
The Influence of Oxygen Activity on Phase Composition, Crystal Structure, and Electrical Conductivity of CaV1–xMoxO3±δ


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
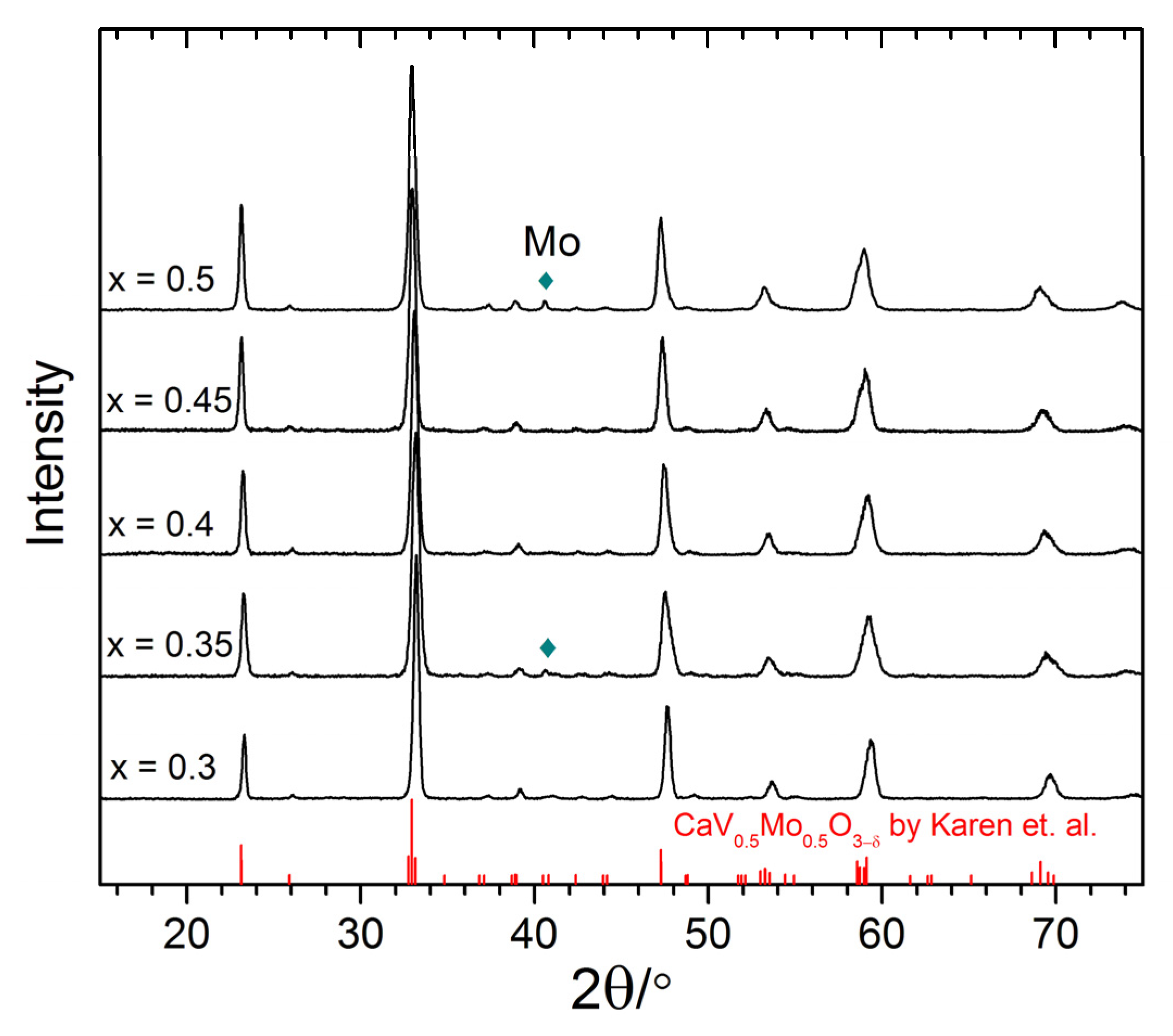
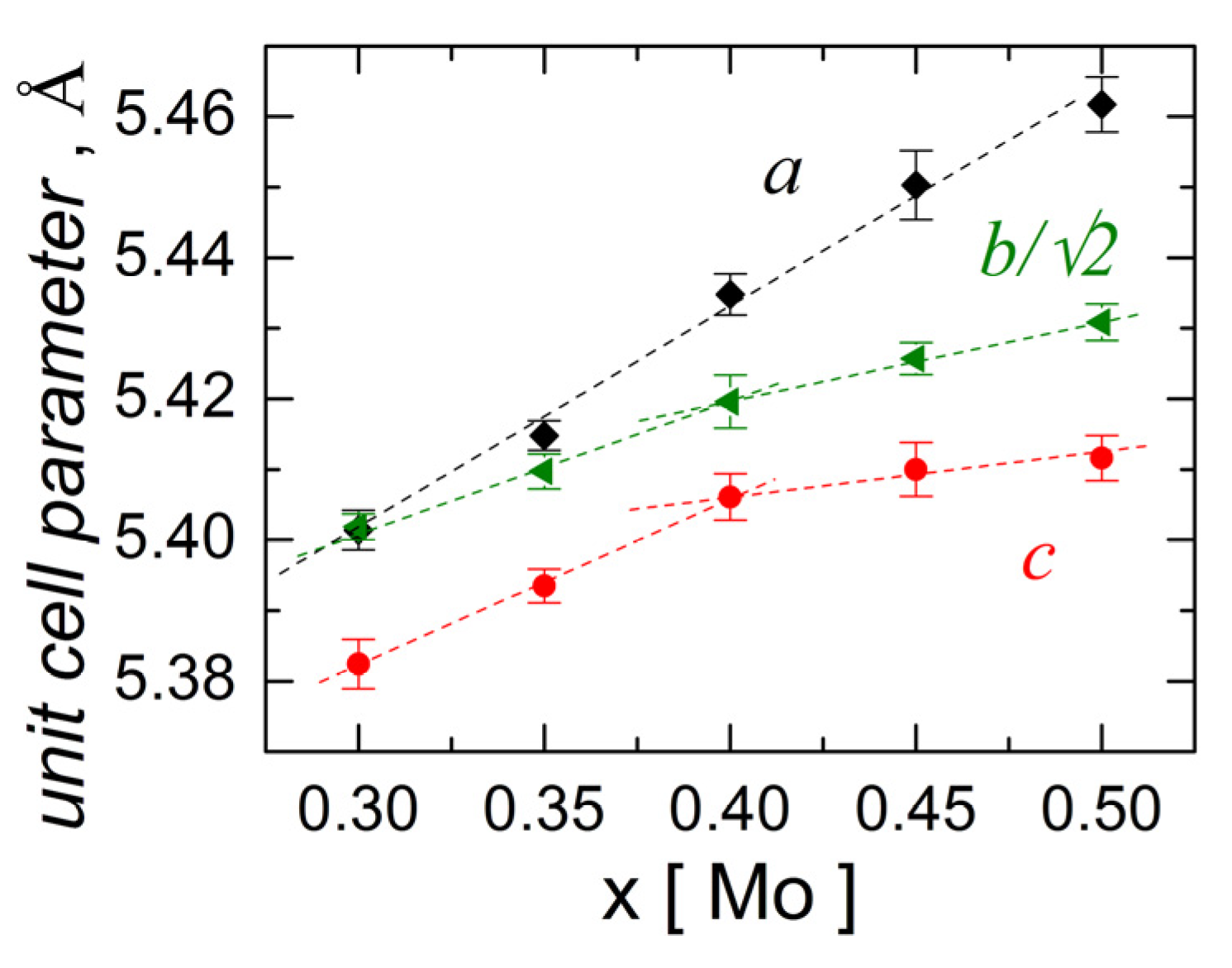
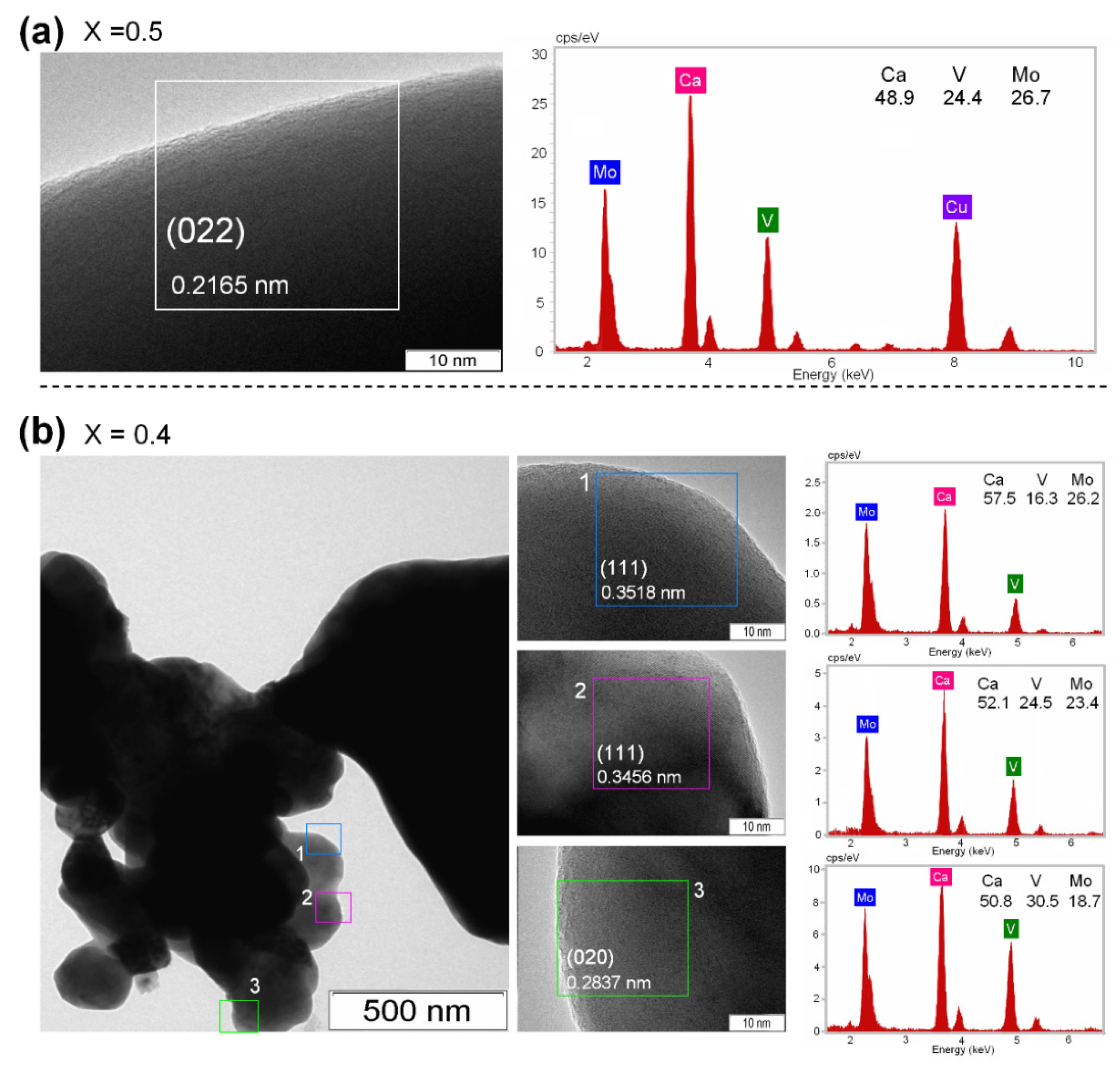
3.1. Materials Characterization
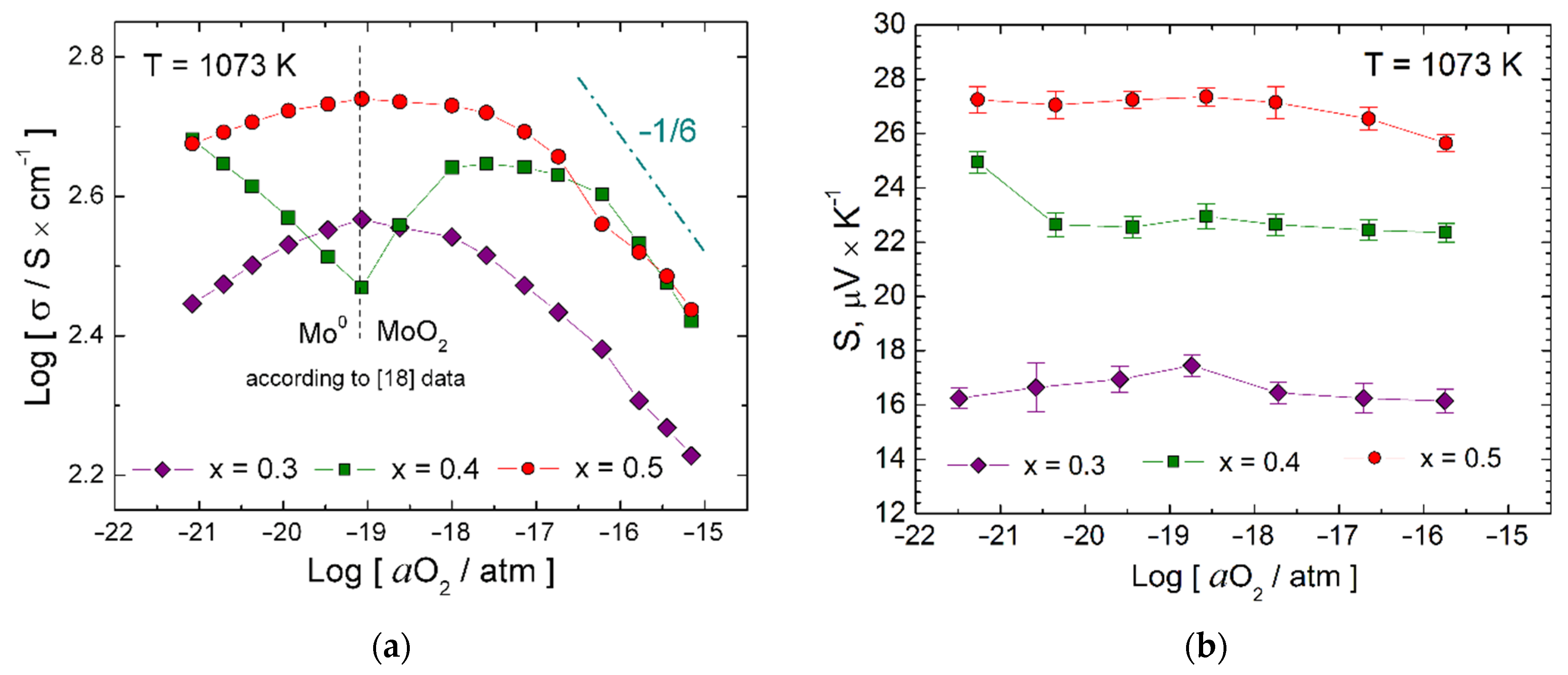
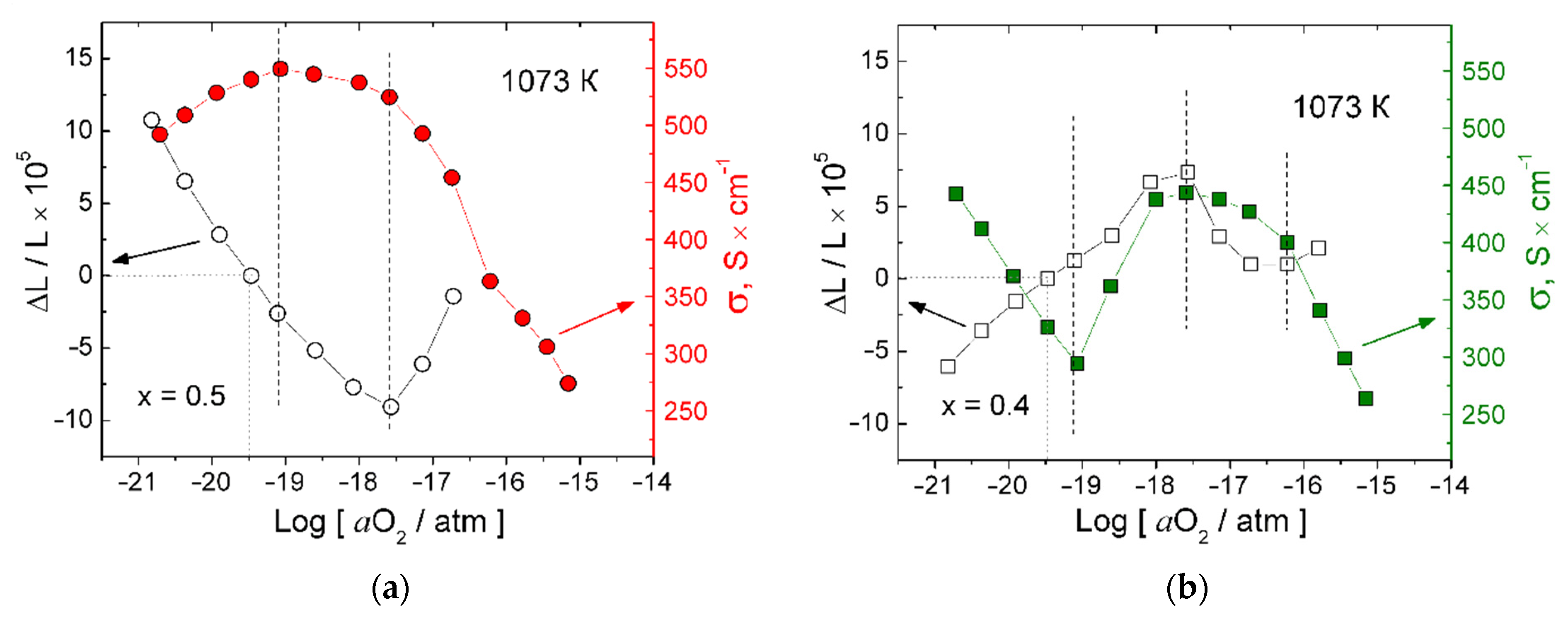
3.2. Impact of aO2 on Properties of the Materials
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hayashi, S.; Aoki, R.; Nakamura, T. Metallic conductivity in perovskite-type compounds AMoO3 (A = Ba, Sr, Ca) down to 2.5 K. Mater. Res. Bull. 1979, 14, 409–413. [Google Scholar] [CrossRef]
- Inoue, I.H.; Goto, O.; Makino, H.; Hussey, N.E.; Ishikawa, M. Bandwidth control in a perovskite-type 3d1-correlated metal Ca1-xSrxVO3. I. Evolution of the electronic properties and effective mass. Phys. Rev. B 1998, 58, 4372–4383. [Google Scholar] [CrossRef] [Green Version]
- Falcón, H.; Alonso, J.A.; Casais, M.T.; Martínes-Lope, M.J.; Sánches-Benítes, J. Neutron diffraction study and magnetotransport properties of stoichiometric CaVO3 perovskite with positive magnetoresistance. J. Solid State Chem. 2004, 177, 3099–3104. [Google Scholar] [CrossRef]
- de la Calle, C.; Alonso, J.A.; García-Hernandez, M.; Pomjakushin, V. Neutron diffraction study and magnetotransport properties of stoichiometric CaMoO3 perovskite prepared by a soft-chemistry route. J. Solid State Chem. 2006, 179, 1636–1641. [Google Scholar] [CrossRef]
- Makino, H.; Inoue, I.H.; Rozenberg, M.J.; Aiura, Y.; Hase, I.; Onari, S. Band-width control in a perovskite-type 3d1 correlated metal Ca1-xSrxVO3. II. Optical spectroscopy investigation. Phys. Rev. B 1998, 58, 4384–4393. [Google Scholar] [CrossRef] [Green Version]
- Mossanek, R.J.O.; Abbate, M.; Yoshida, T.; Fujimori, A.; Yoshida, Y.; Shirakawa, N.; Eisaki, H.; Kohno, S.; Vicentin, F.C. Evolution of the spectral weight in the Mott-Hubbard series SrVO3-CaVO3-LaVO3-YVO3. Phys. Rev. B 2008, 78, 54–61. [Google Scholar] [CrossRef]
- Karen, P.; Moodenbaugh, A.R.; Goldberger, J.; Santhosh, P.N.; Woodward, P.M. Electronic, magnetic and structural properties of A2VMoO6 perovskites (A = Ca, Sr). J. Solid State Chem. 2006, 179, 2120–2125. [Google Scholar] [CrossRef]
- Nekrasov, I.A.; Keller, G.; Kondakov, D.E.; Kozhevnikov, A.V.; Pruschke, T.; Held, K.; Vollhardt, D.; Anisimov, V.I. Comparative study of correlation effects in CaVO3 and SrVO3. Phys. Rev. B 2005, 72, 155106. [Google Scholar] [CrossRef] [Green Version]
- Brahlek, M.; Zhang, L.; Lapano, J.; Zhang, H.-T.; Engel-Herbert, R.; Shukla, N.; Datta, S.; Paik, H.; Schlom, D.G. Opportunities in vanadium-based strongly correlated electron systems. MRS Commun. 2017, 7, 27–52. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhou, Y.; Guo, L.; Zhao, W.; Barnes, A.; Zhang, H.-T.; Eaton, C.; Zheng, Y.; Brahlek, M.; Haneef, H.F.; et al. Correlated metals as transparent conductors. Nat. Mater. 2016, 15, 204–210. [Google Scholar] [CrossRef]
- Gu, M.; Laverock, J.; Chen, B.; Smith, K.E.; Wolf, S.A.; Lu, J. Metal-insulator transition induced in CaVO3 thin films. J. Appl. Phys. 2013, 113, 133704. [Google Scholar] [CrossRef] [Green Version]
- Beck, S.; Sclauzero, G.; Chopra, U.; Ederer, C. Metal-insulator transition in CaVO3 thin films: Interplay between epitaxial strain, dimensional confinement, and surface affects. Phys. Rev. B 2018, 97, 075107. [Google Scholar] [CrossRef] [Green Version]
- McNally, D.E.; Lu, X.; Pelliciari, J.; Beck, S.; Dantz, M.; Naamneh, M.; Shang, T.; Medarde, M.; Schneider, C.W.; Strocov, V.N.; et al. Electronic localization in CaVO3 films via bandwidth control. Npj Quant. Mater. 2019, 4, 6. [Google Scholar] [CrossRef]
- Stoner, J.L.; Murgatroyd, P.A.E.; O’Sullivan, M.; Dyer, M.S.; Manning, T.D.; Claridge, J.B.; Rosseinsky, M.J.; Alaria, J. Chemical control of correlated metals as transparent conductors. Adv. Funct. Mater. 2019, 29, 1808609. [Google Scholar] [CrossRef]
- Yaremchenko, A.A.; Brinkmann, B.; Janssen, R.; Frade, J.R. Electrical conductivity, thermal expansion and stability of Y- and Al-substituted SrVO3 as prospective SOFC anode material. Solid State Ion. 2013, 247–248, 86–93. [Google Scholar] [CrossRef]
- Macías, J.; Yaremchenko, A.A.; Frade, J.R. Redox transitions in strontium vanadates: Electrical conductivity and dimentional changes. J. Alloys Compd. 2014, 601, 186–194. [Google Scholar] [CrossRef]
- Macías, J.; Yaremchenko, A.A.; Frade, J.R. Enhanced stability of perovskite-like SrVO3-based anode materials by donor-type substitution. J. Mater. Chem. 2016, A4, 10186–10194. [Google Scholar] [CrossRef]
- Hui, S.Q.; Petric, A. Conductivity and stability of SrVO3 and mixed perovskites at low oxygen partial pressures. Solid State Ion. 2001, 143, 275–283. [Google Scholar] [CrossRef]
- Macías, J.; Yaremchenko, A.A.; Rodríguez-Castellón, E.; Starykevich, M.; Frade, J.R. Compromising between phase stability and electrical performance: SrVO3-SrTiO3 solid solutions as solid oxide fuel cell anode components. ChemSusChem 2019, 12, 240–251. [Google Scholar] [CrossRef]
- Brixner, L.H. X-ray study and electrical properties of system BaxSr(1-x)MoO3. J. lnorg. Nucl. Chem. 1960, 14, 225–230. [Google Scholar] [CrossRef]
- Ha, Y.; Lee, S. Oxygen-vacancy-endurable conductors with enhanced transparency using correlated 4d2 SrMoO3 thin films. Adv. Funct. Mater. 2020, 30, 2001489. [Google Scholar] [CrossRef]
- Kamata, K.; Nakamura, T.; Sata, T. Valence stability of molybdenum in alkaline earth molybdates. Mater. Res. Bull. 1975, 10, 373–378. [Google Scholar] [CrossRef]
- Im, H.-N.; Jeon, S.-Y.; Choi, M.-B.; Kim, H.-S.; Song, S.-J. Chemical stability and electrochemical properties of CaMoO3-δ for SOFC anode. Ceram. Int. 2012, 38, 153–158. [Google Scholar] [CrossRef]
- Graves, C.R.; Reddy Sudireddy, B.; Mogensen, M.B. Molybdate based ceramic negative-electrode materials for solid oxide cells. ECS Trans. 2010, 28, 173–192. [Google Scholar] [CrossRef] [Green Version]
- Osinkin, D.A. Kinetics of CO oxidation and redox cycling of Sr2Fe1.5Mo0.5O6-δ electrode for symmetrical solid state electrochemical devices. J. Power Sources 2019, 418, 17–23. [Google Scholar] [CrossRef]
- Ueda, Y. Oxygen nonstoichiometry, structures, and physical properties of CaVO3-δ: I. A series of new oxygen-deficient phases. J. Solid State Chem. 1998, 135, 36–42. [Google Scholar] [CrossRef]
- Ueda, Y.; Nakayama, N. Electron diffraction study of oxygen-defect perovskite CaVO3-δ. Solid State Ion. 1998, 108, 303–306. [Google Scholar] [CrossRef]
- Iga, F.; Nishihara, Y. Metal-insulator transition with oxygen content in CaVO3-y. J. Phys. Soc. Jpn. 1992, 61, 1867–1870. [Google Scholar] [CrossRef]
- Inoue, I.H.; Morikawa, K.; Fukuchi, H.; Tsujii, T.; Iga, F.; Nishihara, Y. Metal-to-insulator transitions in CaVO3-y. Phys. B Condens. Matter 1994, 194–196, 1067–1068. [Google Scholar] [CrossRef]
- García-Jaca, J.; Mesa, J.L.; Insausti, M.; Larramendi, J.I.R.; Arriortua, M.I.; Rojo, T. Synthesis, crystal structure, stoichiometry and magnetic properties of (Ca1-xSrx)VO3. Mater. Res. Bull. 1999, 34, 289–301. [Google Scholar] [CrossRef]
- Shikano, M.; Sakaebe, H.; Wakabayashi, N.; Higuchi, S. Structure and electrical properties of Sr0.5Ca0.5VOy. Solid State Ion. 1998, 108, 327–332. [Google Scholar] [CrossRef]
- Takeda, Y.; Kanno, K.; Takada, T.; Yamamoto, O.; Takano, M.; Nakayama, N.; Bando, Y. Phase relation in the oxygen nonstoichiometric system, SrFeOx (2.5 ≤ x ≤ 3.0). J. Solid State Chem. 1986, 63, 237–249. [Google Scholar] [CrossRef]
- Nakayama, N.; Takano, M.; Inamura, S.; Nakanishi, N.; Kosuge, K. Electron microscopy study of the “cubic” perovskite phase SrFe1-xVxO2.5+x (0.05 ≤ x ≤ 0.1). J. Solid State Chem. 1987, 71, 403–417. [Google Scholar] [CrossRef]
- Savinskaya, O.; Nemudry, A.P. Oxygen transport properties of nanostructured SrFe1-xMoxO2.5+3/2x (0 < x < 0.1) perovskites. J. Solid State Electrochem. 2011, 15, 269–275. [Google Scholar] [CrossRef]
- Ancharova, U.V.; Cherepanova, S.V. Nano-domain states of strontium ferrites SrFe1-yMyO2.5+x (M = V, Mo; y ≤ 0.1, x ≤ 0.2). J. Solid State Chem. 2015, 225, 410–416. [Google Scholar] [CrossRef]
- Inoue, I.H.; Bergemann, C.; Hase, I.; Julian, S.R. Fermi surface of 3d1 perovskite CaVO3 near the Mott transition. Phys. Rev. Lell. 2002, 88, 236403. [Google Scholar] [CrossRef] [Green Version]
- Backes, S.; Rödel, T.C.; Fortuna, F.; Frantzeskakis, E.; Le Fèvre, P.; Bertran, F.; Kobayashi, M.; Yukawa, R.; Mitsuhashi, T.; Kitamura, M.; et al. Hubbard band versus oxygen vacancy states in the correlated electron metal SrVO3. Phys. Rev. B 2016, 94, 241110. [Google Scholar] [CrossRef] [Green Version]
- Belyakov, S.A.; Shkerin, S.N.; Kellerman, D.G.; Plekhanov, M.S. The effect of Mo concentration on the electrical properties of CaV1-xMoxO3-δ (x = 0.2 ÷ 0.6) anode material for solid oxide fuel cells. Mater. Res. Bull. 2020, 129, 110904. [Google Scholar] [CrossRef]
- Belyakov, S.A.; Kuznetsov, M.V.; Shkerin, S.N. X-ray photoelectron spectroscopy study of CaV1-xMoxO3-δ. J. Solid State Chem. 2018, 262, 301–308. [Google Scholar] [CrossRef]
- Belyakov, S.A.; Shkerin, S.N.; Selezneva, N.V. Synthesis of an anode material based on mixed calcium vanadatomolybdate and int stability in contact with solid electrolytes. Russ. J. Appl. Chem. 2015, 88, 706–710. [Google Scholar] [CrossRef]
- Burkov, A.T.; Heinrich, A.; Konstantinov, P.P.; Nakama, T.; Yagasaki, K. Experimental set-up for thermopower and resistivity measurements at 100–1300 K. Meas. Sci. Technol. 2001, 12, 264–272. [Google Scholar] [CrossRef]
- Aguadero, A.; de la Calle, C.; Alonso, J.A.; Pérez-Coll, D.; Escudero, M.J.; Daza, L. Structure, thermal stability and electrical properties of Ca(V0.5Mo0.5)O3 as solid oxide fuel cell anode. J. Power Sources 2009, 192, 78–83. [Google Scholar] [CrossRef]
- Liu, G.Y.; Rao, G.H.; Feng, X.M.; Yang, H.F.; Ouyang, Z.W.; Liu, W.F.; Liang, J.K. Structural transition and atomic ordering in the non-stoichiometric double perovskite Sr2FexMo2-xO6. J. Alloys Compd. 2003, 353, 42–47. [Google Scholar] [CrossRef]
- Savinskaya, O.A.; Nemudry, A.P.; Nadeev, A.N.; Tsybulya, S.V. Synthesis and study of the thermal stability of SrFe1-xMxO3-z (M = Mo, W) perovskites. Solid State Ion. 2008, 179, 1076–1079. [Google Scholar] [CrossRef]
- Merkulov, O.V.; Markov, A.A.; Naumovich, E.N.; Shalaeva, E.V.; Leonidov, I.A.; Patrakeev, M.V. Non-uniform electron conduction in weakly ordered SrFe1-xMoxO3-δ. Dalton Trans. 2019, 48, 4530–4537. [Google Scholar] [CrossRef]
- Hiroi, Z.; Hayamizu, H.; Yoshida, T.; Muraoka, Y.; Okamoto, Y.; Yamaura, J.; Ueda, Y. Spinodal decomposition in the TiO2-VO2 system. Chem. Mater. 2013, 25, 2202–2210. [Google Scholar] [CrossRef] [Green Version]
- Demond, A.; Sayers, R.; Tsiamtsouri, M.A.; Romani, S.; Chater, P.A.; Niu, H.; Martí-Gastaldo, C.; Xu, Z.; Deng, Z.; Bréard, Y.; et al. Single sublattice endotaxial phase separation driven by change frustration in a complex oxide. J. Am. Chem. Soc. 2013, 135, 10114–10123. [Google Scholar] [CrossRef]
- Mikhailova, D.; Kuratieva, N.N.; Utsumi, Y.; Tsirlin, A.A.; Abakumov, A.M.; Schmidt, M.; Oswald, S.; Fuess, H.; Ehrenberg, H. Composition-dependent charge transfer and phase separation in the V1-xRexO2 solid solution. Dalton Trans. 2016, 46, 1606–1617. [Google Scholar] [CrossRef]
- Smyth, D.M. The effect of dopants on the properties of metal oxides. Solid State Ion. 2000, 129, 5–12. [Google Scholar] [CrossRef]
- Macías, J.; Yaremchenko, A.A.; Fagg, D.P.; Frade, J.R. Structural and defect chemistry guidelines for Sr(V,Nb)O3-based SOFC anode materials. Phys. Chem. Chem. Phys. 2015, 17, 10749–10758. [Google Scholar] [CrossRef]
- Adler, S.B. Chemical expansivity of electrochemical ceramics. J. Am. Ceram. Soc. 2001, 84, 2117–2119. [Google Scholar] [CrossRef]
- Marrocchelli, D.; Perry, N.H.; Bishop, S.R. Understanding chemical expansion in perovskite-structured oxides. Phys. Chem. Chem. Phys. 2015, 17, 10028–10039. [Google Scholar] [CrossRef] [PubMed]
- Sharp, J.H.; Brindley, G.W.; Narahari Achar, B.N. Numerical data for some commonly used solid state reaction equations. J. Am. Ceram. Soc. 1966, 49, 379–382. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belyakov, S.A.; Gerasimov, E.Y.; Kuzmin, A.V. The Influence of Oxygen Activity on Phase Composition, Crystal Structure, and Electrical Conductivity of CaV1–xMoxO3±δ. Crystals 2022, 12, 419. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12030419
Belyakov SA, Gerasimov EY, Kuzmin AV. The Influence of Oxygen Activity on Phase Composition, Crystal Structure, and Electrical Conductivity of CaV1–xMoxO3±δ. Crystals. 2022; 12(3):419. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12030419
Chicago/Turabian StyleBelyakov, Semyon A., Evgeny Yu. Gerasimov, and Anton V. Kuzmin. 2022. "The Influence of Oxygen Activity on Phase Composition, Crystal Structure, and Electrical Conductivity of CaV1–xMoxO3±δ" Crystals 12, no. 3: 419. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12030419