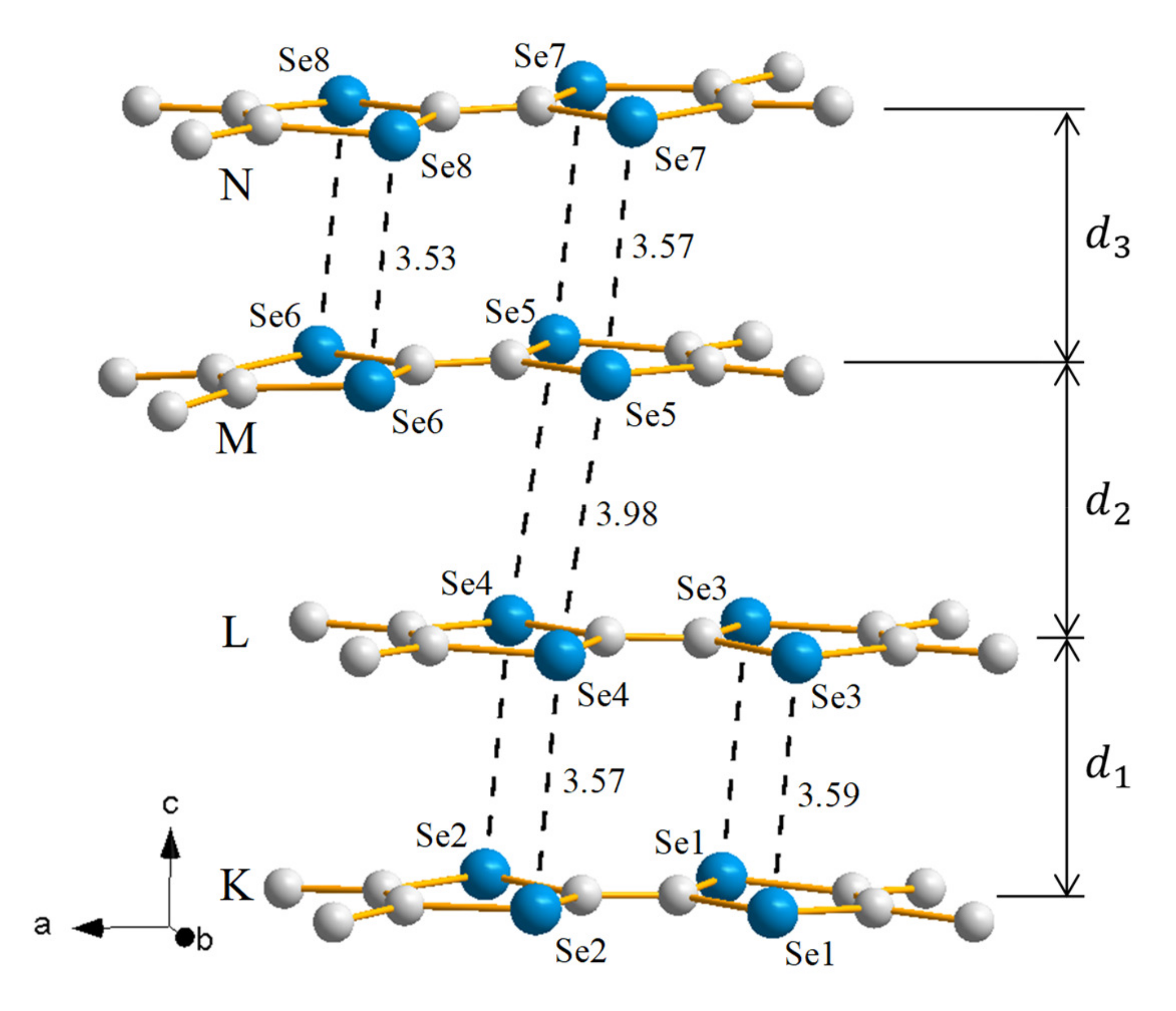
2.1.1. Crystal Structure
2.1.2. Electrical Resistivity
2.2. (TMTSF)(I)
2.2.1. Crystal Structure
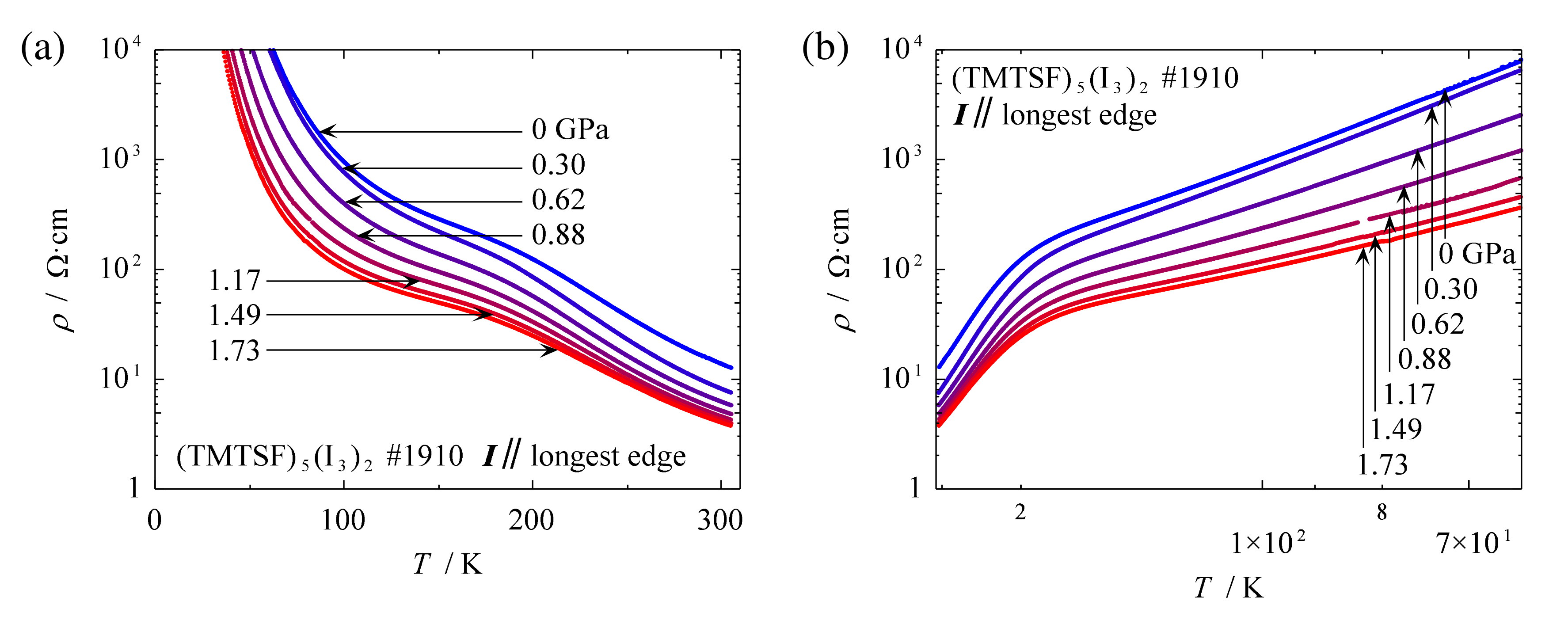
2.2.2. Electrical Resistivity
2.3. (TMTSF)(I)·THF
3. Discussion
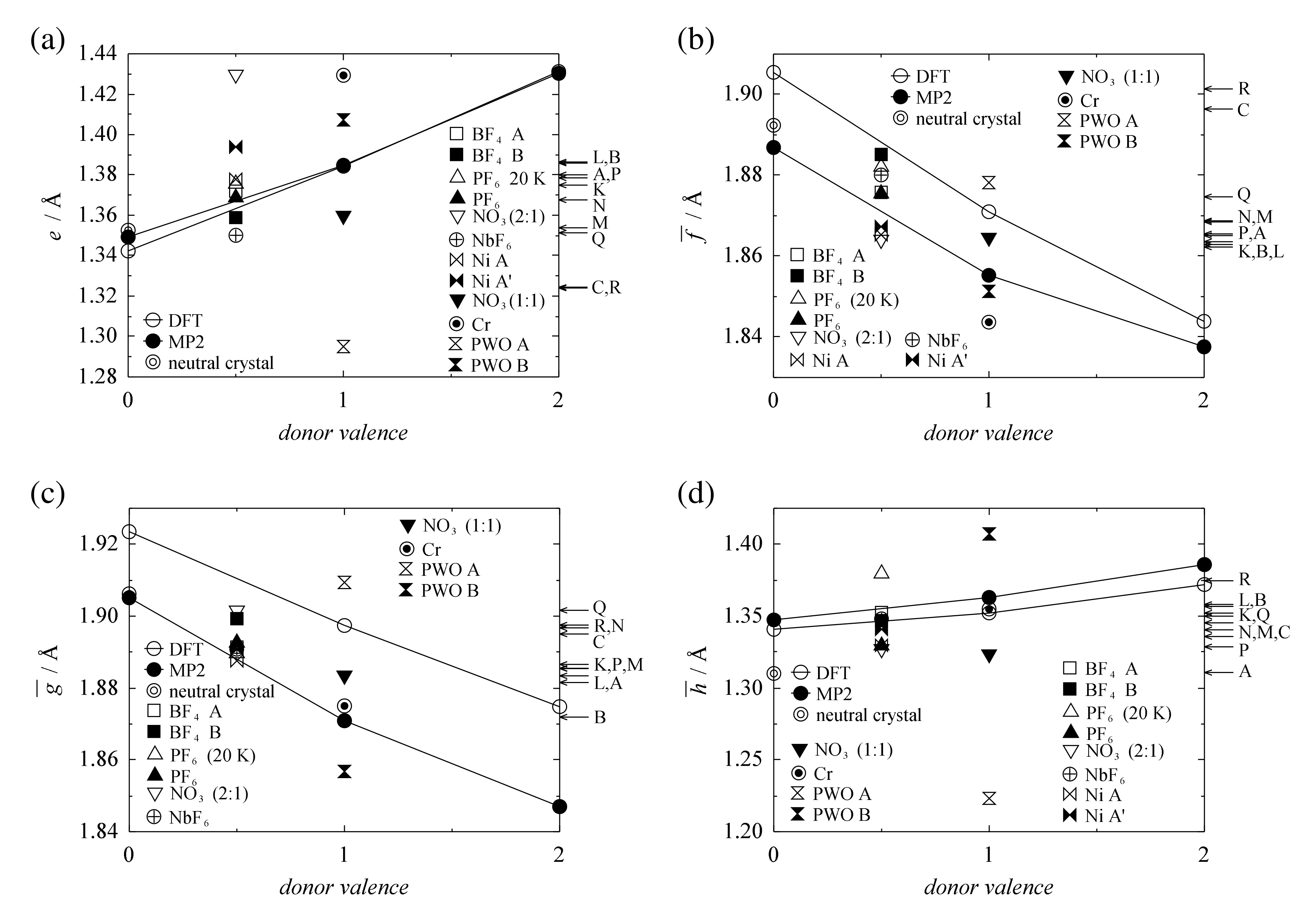
3.1. Valence and Bond Lengths of TMTSF Molecules
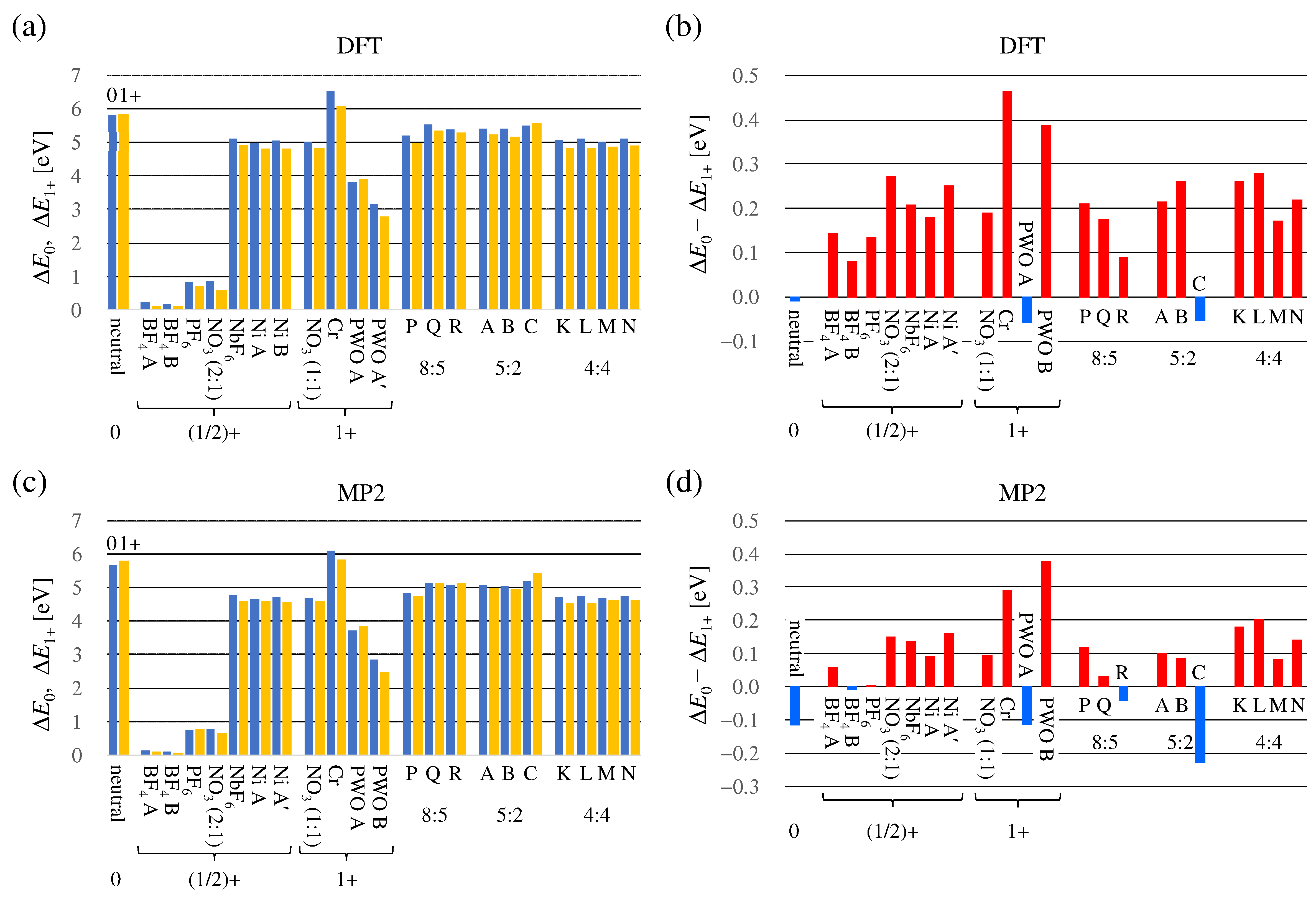
3.2. Valence of TMTSF Molecule Estimated from Total Energy
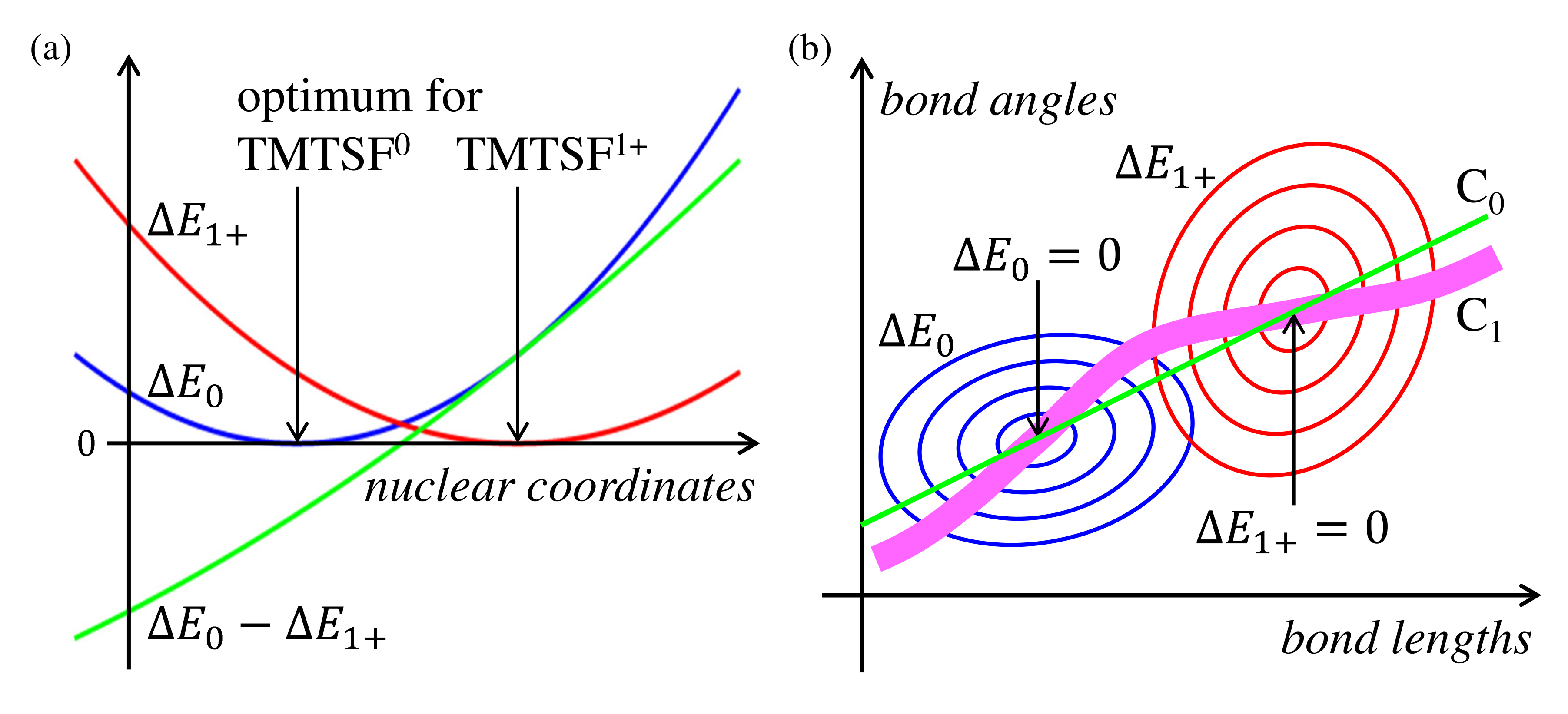
3.2.1. Principles
- The optimized structures and their total energies of TMTSF () and TMTSF () are calculated. Please note that the spin-unrestricted calculations are applied for cations; and all the electrons are considered in the MP2 calculations.
- For example, for the TMTSF A in the 5:2 salt, the total energies of TMTSF () and TMTSF () are calculated using the crystallographic data.
- The energy differences and are calculated. The and should be positive since the conformation of A is not optimum both in the neutral and cationic states.
- The difference is used as a measure of the valence of A in the crystal.
3.2.2. Energy Calculations
3.2.3. (TMTSF)(I)
3.2.4. (TMTSF)(I)
3.2.5. (TMTSF)(I)·THF
3.2.6. Ionization Potentials
4. Materials and Methods
4.1. Sample Preparation
4.2. X-ray Crystal Structure Analysis
4.3. Measurement of Electrical Resistivity
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Jérome, D.; Mazaud, A.; Ribault, M.; Bechgaard, K. Superconductivity in a synthetic organic conductor (TMTSF)2PF6. J. Phys. Lett. 1980, 41, L95–L98. [Google Scholar] [CrossRef] [Green Version]
- Bechgaard, K.; Carneiro, K.; Olsen, M.; Rasmussen, F.B. Zero-pressure organic superconductor: Di-(tetramethyltetraselenafulvalenium)-perchlorate [(TMTSF)2ClO4]. Phys. Rev. Lett. 1981, 46, 852–855. [Google Scholar] [CrossRef] [Green Version]
- Bechgaard, K.; Jacobsen, C.S.; Mortensen, K.; Pedersen, H.J.; Thorup, N. The properties of five highly conducting salts: (TMTSF)2X, X = PF, AsF, SbF, BF and NO, derived from tetramethyltetraselenafulvalene (TMTSF). Solid State Commun. 1980, 33, 1119–1125. [Google Scholar] [CrossRef]
- Parkin, S.S.S.; Ribault, M.; Jérome, D.; Bechgaard, K. Superconductivity in the family of organic salts based on the tetramethyltetraselenafulvalene (TMTSF) molecule: (TMTSF)2X (X = ClO4, PF6, AsF6, SbF6, TaF6). J. Phys. C Solid State Phys. 1981, 14, 5305–5326. [Google Scholar] [CrossRef]
- Sakata, M.; Yoshida, Y.; Maesato, M.; Hagiwara, R. Preparation of superconducting (TMTSF)2NbF6 by electrooxidation of TMTSF using ionic liquid as electrolyte. Mol. Cryst. Liq. Cryst. 2006, 452, 103–112. [Google Scholar] [CrossRef]
- Parkin, S.S.S.; Jérome, D.; Bechgaard, K. Pressure dependence of the metal–insulator and superconducting phase transitions in (TMTSF)2ReO4. Mol. Cryst. Liq. Cryst. 1981, 79, 213–224. [Google Scholar]
- Rindorf, G.; Soling, H.; Thorup, N. The structures of di(2,3,6,7-tetramethyl-1,4,5,8-tetraselenafulvalenium) perrhenate, (TMTSF)2ReO4, and perchlorate, (TMTSF)2ClO4. Acta Cryst. B 1982, 38, 2805–2808. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.M.; Beno, M.A.; Appelman, E.H.; Capriotti, J.M.; Wudl, F.; Aharon-Shalom, E.; Nalewajek, D. New (TMTSF)2X derivatives: A change in the selenium network dimensionality derived from the molecular and crystal structures of (TMTSF)2(FSO3) [T = 298K, 123K] and (TMTSF)2(BrO4) T = 298K. Mol. Cryst. Liq. Cryst. 1982, 79, 319–326. [Google Scholar] [CrossRef]
- Cox, S.; Boysel, R.M.; Moses, D.; Wudl, F.; Chen, J.; Ochsenbein, S.; Heeger, A.J.; Walsh, W.M., Jr.; Rupp, L.W., Jr. (TMTSF)2F2PO2: An unusual member of the (TMTSF)2X family of organic metals. Solid State Commun. 1984, 49, 259–263. [Google Scholar] [CrossRef]
- Soling, H.; Rindorf, G.; Thorup, N. Di(4,4′,5,5′-tetramethyl-Δ2,2′-bi-1,3-diselenolyliden)ium trifluoromethanesulfonate, C21H24F3O3SSe8, (TMTSF)2CF3SO3. Acta Cryst. C 1983, 39, 490–491. [Google Scholar] [CrossRef]
- Thorup, N.; Rindorf, G.; Soling, H.; Johannsen, I.; Mortensen, K.; Bechgaard, K. Structural studies of some (TMTSF)2X compounds. J. Phys.-Paris C 1983, 3, 1017–1020. [Google Scholar] [CrossRef]
- Beno, M.A.; Blackman, G.S.; Williams, J.M.; Bechgaard, K. Synthetic metals based on tetramethyltetraselenafulvalene (TMTSF): Synthesis, structure (T = 298 and 125 K), and novel properties of (TMTSF)2H2F3. Inorg. Chem. 1982, 21, 3860–3862. [Google Scholar] [CrossRef]
- Eriks, K.; Beno, M.A.; Bechgaard, K.; Williams, J.M. The structure of di(3,3′,4,4′-tetramethyl-2,2’,5,5′-tetraselenafulvalenium) pentafluorosilicate, (C10H11Se4)2SiF5 at 293 and 125 K. Acta Cryst. C 1984, 40, 1715–1717. [Google Scholar] [CrossRef] [Green Version]
- Nickel, J.C.; Duprat, R.; Bourbonnais, C.; Dupuis, N. Triplet superconducting pairing and density-wave instabilities in organic conductors. Phys. Rev. Lett. 2005, 95, 247001. [Google Scholar] [CrossRef] [Green Version]
- Rozhkov, A.V. Superconductivity without attraction in a quasi-one-dimensional metal. Phys. Rev. B 2009, 79, 224520. [Google Scholar] [CrossRef] [Green Version]
- Lebed, A.G. d-Wave-like nodal superconductivity in the organic conductor (TMTSF)2ClO4. Physica B 2012, 404, 1803–1805. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Kuroki, K. Microscopic theory of spin-triplet f-wave pairing in quasi-one-dimensional organic superconductors. Phys. Rev. B 2004, 70, 060502R. [Google Scholar] [CrossRef] [Green Version]
- Fuseya, Y.; Suzumura, Y. Superconductivity and density wave in the quasi-one-dimensional systems: Renormalization group study. J. Phys. Soc. Jpn. 2005, 74, 1263–1269. [Google Scholar] [CrossRef] [Green Version]
- Pouget, J.P.; Moret, R.; Comes, R.; Bechgaard, K.; Fabre, J.M.; Giral, L. X-ray diffuse scattering study of some (TMTSF)2X and (TMTTF)2X salts. Mol. Cryst. Liq. Cryst. 1982, 79, 129–143. [Google Scholar] [CrossRef]
- Andrieux, A.; Jérome, D.; Bechgaard, K. Spin-density wave ground state in the one-dimensional conductor (TMTSF)2PF6: Microscopic evidence from 77Se and 1H NMR experiments. J. Phys. Lett. 1981, 42, L87–L90. [Google Scholar] [CrossRef]
- Scott, J.C.; Pedersen, H.J.; Bechgaard, K. Proton NMR in the organic conductor tetramethyltetraselenafulvalinium hexafluorophosphate. Phys. Rev. B 1981, 24, 475–477. [Google Scholar] [CrossRef]
- Mortensen, K.; Tomkiewicz, Y.; Bechgaard, K. Antiferromagnetism in the organic conductor bis-tetramethyltetraselenafulvalene hexafluoroarsenate [(TMTSF)2AsF6]: Static magnetic susceptibility. Phys. Rev. B 1982, 25, 3319–3325. [Google Scholar] [CrossRef] [Green Version]
- Torrance, J.B.; Pedersen, H.J.; Bechgaard, K. Observation of antiferromagnetic resonance in an organic superconductor. Phys. Rev. Lett. 1982, 49, 881–884. [Google Scholar] [CrossRef] [Green Version]
- Walsh, W.M., Jr.; Wudl, F.; Aharon-Shalom, E.; Rupp, L.W., Jr.; Vandenberg, J.M.; Andres, K.; Torrance, J.B. Itinerant-electron antiferromagnetism precursor to superconductivity in an organic conductor. Phys. Rev. Lett. 1982, 49, 885–888. [Google Scholar] [CrossRef]
- Delrieu, J.M.; Roger, M.; Toffano, Z.; Moradpour, A.; Bechgaard, K. NMR proton lineshape in (TMTSF)2X: Incommensurability of nesting vector and order parameter. J. Phys. 1986, 47, 839–861. [Google Scholar] [CrossRef]
- Takahashi, T.; Maniwa, Y.; Kawamura, H.; Saito, G. Determination of SDW characteristics in (TMTSF)2PF6 by 1H-NMR analysis. J. Phys. Soc. Jpn. 1986, 55, 1364–1373. [Google Scholar] [CrossRef]
- Takahashi, T.; Maniwa, Y.; Kawamura, H.; Saito, G. Pressure dependence of SDW properties in the organic conductor, (TMTSF)2PF6. J. Phys. Soc. Jpn. 1989, 58, 703–709. [Google Scholar] [CrossRef]
- Kwak, J.F.; Schirber, J.E.; Greene, R.L.; Engler, E.M. Magnetic quantum oscillations in tetramethyltetraselenafulvalenium hexafluorophosphate [(TMTSF)2PF6]. Phys. Rev. Lett. 1981, 46, 1296–1299. [Google Scholar] [CrossRef]
- Bando, H.; Oshima, K.; Suzuki, M.; Kobayashi, H.; Saito, G. Shubnikov–de Haas effect in (TMTSF)2ClO4. J. Phys. Soc. Jpn. 1982, 51, 2711–2712. [Google Scholar] [CrossRef]
- Chaikin, P.M.; Choi, M.-Y.; Kwak, J.F.; Brooks, J.S.; Martin, K.P.; Naughton, M.J.; Engler, E.M.; Greene, R.L. Tetramethyltetraselenafulvalenium perchlorate, (TMTSF)2ClO4, in high magnetic fields. Phys. Rev. Lett. 1983, 51, 2333–2336. [Google Scholar] [CrossRef]
- Azevedo, L.J.; Williams, J.M.; Compton, S.J. Field-induced magnetic order in bis-tetramethyltetraselenafulvalenium perchlorate (TMTSF)2ClO4. Phys. Rev. B 1983, 28, 6600–6602. [Google Scholar] [CrossRef]
- Takahashi, T.; Jérome, D.; Bechgaard, K. An NMR study of the organic superconductor: (TMTSF)2ClO4. J. Phys. 1984, 45, 945–952. [Google Scholar] [CrossRef] [Green Version]
- Montambaux, G.; Héritier, H.; Lederer, P. Spin susceptibility of the two-dimensional electron gas with open Fermi surface under magnetic field. Phys. Rev. Lett. 1985, 55, 2078–2081. [Google Scholar] [CrossRef] [PubMed]
- Yamaji, K. Theory of field-induced spin-density-wave in Bechgaard salts. Synth. Met. 1986, 13, 29–43. [Google Scholar] [CrossRef]
- Maki, K. Thermodynamics of field-induced spin-density-wave states in Bechgaard salts. Phys. Rev. B 1986, 33, 4826–4829. [Google Scholar] [CrossRef]
- Lebed, A.G.; Bak, P. Theory of unusual anisotropy of magnetoresistance in organic superconductors. Phys. Rev. Lett. 1989, 63, 1315–1317. [Google Scholar] [CrossRef]
- Boebinger, G.S.; Montambaux, G.; Kaplan, M.L.; Haddon, R.C.; Chichester, S.V.; Chiang, L.Y. Anomalous magnetoresistance anisotropy in metallic and spin-density-wave phases of the quasi-one-dimensional organic conductor (TMTSF)2ClO4. Phys. Rev. Lett. 1990, 64, 591–594. [Google Scholar] [CrossRef]
- Osada, T.; Kawasumi, A.; Kagoshima, S.; Miura, N.; Saito, G. Commensurability effect of magnetoresistance anisotropy in the quasi-one-dimensional conductor tetramethyltetraselenafulvalenium perchlorate, (TMTSF)2ClO4. Phys. Rev. Lett. 1991, 66, 1525–1528. [Google Scholar] [CrossRef]
- Osada, T.; Kagoshima, S.; Miura, N. Resonance effect in magnetotransport anisotropy of quasi-one- dimensional conductors. Phys. Rev. B 1992, 46, 1812–1815. [Google Scholar] [CrossRef]
- Danner, G.M.; Kang, W.; Chaikin, P.M. Measuring the Fermi surface of quasi-one-dimensional metals. Phys. Rev. Lett. 1994, 72, 3714–3717. [Google Scholar] [CrossRef]
- Lee, I.J.; Naughton, M.J. Effective electrons and angular oscillations in quasi-one-dimensional conductors. Phys. Rev. B 1998, 57, 7423–7426. [Google Scholar] [CrossRef]
- Osada, T. Novel angular effect and electron orbital motion in quasi-one-dimensional conductors. Synth. Met. 1997, 86, 2143–2144. [Google Scholar] [CrossRef]
- Lebed, A.G.; Bagmet, N.N. Nonanalytical magnetoresistance, the third angular effect, and a method to investigate Fermi surfaces in quasi-one-dimensional conductors. Phys. Rev. B 1997, 55, R8654–R8657. [Google Scholar] [CrossRef]
- Yoshino, H.; Murata, K. Origin of the third angular effect of magnetoresistance of quasi-one-dimensional metals. J. Phys. Soc. Jpn. 1999, 68, 3027–3033. [Google Scholar] [CrossRef]
- Bechgaard, K.; Kistenmacher, T.J.; Bloch, A.N.; Cowan, D.O. The crystal and molecular structure of an organic conductor from 4,4′,5,5′-tetramethyl-Δ2,2′-bis-1,3-diselenole and 7,7,8,8,-tetracyano-p-quinodimethane [TMTSF–TCNQ]. Acta Cryst. B 1977, 33, 417–422. [Google Scholar] [CrossRef]
- Kistenmacher, T.J.; Emge, T.J.; Bloch, A.N.; Cowan, D.O. Structure of the red, semiconducting form of 4,4′5,5′-tetramethyl-Δ2,2′-bi-1,3-deselenole-7,7,8,8,-tetracyano-p-quinodimethane, TMTSF–TCNQ. Acta Cryst. B 1982, 38, 1193–1199. [Google Scholar] [CrossRef]
- Andersen, J.R.; Bechgaard, K.; Jacobsen, C.S.; Rindorf, G.; Soling, H.; Thorup, N. The crystal and molecular structure of the organic conductor 2,3,6,7-tetramethyl-1,3,5,8-tetraselenafulvalenium 2,5-dimethyl-7,7,8,8-tetracyano-p-quinodimethanide (TMTSF–DMTCNQ). Acta Cryst. B 1978, 34, 1901–1905. [Google Scholar] [CrossRef]
- Stokes, J.P.; Emge, T.J.; Bryden, W.A.; Chappell, J.S.; Cowan, D.O.; Poehler, T.O.; Bloch, A.N.; Kistenmacher, T.J. (TMTSF)2(2,5-TCNQBr2): Structure and physical properties. Mol. Cryst. Liq. Cryst. 1982, 79, 327–336. [Google Scholar] [CrossRef]
- Konovalikhin, S.V.; D’yachenko, O.A.; Shilov, G.V.; Spitsyna, N.G.; Van, K.V.; Yagubskii, E.B. Crystal structure of a new molecular complex of fullerene with tetramethyltetraselenafulvalene: C60·TMTSF·2CS2. Russ. Chem. B+ 1997, 46, 1415–1420. [Google Scholar] [CrossRef]
- Konovalikhin, S.V.; Dyachenko, O.A.; Shilov, G.V.; Spitsyna, N.G.; Yagubskii, E.B. Crystal and molecular structure of a new complex of [60]fullerene, 2(C60)·2(TMTSF)·(C6H6). Fullerene Sci. Techn. 1998, 6, 563–575. [Google Scholar] [CrossRef]
- Heuer, W.B.; Hoffman, B.M. A novel phase transition in conductive molecular crystals based on metal bis-diselenoalkene complexes: [TMTSF]2[M{Se2C2(CF3)2}2]; M = Ni, Pt; TMTSF = tetremethyltetraselenafulvalene. J. Chem. Soc. Chem. Commun. 1986, 174–175. [Google Scholar] [CrossRef]
- Heuer, W.B.; Squattrito, P.J.; Hoffman, B.M.; Ibers, J.A. Phase transition in the conductive molecular crystals bis(tetramethyltetraselenafulvalene)[bis(trifluoromethyl)ethylene]diselenolatonickel and -platinum, [TMTSF]2[M(tds)2], M = Ni, Pt. Relation to the Copper Analogue. J. Am. Chem. Soc. 1988, 110, 792–803. [Google Scholar] [CrossRef]
- Morgado, J.; Alcácer, L.; Almeida, M. Electrical resistivity and thermoelectric power of (TMTSF)2M(tds)2, M = Pt, Cu and Ni; evidence for the existence of two different phases. Solid State Commun. 1994, 89, 755–759. [Google Scholar] [CrossRef]
- Kazheva, O.N.; Chekhlov, A.N.; Alexandrov, G.G.; Buravov, L.I.; Kravchenko, A.V.; Starodub, V.A.; Sivaev, I.B.; Bregadze, V.I.; Dyachenko, O.A. Synthesis, structure and electrical conductivity of fulvalenium salts of cobalt bis(dicarbollide) anion. J. Organomet. Chem. 2006, 691, 4225–4233. [Google Scholar] [CrossRef]
- Kazheva, O.N.; Alexandrov, G.G.; Kravchenko, A.V.; Starodub, V.A.; Sivaev, I.B.; Lobanova, I.A.; Bregadze, V.I.; Bravov, L.I.; Dyachenko, O.A. New fulvalenium salts of bis(dicarbollide) cobalt and iron: Synthesis, crystal structure and electrical conductivity. J. Organomet. Chem. 2007, 692, 5033–5043. [Google Scholar] [CrossRef]
- Penicaud, A.; Batail, P.; Bechgaard, K.; Sala-Pala, J. A conductive tetramethyltetraselenafulvalenium salt based on a divalent oxalato-bridged bis-(tetrafluorometallate) complex: (TMTSF)3[Ti2F8(C2O4)]. Synth. Met. 1988, 22, 201–207. [Google Scholar] [CrossRef]
- Ouahab, L.; Paiou, J.; Grandjean, D.; Garrigou–Langrange, C.; Delhaes, P.; Bencharif, M. Charge transfer salts obtained with organic donors (TTF, TMTTF, TMTSF, and BEDT-TTF) and tetracyanometallate planar dianions [M(CN), M = PtII, NiII]. J. Chem. Soc. Chem. Commun. 1998, 1038–1041. [Google Scholar] [CrossRef]
- Triki, S.; Ouahab, L.; Paidou, J.; Grandjean, D. The use of polyoxometallates as acceptors in charge transfer salts: Preparation, X-ray crystal structures, and preliminary spectroscopic characterizations of D3M6O19, D = TTF, TMTSF; M = Mo, W. J. Chem. Soc. Chem. Commun. 1998, 1068–1070. [Google Scholar] [CrossRef]
- Turner, S.S.; Le Pevelen, D.; Day, P.; Prout, K. TTF based charge transfer salts of [Cr(NCS)4(phen)]−: Bulk magnetic order and crystal structures of the TTF, TMTTF (tetramethyltetrathiafulvalene) and TMTSF (tetramethyltetraselenafulvalene) derivatives. J. Chem. Soc. Dalton Trans. 2000, 2739–2744. [Google Scholar] [CrossRef]
- Rosokha, S.V.; Stern, C.L.; Ritzert, J.T. π-bonded molecular wires: Self-assembly of mixed-valence cation-radical stacks within the nanochannels formed by inert tetrakis[3,5-bis(trifluoromethyl)phenyl]borate anions. CrystEngComm 2013, 15, 10638. [Google Scholar] [CrossRef]
- Mroweh, N.; Mézière, C.; Allain, M.; Auban–Senzier, P.; Canadell, E.; Avarvari, N. Conservation of structural arrangements and 3:1 stoichiometry in a series of crystalline conductors of TMTTF, TMTSF, BEDT-TTF, and chiral DM-EDT-TTF with the oxo-bis[pentafluorotantalate(V)] dianion. Chem. Sci. 2020, 11, 10078–10091. [Google Scholar] [CrossRef]
- Penicaud, A.; Batail, P.; Perrin, C.; Coulon, C.; Parkin, S.S.P.; Torrance, J.B. First cation radical mixed-valence hybrid salts of the paramagnetic octahedral cluster Nb6Cl183−. Preparation, crystal structures, and conducting and magnetic properties of pentakis[2,3,6,7-tetramethyl-1,4,5,8-tetra-selena- and -thia-fulvalenium) hexachloro(dodeca-μ2-chloro-octahedro-hexaniobate). J. Chem. Soc. Chem. Commun. 1987, 330–332. [Google Scholar] [CrossRef]
- Pénicaud, A.; Batail, P.; Coulon, C.; Canadell, E.; Perrin, C. Novel redox properties of the paramagnetic hexanuclear niobium cluster halide Nb6Cl183− and the preparation, structures, and conducting and magnetic properties of its one-dimensional mixed-valence tetramethyltetra(selena and thia)fulvalenium salts: [TMTSF and TMTTF]5[Nb6Cl18]·(CH2Cl2)0.5. Chem. Mater. 1990, 2, 123–132. [Google Scholar]
- Reinheimer, E.W.; Galán-Mascarós, J.R.; Cómez-García, C.J.; Zhao, H.; Fourmigué, M.; Dunbar, K.R. Radical salts of TTF derivatives with the metal–metal bonded [Re2Cl8]2− anion. J. Mol. Struct. 2008, 890, 81–89. [Google Scholar] [CrossRef]
- Kobayashi, H.; Kobayashi, A.; Sasaki, Y.; Saito, G.; Inokuchi, H. The crystal structure of tetramethyltetraselenafulvalenium perrheneate, (TMTSF)(ReO4)(C2H3Cl3)0.25. Bull. Chem. Soc. Jpn. 1983, 56, 2894–2898. [Google Scholar] [CrossRef]
- Batail, P.; Ouahab, L.; Torrance, J.B.; Pylman, M.L.; Parkin, S.S.P. Cation radical salts with magnetic anions: Preparation and characterization of FeCl4 salts of TMTTF and TMTSF. Solid State Commun. 1985, 55, 597–600. [Google Scholar] [CrossRef]
- Batail, P.; Ouahab, L. Effect of anion volume on dimensionality of radical cation salts. Mol. Cryst. Liq. Cryst. 1985, 125, 205–214. [Google Scholar] [CrossRef]
- Ouahab, L.; Grandjean, D. Structure of tris(3,3′,4,4′-tetramethyl-2,2’5,5′-tetraselenafulvalenium) phosphododecatungstate: (TMTSF)3PW12O40. Acta Cryst. C 1991, 47, 2670–2672. [Google Scholar] [CrossRef]
- Wu, L.Y.; Coppens, P.; Bu, X.H. Crystal structure of tetramethyltetraselenafulvalene nitrate, (C10H12Se4)NO3. Z. Krist.-New Cryst. St. 1997, 212, 101–102. [Google Scholar] [CrossRef]
- Chang, H.-C.; Kitagawa, S.; Kondo, M.; Ishii, T. Structural, spectroscopic and magnetic properties of charge-transfer complex, (TMTSF)[Cr(Cl4SQ)2(Cl4Cat)]·0.5CH2Cl2. Mol. Cryst. Liq. Cryst. 1999, 335, 183–192. [Google Scholar] [CrossRef]
- Chang, H.-C.; Ishii, T.; Kondo, M.; Kitagawa, S. Synthesis, X-ray crystal structures and properties of chromium complexes with semiquinonate and catecholate. J. Chem. Soc. Dalton Trans. 1999, 2467–2476. [Google Scholar] [CrossRef]
- Pauling, L. The Nature of the Chemical Bond, 3rd ed.; Cornell Univ. Press: Ithaca, NY, USA, 1960; p. 260. [Google Scholar]
- Bondi, A. van der Waals volumes and radii. J. Phys. Chem. 1964, 441–448. [Google Scholar] [CrossRef]
- Murata, K.; Yoshino, H.; Yadav, H.O.; Honda, Y.; Shirakawa, N. Pt resistor thermometry and pressure calibration in a clamped pressure cell with the medium, Daphne 7373. Rev. Sci. Instrum. 1997, 68, 2490–2493. [Google Scholar] [CrossRef]
- Yokogawa, K.; Murata, K.; Yoshino, H.; Aoyama, S. Solidification of high-pressure medium Daphne 7373. Jpn. J. Appl. Phys. 2007, 46, 3636–3639. [Google Scholar] [CrossRef]
- Kistenmacher, T.J.; Emge, T.J.; Shu, P.; Cowan, D.O. 4,4′,5,5′-tetramethyl-Δ2,2′-bis-1,3-diselenole, TMTSF. Acta Cryst. B 1979, 35, 772–775. [Google Scholar] [CrossRef]
- Emge, T.J.; Wang, H.H.; Beno, M.A.; Williams, J.M.; Whangbo, M.-H.; Evain, E. Effect of anion ordering on the H-anion interactions and band electronic structure of (TMTSF)2BF4 at 20 K. J. Am. Chem. Soc. 1986, 108, 8215–8223. [Google Scholar] [CrossRef]
- Gallois, B.; Gaultier, J.; Hauw, C.; Lamcharfi, T.D.; Filhol, A. Neutron low-temperature (4 and 20 K) and X-ray high-pressure (6.5×102 and 9.8×102 MPa) structures of the organic superconductor di(2,3,6.7-tetramethyl-1,4,5,8-tetraselenafulvalenium) hexafluorophosphate, (TMTSF)2PF6. Acta Cryst. B 1986, 42, 564–575. [Google Scholar] [CrossRef]
- Thorup, N.; Rindorf, G.; Soling, H.; Bechgaard, K. The structure of di(2,3,6,7-tetramethyl-1,4,5,8- tetraselenafulvalenium) hexafluorophosphate, (TMTSF)2PF6, the first superconducting organic solid. Acta Cryst. B 1981, 37, 1236–1240. [Google Scholar] [CrossRef]
- Beno, M.A.; Daws, C.A.; Wang, H.H.; Williams, J.M. Novel structural features, and their relationship to the electrical properties, of the organic conductor (TMTSF)2NO3 at 298 K and 125 K. Mol. Cryst. Liq. Cryst. 1984, 116, 153–171. [Google Scholar] [CrossRef]
- Kistenmacher, T.J. Anion symmetry and the separability of structural parameters for tetramethyltetraselenafulvalenium salts, (TMTSF)2X. Mol. Cryst. Liq. Cryst. 1986, 136, 361–382. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Emery, V.J.; Bruinsma, R.; Barišić, S. Electron-electron Umklapp scattering in organic superconductors. Phys. Rev. Lett. 1982, 48, 1039–1043. [Google Scholar] [CrossRef]
- Gleiter, R.; Kobayashi, M.; Spanget-Larsen, J.; Ferraris, J.P.; Bloch, A.N.; Bechgaard, K.; Cowan, D.O. Photoelectron and electronic absorption spectra of tetrathiafulvalene and related compounds. Ber. Bursenges. Phys. Chem. 1975, 79, 1218. [Google Scholar] [CrossRef]
- Kakiuchi, T.; Wakabayashi, Y.; Sawa, H.; Takahashi, T.; Nakamura, T. Charge ordering in α-(BEDT-TTF)2I3 by synchrotron X-ray diffraction. J. Phys. Soc. Jpn. 2007, 76, 113702. [Google Scholar] [CrossRef] [Green Version]
- Kitou, S.; Fujii, T.; Kawamoto, T.; Katayama, N.; Maki, S.; Nishibori, E.; Sugimoto, K.; Takata, M.; Nakamura, T.; Sawa, H. Sawa, Successive dimensional transition in (TMTTF)2PF6 revealed by synchrotron X-ray diffraction. Phys. Rev. Lett. 2017, 119, 065701. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | (TMTSF)(I) | (TMTSF)(I) | (TMTSF)(I)·THF |
|---|---|---|---|
| Formula | CHSeI | CHSeI | CHOSeI |
| Formula weight | 5488.21 | 2054.18 | 3387.28 |
| Crystal system | Orthorhombic | Monoclinic | Orthorhombic |
| Space group | (No.63) | (No.14) | (No.62) |
| a (Å) | 30.362(5) | 11.263(9) | 26.900(6) |
| b (Å) | 20.389(3) | 18.174(14) | 9.914(2) |
| c (Å) | 21.346(3) | 18.166(14) | 28.547(7) |
| () | 90 | 90 | 90 |
| () | 90 | 90.355(9) | 90 |
| () | 90 | 90 | 90 |
| Volume (Å) | 13,214.26 | 3718.40 | 7613.10 |
| Z | 4 | 2 | 4 |
| (Å) | 0.71070 | 0.71070 | 0.71070 |
| (mm) | 12.364 | 12.328 | 12.562 |
| Temperature (K) | 296(2) | 296(2) | 296(2) |
| Number of reflections collected | 7935 | 8512 | 9107 |
| Number of reflections with [] | 7216 | 5621 | 7309 |
| Number of parameters refined | 361 | 337 | 368 |
| () () | 25.11 | 25.36 | 25.08 |
| final R indices [] | 0.0997 | 0.1018 | 0.0656 |
| 0.2401 | 0.2559 | 0.1629 | |
| R indices (all data) | 0.1062 | 0.1376 | 0.0812 |
| 0.2431 | 0.2918 | 0.1749 |
 | |||||
| (Average) Valence | Material/Calculation | /Å | /Å | /Å | /Å |
| 0 | neutral | 1.3526 | 1.8924 | 1.9062 | 1.3101 |
| BF (A, 20 K) | 1.3720 | 1.8758 | 1.8914 | 1.3525 | |
| BF (B, 20 K) | 1.3589 | 1.8852 | 1.8993 | 1.3469 | |
| PF (20 K) | 1.3756 | 1.8821 | 1.8896 | 1.3796 | |
| PF | 1.3688 | 1.8755 | 1.8926 | 1.3295 | |
| NO (2:1, 125 K) | 1.4297 | 1.8641 | 1.9016 | 1.3271 | |
| NbF | 1.3501 | 1.8801 | 1.8908 | 1.3481 | |
| Ni (A) | 1.3779 | 1.8653 | 1.8876 | 1.3297 | |
| Ni (A’) | 1.3939 | 1.8673 | 1.8908 | 1.3409 | |
| NO (1:1) | 1.3598 | 1.8646 | 1.8835 | 1.3235 | |
| Cr | 1.4293 | 1.8437 | 1.8750 | 1.3550 | |
| PWO (A) | 1.2951 | 1.8782 | 1.9094 | 1.2232 | |
| PWO (B) | 1.4072 | 1.8513 | 1.8568 | 1.3392 | |
| 8:5 (P) | 1.3786 | 1.8655 | 1.8855 | 1.3287 | |
| 8:5 (Q) | 1.3513 | 1.8747 | 1.9016 | 1.3499 | |
| 8:5 (R) | 1.3241 | 1.9014 | 1.8975 | 1.3747 | |
| 5:2 (A) | 1.3801 | 1.8651 | 1.8815 | 1.3107 | |
| 5:2 (B) | 1.3858 | 1.8629 | 1.8719 | 1.3568 | |
| 5:2 (C) | 1.3244 | 1.8964 | 1.8950 | 1.3359 | |
| 4:4 (K) | 1.3750 | 1.8636 | 1.8866 | 1.3522 | |
| 4:4 (L) | 1.3864 | 1.8623 | 1.8834 | 1.3582 | |
| 4:4 (M) | 1.3539 | 1.8685 | 1.8854 | 1.3404 | |
| 4:4 (N) | 1.3676 | 1.8688 | 1.8967 | 1.3453 | |
| 0 | DFT | 1.3424 | 1.9055 | 1.9235 | 1.3409 |
| DFT | 1.3843 | 1.8710 | 1.8974 | 1.3522 | |
| DFT | 1.4312 | 1.8439 | 1.8748 | 1.3720 | |
| 0 | MP2 | 1.3493 | 1.8869 | 1.9051 | 1.3474 |
| MP2 | 1.3848 | 1.8553 | 1.8709 | 1.3630 | |
| MP2 | 1.4303 | 1.8376 | 1.8470 | 1.3859 | |
| Conformation | /Hartree | /Hartree | /eV | /eV | /eV |
|---|---|---|---|---|---|
| optimized for 0 | |||||
| optimized for | − | ||||
| neutral | |||||
| BF (A, 20 K) | − | ||||
| BF (B, 20 K) | − | ||||
| PF (20 K) | − | ||||
| NO (2:1) | − | ||||
| NbF | − | ||||
| Ni (A) | − | ||||
| Ni (A’) | − | ||||
| NO (1:1) | − | ||||
| Cr | − | ||||
| PWO (A) | |||||
| PWO (B) | − | ||||
| 8:5 (P) | − | ||||
| 8:5 (Q) | − | ||||
| 8:5 (R) | − | ||||
| 5:2 (A) | − | ||||
| 5:2 (B) | − | ||||
| 5:2 (C) | |||||
| 4:4 (K) | − | ||||
| 4:4 (L) | − | ||||
| 4:4 (M) | − | ||||
| 4:4 (N) | − |
| Conformation | /Hartree | /Hartree | /eV | /eV | /eV |
|---|---|---|---|---|---|
| optimized for 0 | |||||
| optimized for | − | ||||
| neutral | |||||
| BF (A, 20 K) | − | ||||
| BF (B, 20 K) | |||||
| PF (20 K) | − | ||||
| NO (2:1) | − | ||||
| NbF | − | ||||
| Ni (A) | − | ||||
| Ni (A’) | − | ||||
| NO (1:1) | − | ||||
| Cr | − | ||||
| PWO (A) | |||||
| PWO (B) | − | ||||
| 8:5 (P) | − | ||||
| 8:5 (Q) | − | ||||
| 8:5 (R) | |||||
| 5:2 (A) | − | ||||
| 5:2 (B) | − | ||||
| 5:2 (C) | |||||
| 4:4 (K) | − | ||||
| 4:4 (L) | − | ||||
| 4:4 (M) | − | ||||
| 4:4 (N) | − |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoshino, H.; Iwasaki, Y.; Tanaka, R.; Tsujimoto, Y.; Matsuoka, C.
Crystal Structures and Electrical Resistivity of Three Exotic TMTSF Salts with I
Yoshino H, Iwasaki Y, Tanaka R, Tsujimoto Y, Matsuoka C.
Crystal Structures and Electrical Resistivity of Three Exotic TMTSF Salts with I
Yoshino, Harukazu, Yoshiki Iwasaki, Rika Tanaka, Yuka Tsujimoto, and Chiaki Matsuoka.
2020. "Crystal Structures and Electrical Resistivity of Three Exotic TMTSF Salts with I