
Pressure Tuned Structural, Electronic and Elastic Properties of U3Si2C2: A First Principles Study

 and
and
Abstract
:1. Introduction
2. Methodology
3. Results and Discussions
3.1. Ground State Structural Parameters
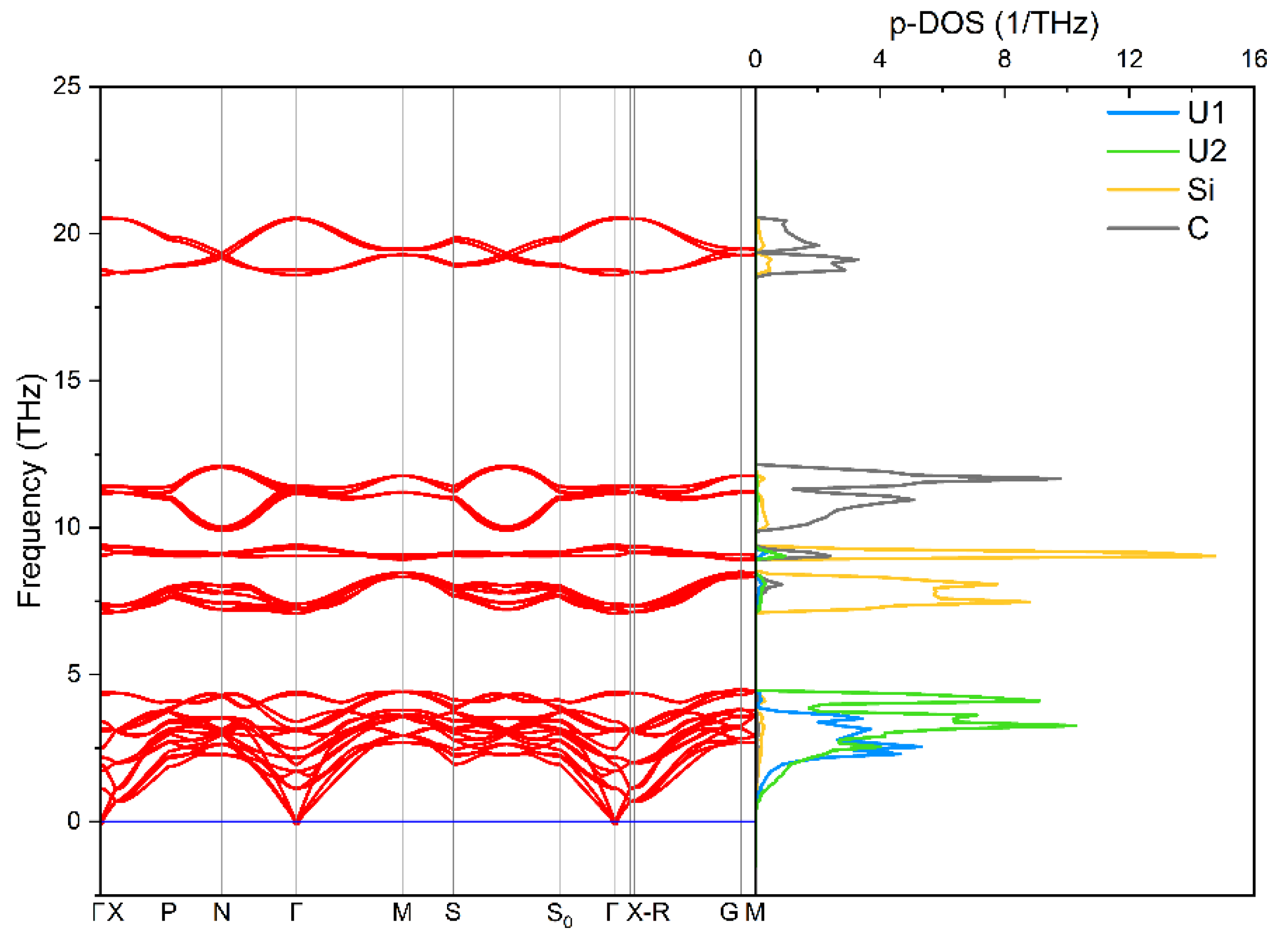
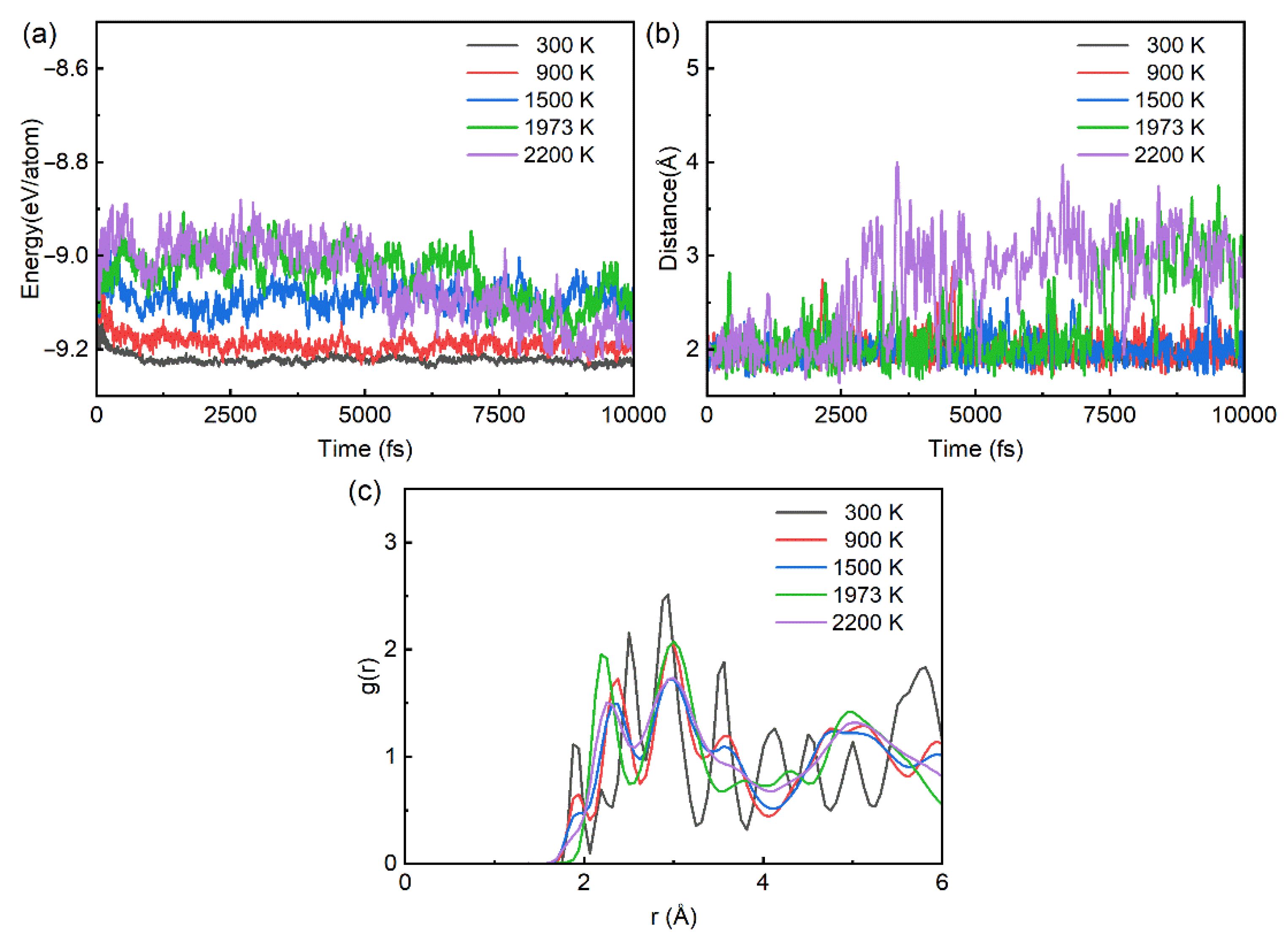
3.2. Stability of Ground State Structure
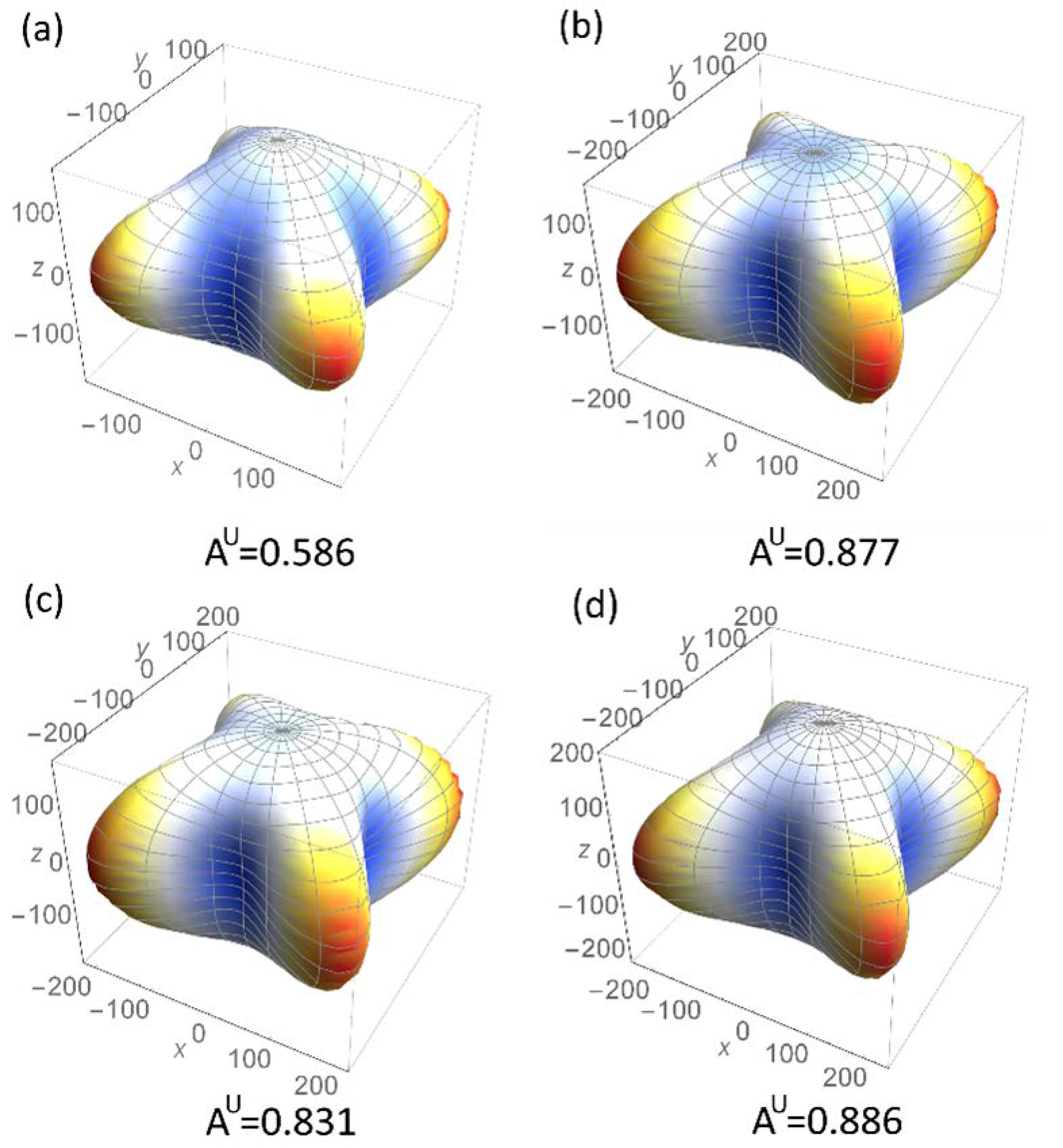
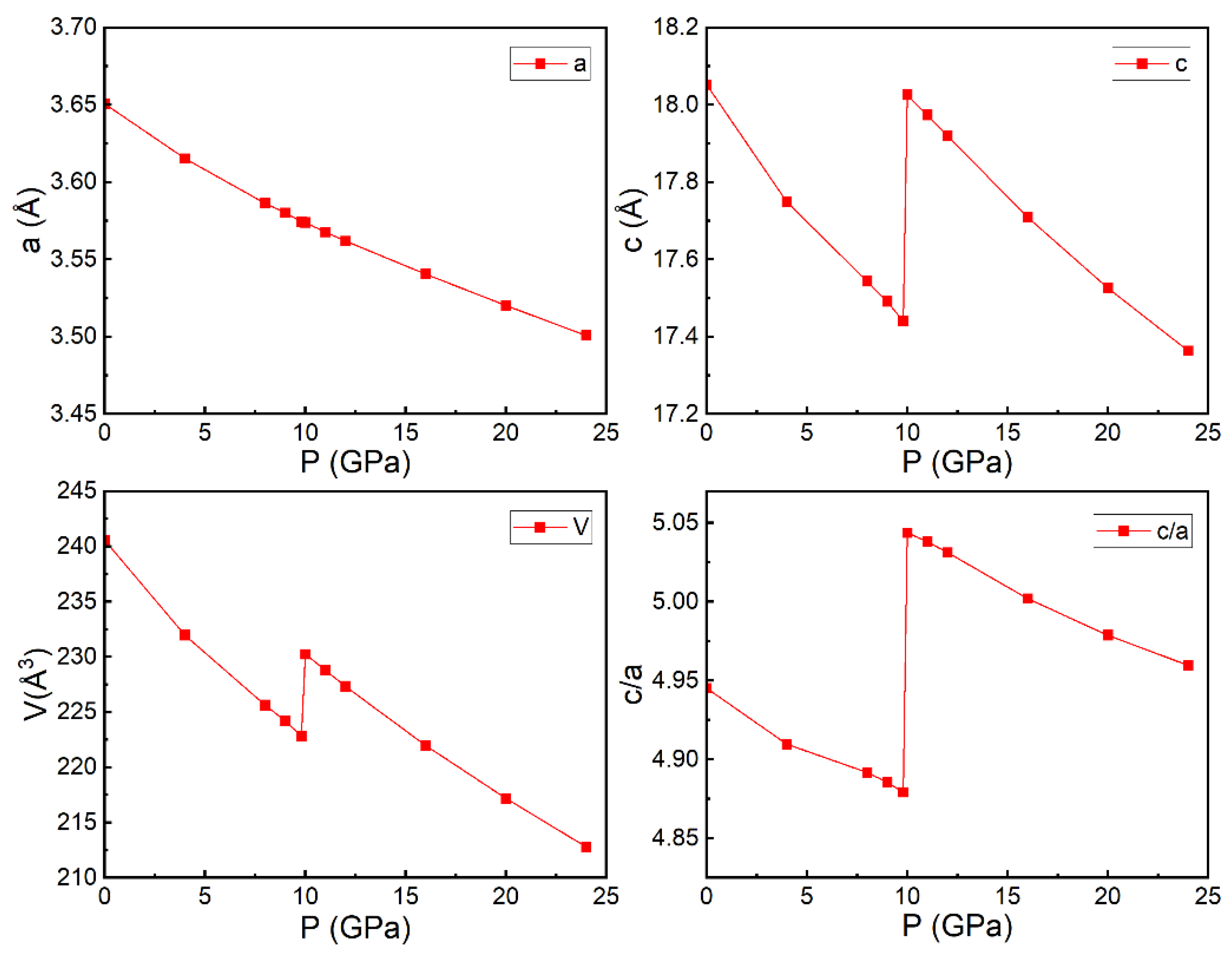
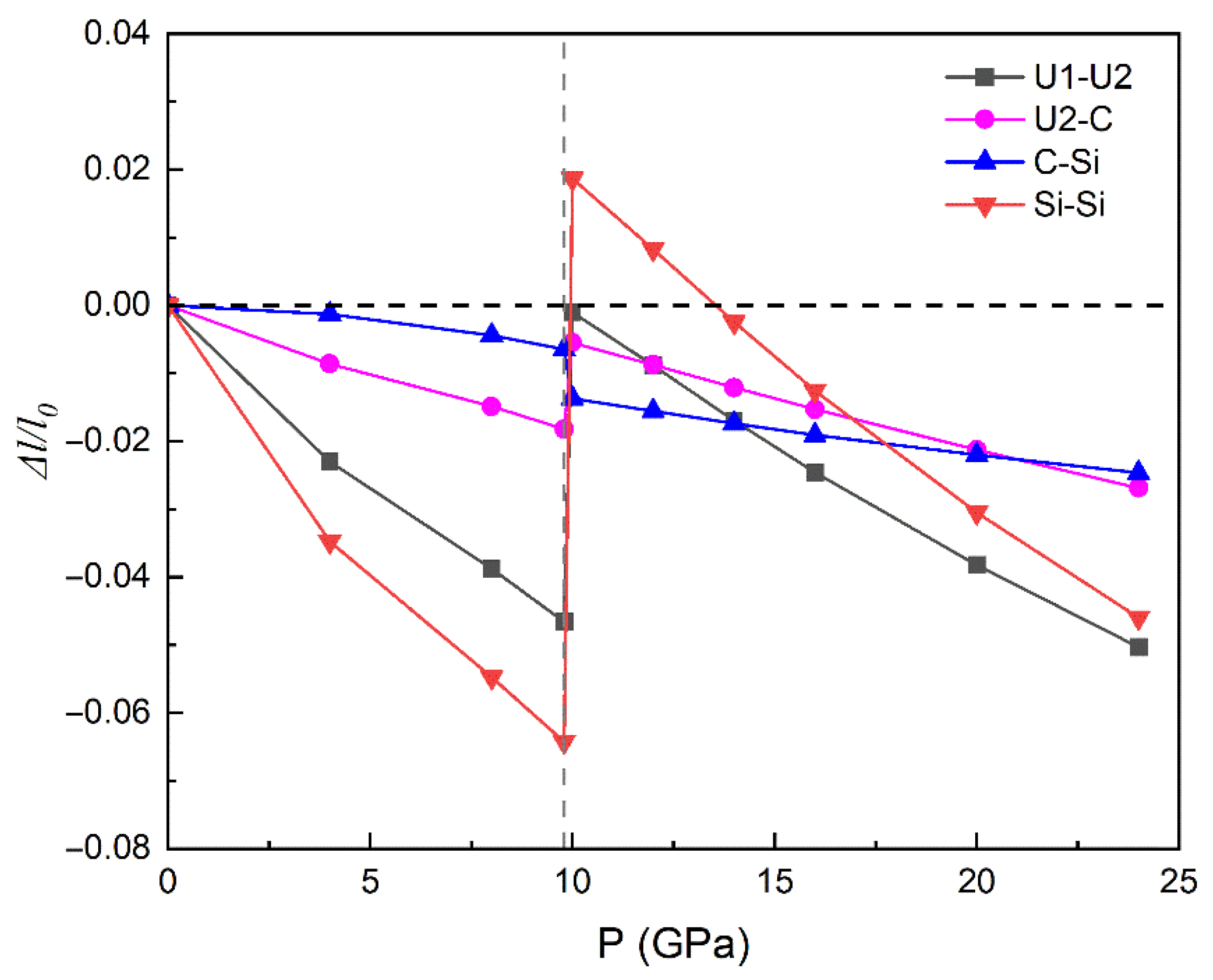
3.3. Properties under High Pressure
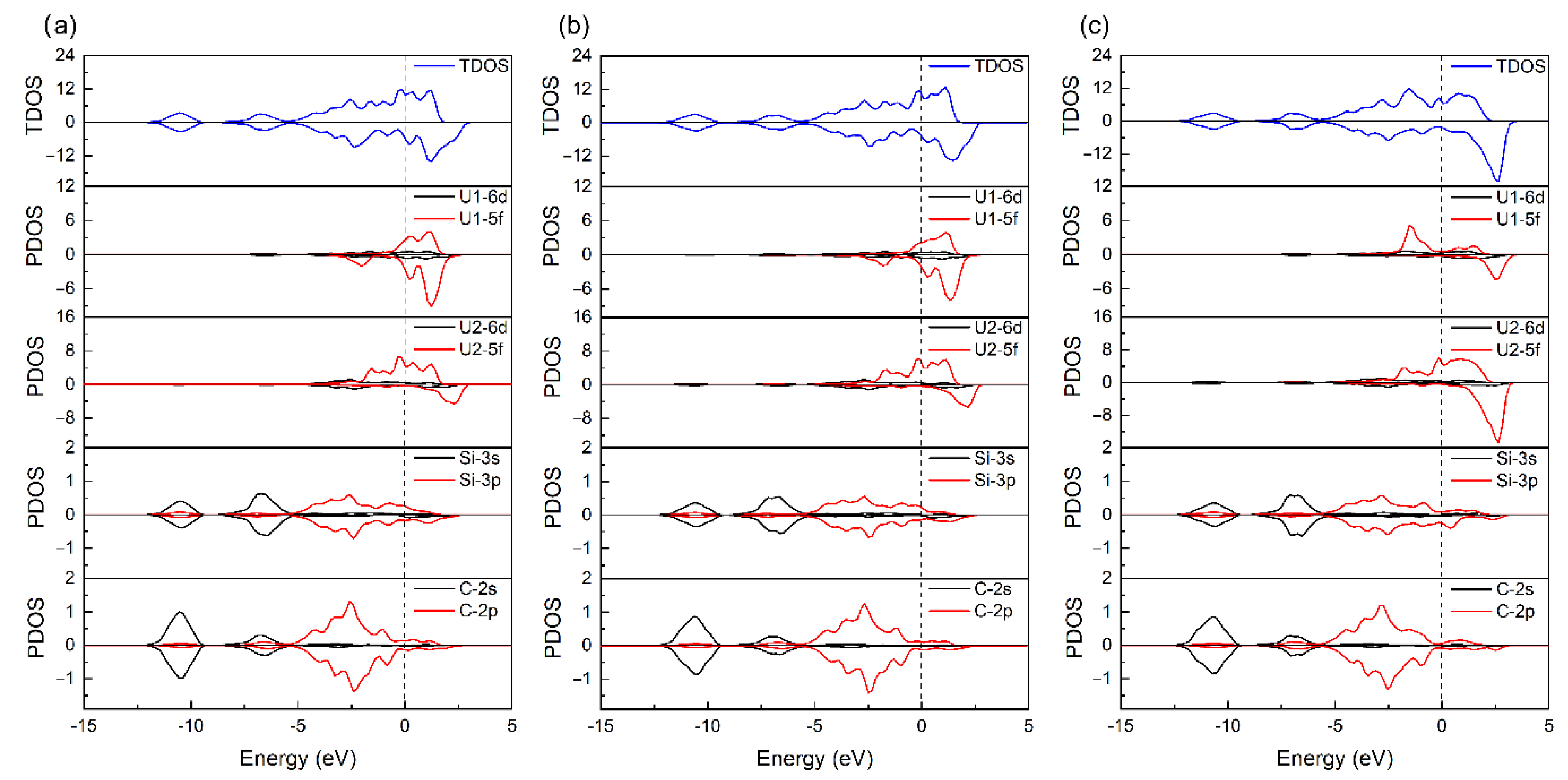
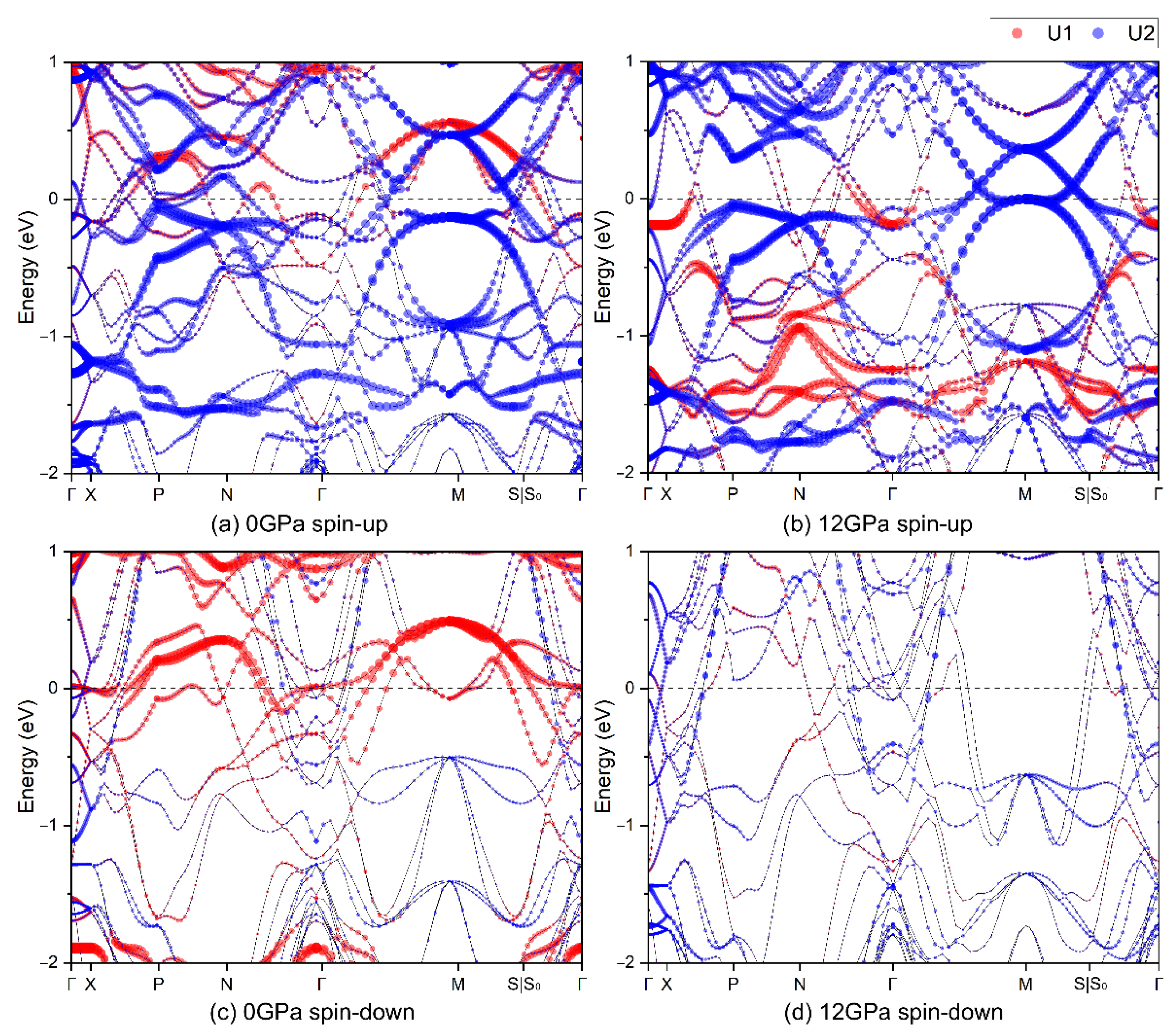
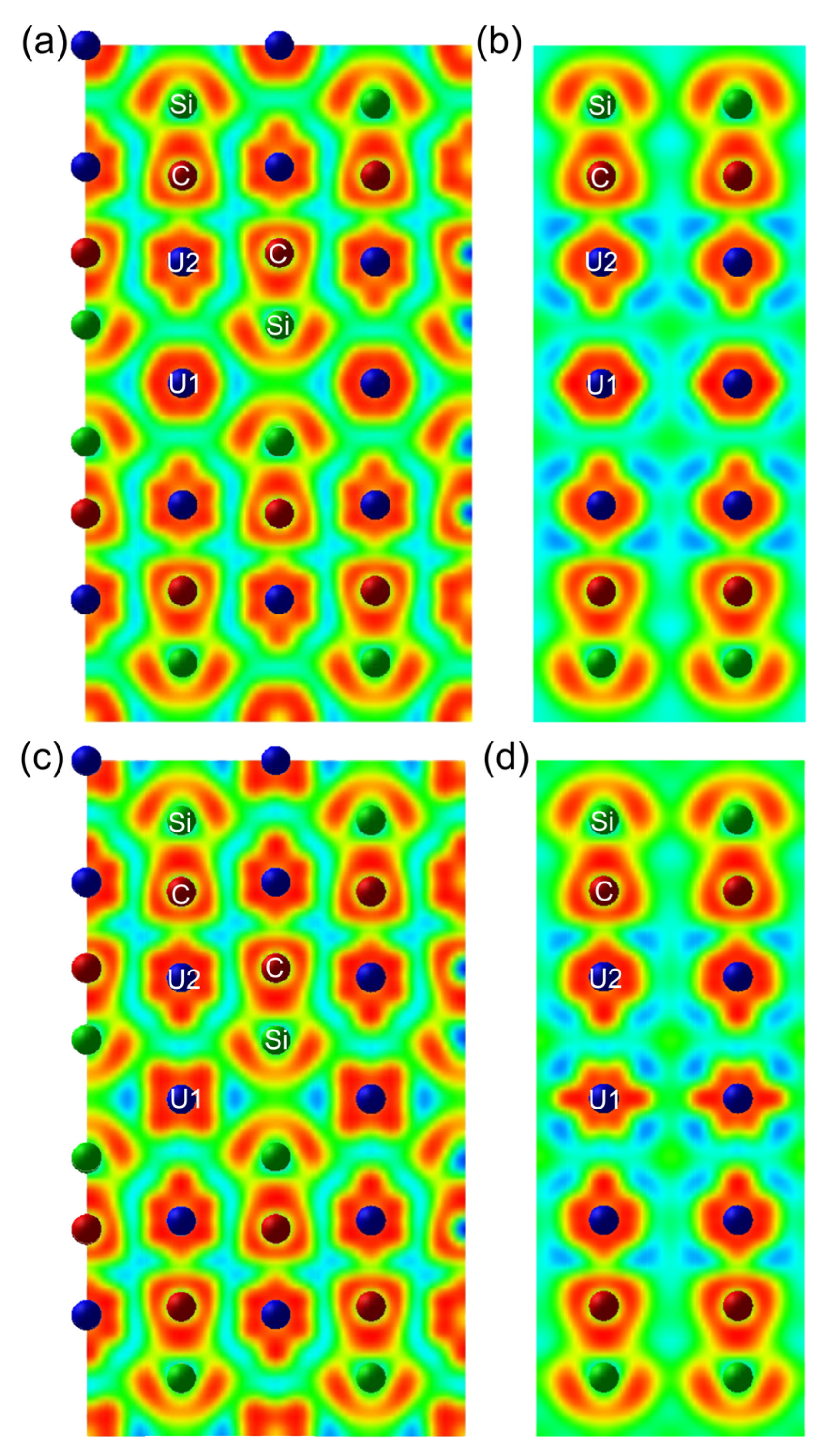
3.3.1. Structural Parameters and Electronic Behaviors
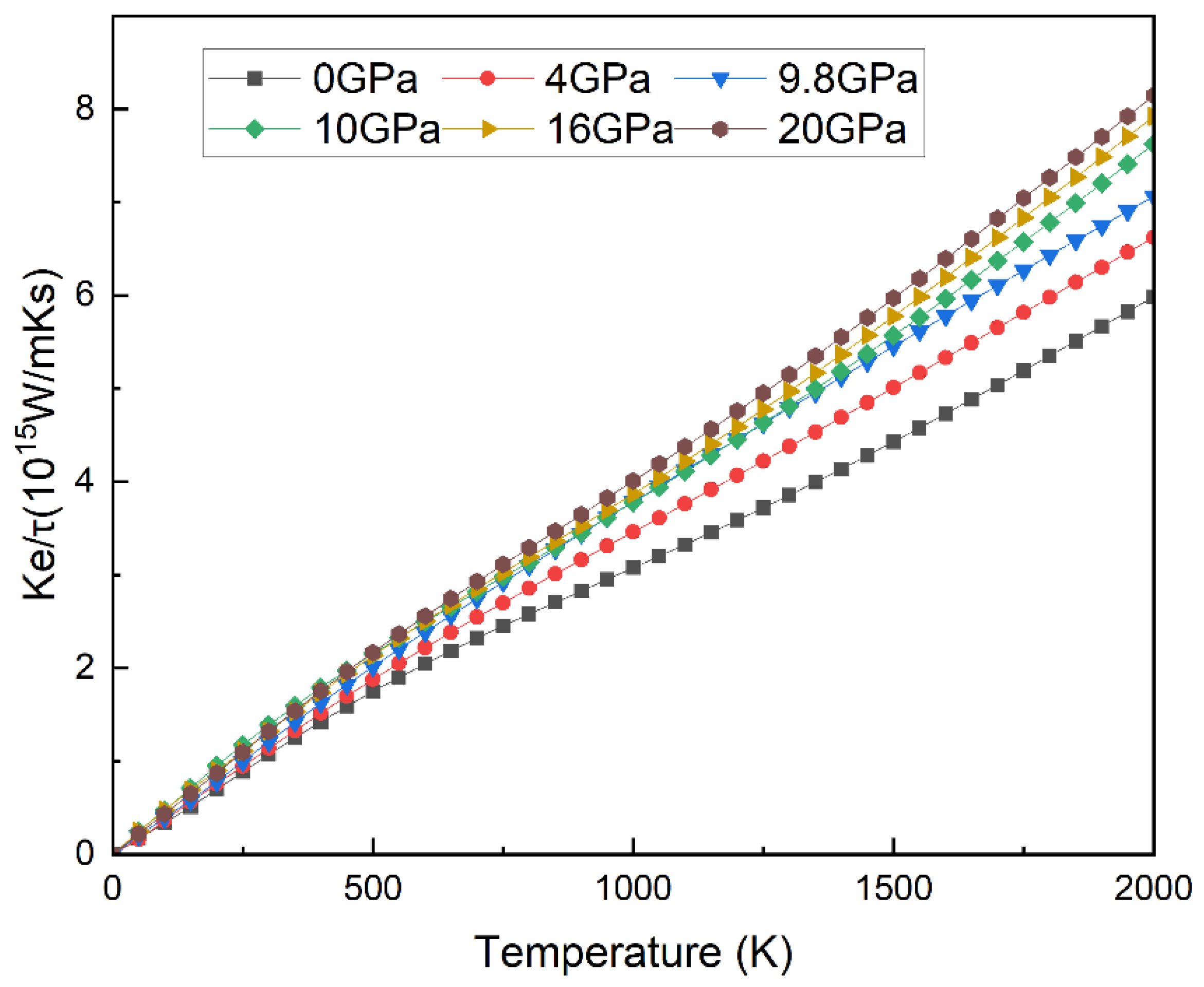
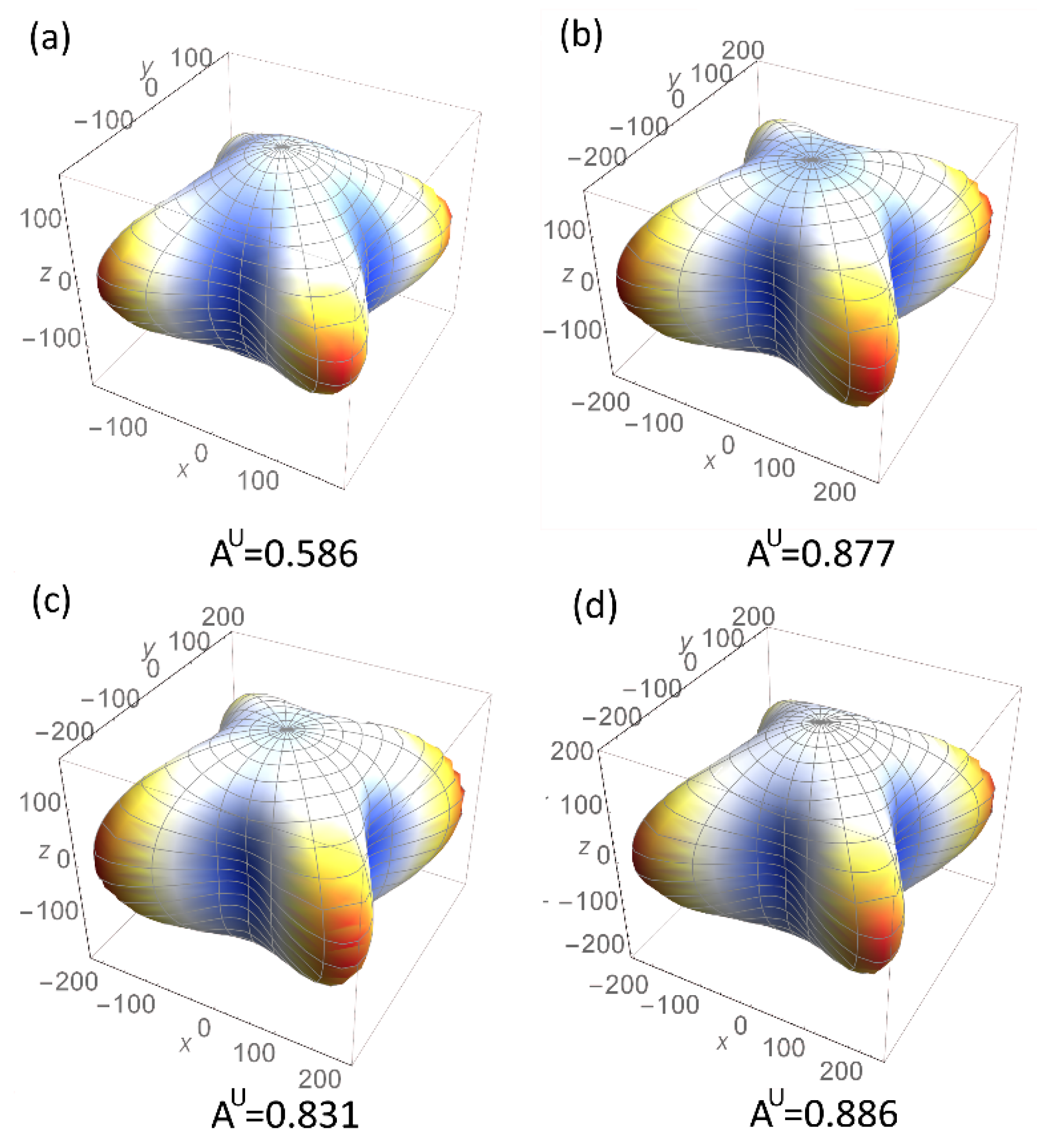
3.3.2. Thermal and Mechanical Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zinkle, S.J.; Terrani, K.A.; Gehin, J.C.; Ott, L.J.; Snead, L.L. Accident tolerant fuels for LWRs: A perspective. J. Nucl. Mater. 2014, 448, 374–379. [Google Scholar] [CrossRef]
- Lopes, D.A.; Kocevski, V.; Wilson, T.L.; Moore, E.E.; Besmann, T.M. Stability of U5Si4 phase in U-Si system: Crystal structure prediction and phonon properties using first-principles calculations. J. Nucl. Mater. 2018, 510, 331–336. [Google Scholar] [CrossRef]
- Ortega, L.H.; Blamer, B.J.; Evans, J.A.; McDeavitt, S.M. Development of an accident-tolerant fuel composite from uranium mononitride (UN) and uranium sesquisilicide (U3Si2) with increased uranium loading. J. Nucl. Mater. 2016, 471, 116–121. [Google Scholar] [CrossRef] [Green Version]
- White, J.T.; Nelson, A.T.; Byler, D.D.; Safarik, D.J.; Dunwoody, J.T.; McClellan, K.J. Thermophysical properties of U3Si5 to 1773K. J. Nucl. Mater. 2015, 456, 442–448. [Google Scholar] [CrossRef] [Green Version]
- White, J.T.; Nelson, A.T.; Byler, D.D.; Valdez, J.A.; McClellan, K.J. Thermophysical properties of U3Si to 1150K. J. Nucl. Mater. 2014, 452, 304–310. [Google Scholar] [CrossRef]
- White, J.T.; Nelson, A.T.; Dunwoody, J.T.; Byler, D.D.; McClellan, K.J. Thermophysical properties of USi to 1673 K. J. Nucl. Mater. 2016, 471, 129–135. [Google Scholar] [CrossRef] [Green Version]
- White, J.T.; Nelson, A.T.; Dunwoody, J.T.; Byler, D.D.; Safarik, D.J.; McClellan, K.J. Thermophysical properties of U3Si2 to 1773K. J. Nucl. Mater. 2015, 464, 275–280. [Google Scholar] [CrossRef] [Green Version]
- White, J.T.; Nelson, A.T.; Dunwoody, J.T.; Safarik, D.J.; McClellan, K.J. Corrigendum to “Thermophysical properties of U3Si2 to 1773 K” [J. Nucl. Mater. 464 (2015) 275–280]. J. Nucl. Mater. 2017, 484, 386–387. [Google Scholar] [CrossRef]
- Klipfel, M.; Di Marcello, V.; Schubert, A.; van de Laar, J.; Van Uffelen, P. Towards a multiscale approach for assessing fission product behaviour in UN. J. Nucl. Mater. 2013, 442, 253–261. [Google Scholar] [CrossRef]
- Frost, B.R.T. The carbides of uranium. J. Nucl. Mater. 1963, 10, 265–300. [Google Scholar] [CrossRef]
- De Coninck, R.; Van Lierde, W.; Gijs, A. Uranium carbide: Thermal diffusivity, thermal conductivity and spectral emissivity at high temperatures. J. Nucl. Mater. 1975, 57, 69–76. [Google Scholar] [CrossRef]
- Corradetti, S.; Manzolaro, M.; Andrighetto, A.; Zanonato, P.; Tusseau-Nenez, S. Thermal conductivity and emissivity measurements of uranium carbides. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2015, 360, 46–53. [Google Scholar] [CrossRef]
- Mankad, V.H.; Jha, P.K. Thermodynamic properties of nuclear material uranium carbide using density functional theory. J. Therm. Anal. Calorim. 2016, 124, 11–20. [Google Scholar] [CrossRef]
- Song, J.; Guo, Y.; Bu, M.; Liu, Z.; Shi, D.; Huang, Q.; Du, S. Theoretical investigations on the U2Mo3Si4 compound from first-principles calculations. Prog. Nucl. Energy 2020, 118, 103121. [Google Scholar] [CrossRef]
- Ugajin, M.; Itoh, A. Experimental investigations on the chemical state of solid fission-product elements in U3Si2. J. Alloys Compd. 1994, 213–214, 369–371. [Google Scholar] [CrossRef]
- Ugajin, M.; Itoh, A.; Okayasu, S.; Kazumata, Y. Uranium molybdenum silicide U3MoSi2 and phase equilibria in the U–Mo–Si system. J. Nucl. Mater. 1998, 257, 145–151. [Google Scholar] [CrossRef]
- Rabin, D.; Shneck, R.Z.; Rafailov, G.; Dahan, I.; Meshi, L.; Brosh, E. Thermodynamic modeling of Al–U–X (X=Si,Zr). J. Nucl. Mater. 2015, 464, 170–184. [Google Scholar] [CrossRef]
- Chen, X.; Qin, Y.; Shi, D.; Guo, Y.; Song, J.; Bu, M.; Zhang, Y.; Huang, Q.; Liu, G.; Chai, Z.; et al. Investigations of the stability and electronic structures of U3Si2-Al: A first-principles study. Chem. Phys. 2021, 543, 111088. [Google Scholar] [CrossRef]
- Chen, X.; Qin, Y.; Shi, D.; Guo, Y.; Bu, M.; Yan, T.; Song, J.; Liu, G.; Zhang, Y.; Du, S. First-principles investigations on the anisotropic elasticity and thermodynamic properties of U3Si2–Al. RSC Adv. 2020, 10, 35049–35056. [Google Scholar] [CrossRef]
- Wu, E.; Qiu, N.; Luo, K.; Chen, X.; Shi, D.; Bu, M.; Du, S.; Chai, Z.; Huang, Q.; Zhang, Y. The studies of electronic structure, mechanical properties and ideal fracture behavior of U3Si1.75Al0.25: First-principle investigations. J. Mater. Res. Technol. 2021, 15, 1356–1369. [Google Scholar] [CrossRef]
- Duan, L.; Gao, R.; Huang, Q.; Jia, J.; Li, B.; Liu, X.; Tang, H.; Wang, Z.; Yang, Z.; Zhong, Y. Preparing Imitated MAX Phase Fault-tolerant Nuclear Fuel Pellet Comprises e.g. Wet Mixing Uranium Dioxide, Silicon-Containing Phase, Carbon Powder, Binder and Sintering Aid Using Ethanol as Wet Mixed Solvent, Mixing, and Processing. China Patent CN106927832, 13 April 2017. [Google Scholar]
- Duan, L.; Gao, R.; Huang, Q.; Jia, J.; Li, B.; Liu, X.; Tang, H.; Wang, Z.; Yang, Z.; Zhong, Y.; et al. Uranium-Silicon-Carbon Ternary Compound Fuel Pellet for Preparing Nuclear Fuel, Comprises Uranium-Silicon-Carbon Ternary Compound with Tetragonal Crystal Structure. China Patent CN107082430, 27 May 2017. [Google Scholar]
- Garcia, P.; Carlot, G.; Dorado, B.; Maillard, S.; Sabathier, C.; Martin, G.; Oh, J.Y.; Welland, M.J. Mechanisms of Microstructural Changes of Fuel under Irradiation; Nuclear Energy Agency of the OECD (NEA): Paris, France, 2015; pp. 24–60. [Google Scholar]
- Wang, T.; Li, R.; Quan, Z.; Loc, W.S.; Bassett, W.A.; Xu, H.; Cao, Y.C.; Fang, J.; Wang, Z. Pressure Processing of Nanocube Assemblies Toward Harvesting of a Metastable PbS Phase. Adv. Mater. 2015, 27, 4544–4549. [Google Scholar] [CrossRef] [PubMed]
- Lü, X.; Wang, Y.; Stoumpos, C.C.; Hu, Q.; Guo, X.; Chen, H.; Yang, L.; Smith, J.S.; Yang, W.; Zhao, Y.; et al. Enhanced Structural Stability and Photo Responsiveness of CH3NH3SnI3 Perovskite via Pressure-Induced Amorphization and Recrystallization. Adv. Mater. 2016, 28, 8663–8668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sears, V.F. Neutron scattering lengths and cross sections. Neutron News 1992, 3, 26–37. [Google Scholar] [CrossRef]
- Smith, G.V.; Smith, R.G.; Thomas, A.G. A Study of the Phase Relationships in the Uranium-Silicon-Carbon System. In Proceedings of the Symposium on Carbides in Nuclear Energy, Harwell, England, 5–7 November 1963; pp. 261–265. [Google Scholar]
- Blum, P.L.; Guinet, P.; Silvestr, G. Structure d’une phase nouvelle, U3C3Si2, dans le système uranium-carbone-silicium. Comptes Rendus Hebd. Des Seances De L Acad. Des Sci. 1965, 260, 1911–1913. [Google Scholar]
- Blum, P.L.; Silvestre, G. La structure cristalline du composé U20C3Si16. Comptes Rendus Hebd. Des Seances De L Acad. Des Sci. Ser. B 1966, 263, 709–711. [Google Scholar]
- Pöttgen, R.; Kaczorowski, D.; Jeitschko, W. Crystal structure, magnetic susceptibility and electrical conductivity of the uranium silicide carbides U3Si2C2 and U20Si16C3. J. Mater. Chem. 1993, 3, 253–258. [Google Scholar] [CrossRef]
- Matar, S.F.; Pöttgen, R. First principles investigations of the electronic structure and chemical bonding of U3Si2C2—A uranium silicide–carbide with the rare [SiC] unit. Chem. Phys. Lett. 2012, 550, 88–93. [Google Scholar] [CrossRef]
- Sahoo, B.D.; Joshi, K.D.; Kaushik, T.C. Structural stability of uranium carbide (UC) under high pressure: Ab-initio study. Comput. Condens. Matter 2019, 21, e00431. [Google Scholar] [CrossRef]
- Staun Olsen, J.; Gerward, L.; Benedict, U.; Itié, J.P.; Richter, K. High-pressure structural studies of UC by v-ray diffraction and synchrotron radiation. J. Less Common Met. 1986, 121, 445–453. [Google Scholar] [CrossRef]
- Sahoo, B.D.; Mukherjee, D.; Joshi, K.D.; Kaushik, T.C. High pressure behaviour of uranium dicarbide (UC2): Ab-initio study. J. Appl. Phys. 2016, 120, 085902. [Google Scholar] [CrossRef]
- Guo, X.; Lü, X.; White, J.T.; Benmore, C.J.; Nelson, A.T.; Roback, R.C.; Xu, H. Bulk moduli and high pressure crystal structure of U3Si2. J. Nucl. Mater. 2019, 523, 135–142. [Google Scholar] [CrossRef]
- Baker, J.L.; Wang, G.; Ulrich, T.; White, J.T.; Batista, E.R.; Yang, P.; Roback, R.C.; Park, C.; Xu, H. High-pressure structural behavior and elastic properties of U3Si5: A combined synchrotron XRD and DFT study. J. Nucl. Mater. 2020, 540, 152373. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505–1509. [Google Scholar] [CrossRef]
- Wang, T.; Qiu, N.; Wen, X.; Tian, Y.; He, J.; Luo, K.; Zha, X.; Zhou, Y.; Huang, Q.; Lang, J.; et al. First-principles investigations on the electronic structures of U3Si2. J. Nucl. Mater. 2016, 469, 194–199. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Press, W.H.; Vetterling, W.T.; Teukolsky, S.A.; Flannery, B.P. Numerical Recipes; Cambridge University Press: Cambridge, UK, 1986; Volume 818. [Google Scholar]
- Giannozzi, P.; de Gironcoli, S.; Pavone, P.; Baroni, S. Ab initio calculation of phonon dispersions in semiconductors. Phys. Rev. B 1991, 43, 7231–7242. [Google Scholar] [CrossRef]
- Gonze, X.; Lee, C. Dynamical matrices, Born effective charges, dielectric permittivity tensors, and interatomic force constants from density-functional perturbation theory. Phys. Rev. B 1997, 55, 10355–10368. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Ranganathan, S.I.; Ostoja-Starzewski, M. Universal Elastic Anisotropy Index. Phys. Rev. Lett. 2008, 101, 055504. [Google Scholar] [CrossRef] [Green Version]
- Hill, R. The Elastic Behaviour of a Crystalline Aggregate. Proc. Phys. Society. Sect. A 1952, 65, 349–354. [Google Scholar] [CrossRef]
- Reuss, A. Berechnung der Fließgrenze von Mischkristallen auf Grund der Plastizitätsbedingung für Einkristalle. Z. angew. Math. Mech. 1929, 9, 49–58. [Google Scholar] [CrossRef]
- Voigt, W. A determination of the elastic constants for beta-quartz lehrbuch de kristallphysik. Terubner Leipz. 1928, 40, 2856–2860. [Google Scholar]
- Dorado, B.; Freyss, M.; Martin, G. GGA+U study of the incorporation of iodine in uranium dioxide. Eur. Phys. J. B 2009, 69, 203–209. [Google Scholar] [CrossRef]
- Obodo, K.O.; Chetty, N. GGA+U studies of the early actinide mononitrides and dinitrides. J. Nucl. Mater. 2013, 442, 235–244. [Google Scholar] [CrossRef] [Green Version]
- Mouhat, F.; Coudert, F.-X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B 2014, 90, 224104. [Google Scholar] [CrossRef] [Green Version]
- Guinet, P.; Vaugoyeau, H.; Laugier, J.; Blum, P.L. Etude d’un equilibre a 4 phases solides dans le systeme ternaire U-C-Si. J. Nucl. Mater. 1967, 21, 21–31. [Google Scholar] [CrossRef]
- Rogl, P.; Nol, H. The C-Si-U system (carbon-silicon-uranium). J. Phase Equilibria 1995, 16, 66–72. [Google Scholar] [CrossRef]
- Guéneau, C.; Sundman, B.; Dupin, N. Thermodynamic Modelling of the U-Pu-Si-C System; European Commission: Brussels, Belgium.
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Madsen, G.K.H.; Singh, D.J. BoltzTraP. A code for calculating band-structure dependent quantities. Comput. Phys. Commun. 2006, 175, 67–71. [Google Scholar] [CrossRef] [Green Version]
- Haines, J.; Léger, J.M.; Bocquillon, G. Synthesis and Design of Superhard Materials. Annu. Rev. Mater. Res. 2001, 31, 1–23. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| U3Si2C2 | a (Å) | c (Å) | V (Å3) | |
|---|---|---|---|---|
| Exp. [30] | 3.5735 | 18.882 | 241.12 | |
| GGA + U (U = 4 eV) [31] | 3.672 | 17.60 | 237.5 | |
| PAW-GGA | 3.645 | 16.790 | 222.24 | |
| GGA + U | U = 1 eV | 3.634 | 17.372 | 229.43 |
| U = 2 eV | 3.628 | 17.664 | 232.55 | |
| U = 3 eV | 3.639 | 17.900 | 237.04 | |
| U = 3.5 eV | 3.651 | 18.052 | 240.58 | |
| U = 4 eV | 3.705 | 17.903 | 245.75 | |
| Compound | C11 | C12 | C13 | C33 | C44 | C66 |
|---|---|---|---|---|---|---|
| U3Si2C2 | 212.268 | 130.170 | 61.994 | 188.622 | 63.394 | 108.233 |
| Pressure (GPa) | E (GPa) | B (GPa) | G (GPa) | ν |
|---|---|---|---|---|
| 0 | 170.895 | 122.622 | 67.402 | 0.268 |
| 8 | 208.740 | 157.157 | 81.626 | 0.279 |
| 12 | 197.671 | 141.395 | 78.008 | 0.267 |
| 20 | 210.763 | 162.239 | 82.106 | 0.283 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bu, M.; Guo, Y.; Shi, D.; Liu, Z.; Song, J.; Li, Y.; Wu, E.; Chen, X.; Qin, Y.; Yang, Y.; et al. Pressure Tuned Structural, Electronic and Elastic Properties of U3Si2C2: A First Principles Study. Crystals 2021, 11, 1420. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11111420
Bu M, Guo Y, Shi D, Liu Z, Song J, Li Y, Wu E, Chen X, Qin Y, Yang Y, et al. Pressure Tuned Structural, Electronic and Elastic Properties of U3Si2C2: A First Principles Study. Crystals. 2021; 11(11):1420. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11111420
Chicago/Turabian StyleBu, Moran, Yaolin Guo, Diwei Shi, Zhen Liu, Jiexi Song, Yifan Li, Erxiao Wu, Xinyu Chen, Yanqing Qin, Yang Yang, and et al. 2021. "Pressure Tuned Structural, Electronic and Elastic Properties of U3Si2C2: A First Principles Study" Crystals 11, no. 11: 1420. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst11111420