
Local Crystalline Structure of Doped Semiconductor Oxides Characterized by Perturbed Angular Correlations: Experimental and Theoretical Insights

 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ab Initio Method in Doped Systems
2.2. Details of Ab Initio Calculations in Oxides
3. Results and Discussion
3.1. Cd Doping in TiO, SnO, CeO, ZnO, and SnO
3.2. Doping in MO, M = Sc, Y, In
3.2.1. Coordination Geometry of the Dopant
3.2.2. Coordination Chemistry and Hyperfine Parameters
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bartos, A.; Lieb, K.P.; Uhrmacher, M.; Wiarda, D. Refinement of Atomic Positions in Bixbyite Oxides using Perturbed Angular Correlation Spectroscopy. Acta Cryst. 1993, B49, 165–169. [Google Scholar] [CrossRef]
- Errico, L.A.; Rentería, M.; Petrilli, H.M. Cd in SnO: Probing structural effects on the electronic structure of doped oxide semiconductors through the electric field gradient at the Cd nucleus. Phys. Rev. B 2007, 75, 155209. [Google Scholar] [CrossRef]
- Muñoz, E.L.; Mercurio, M.E.; Cordeiro, M.R.; Pereira, L.F.D.; Carbonari, A.W.; Rentería, M. Dynamic hyperfine interactions in 111In(111Cd)-doped ZnO semiconductor: PAC results supported by ab initio calculations. Phys. B 2012, 407, 3121–3124. [Google Scholar] [CrossRef]
- Richard, D.; Rentería, M.; Carbonari, A.W. Substitutional Ta-doping in Y2O3 semiconductor by sol-gel synthesis: Experimental and theoretical studies. Semicond. Sci. Technol. 2017, 32, 085010. [Google Scholar] [CrossRef]
- Muñoz, E.L.; Richard, D.; Errico, L.A.; Rentería, M. Ab initio study of the EFG tensor at Cd impurities in Sc2O3 semiconductor. Phys. B 2009, 404, 2757–2759. [Google Scholar] [CrossRef]
- Muñoz, E.L.; Richard, D.; Carbonari, A.W.; Errico, L.A.; Rentería, M. PAC study of dynamic hyperfine interactions at 111In-doped Sc2O3 semiconductor and comparison with ab initio calculations. Hyperfine Interact. 2010, 197, 199–205. [Google Scholar] [CrossRef]
- Richard, D.; Muñoz, E.L.; Errico, L.A.; Rentería, M. Electric-field gradients at Ta impurities in Sc2O3 semiconductor. Phys. B 2012, 407, 3134–3136. [Google Scholar] [CrossRef]
- Errico, L.A.; Rentería, M.; Fabricius, G.; Darriba, G.N. FLAPW Study of the EFG Tensor at Cd Impurities in In2O3. Hyperfine Interact. 2004, 158, 63–69. [Google Scholar] [CrossRef]
- Errico, L.A.; Rentería, M.; Bibiloni, A.G.; Freitag, K. Nonionic contributions to the electric-field gradient at 181Ta and 111Cd impurity sites in R2O3 (R = Sc, In, Lu, Yb, Tm, Er, Y, Ho, Dy, Gd, Eu, Sm) bixbyites. arXiv 2005. [Google Scholar] [CrossRef]
- Richard, D.; Darriba, G.N.; Muñoz, E.L.; Errico, L.A.; Eversheim, P.D.; Rentería, M. Experimental and First-Principles Theoretical Study of Structural and Electronic Properties in Tantalum-Doped In2O3 Semiconductor: Finding a Definitive Hyperfine Interaction Assignment. J. Phys. Chem. C 2016, 120, 5640–5650. [Google Scholar] [CrossRef]
- Darriba, G.N.; Muños, E.L.; Carbonari, A.W.; Rentería, M. Experimental TDPAC and Theoretical DFT Study of Structural, Electronic, and Hyperfine Properties in (111In →)111Cd-Doped SnO2 Semiconductor: Ab Initio Modeling of the Electron-Capture-Decay After-Effects Phenomenon. J. Phys. Chem. C 2018, 122, 17423. [Google Scholar] [CrossRef]
- Ferreira, W.L.; Pereira, L.F.D.; Leite Neto, O.F.S.; Maciel, L.S.; Gonçalves, V.C.; Saxena, R.N.; Carbonari, A.W.; Costa, M.S.; Cabrera-Pasca, G.A. Locally symmetric oxygen vacancy around Cd impurities in CeO2. Phys. Rev. B 2021, 104, 035146. [Google Scholar] [CrossRef]
- Cottenier, S. Density Functional Theory and the Family of (L)APW-Methods: A Step-by-Step Introduction, 2nd ed.; Ghent University: Ghent, Belgium, 2013; Available online: http://www.wien2k.at/reg_user/textbooks/ (accessed on 10 August 2022).
- Andersen, O.K. Linear methods in band theory. Phys. Rev. B 1975, 12, 3060. [Google Scholar] [CrossRef]
- Blaha, P.; Schwarz, K.; Tran, F.; Laskowski, R.; Madsen, G.K.H.; Marks, L.D. WIEN2k: An APW+lo program for calculating the properties of solids. J. Chem. Phys. 2020, 152, 074101. [Google Scholar] [CrossRef] [PubMed]
- Koch, K.; Cottenier, S. Analysis of an Electric-Field Gradient (EFG). Technical Report. 2011. Available online: http://www.wien2k.at/reg_user/faq/efg2.pdf (accessed on 10 August 2022).
- Novák, P. Calculation of Hyperfine Field in WIEN2k. Technical Report. 2006. Available online: http://www.wien2k.at/reg_user/textbooks/Bhf_3.pdf (accessed on 10 August 2022).
- Sena, C.; Costa, M.S.; Muñoz, E.L.; Cabrera-Pasca, G.A.; Pereira, L.F.D.; Mestnik-Filho, J.; Carbonari, A.W.; Coaquira, J.A.H. Charge distribution and hyperfine interactions in the vicinity of impurity sites in In2O3 doped with Fe, Co, and Ni. J. Magn. Magn. Mater. 2015, 387, 165–178. [Google Scholar] [CrossRef]
- Richard, D.; Muñoz, E.L.; Butz, T.; Errico, L.A.; Rentería, M. Electronic and structural properties, and hyperfine interactions at Sc sites in the semiconductor Sc2O3: TDPAC and ab initio study. Phys. Rev. B 2010, 82, 035206. [Google Scholar] [CrossRef]
- Schell, J.; Lupascu, D.C.; Correia, J.G.M.; Carbonari, A.W.; Deicher, M.; Barbosa, M.B.; Mansano, R.D.; Johnston, K.; Ribeiro, I.S., Jr. In and Cd as defect traps in titanium dioxide. Hyperfine Interact. 2017, 238, 2. [Google Scholar] [CrossRef]
- Schell, J.; Lupascu, D.C.; Carbonari, A.W.; Mansano, R.D.; Ribeiro Junior, I.S.; Dang, T.T.; Anusca, I.; Trivedi, H.; Johnston, K.; Vianden, R. Ion implantation in titanium dioxide thin films studied by perturbed angular correlations. J. Appl. Phys. 2017, 121, 145302. [Google Scholar] [CrossRef]
- Wenzel, T.; Bartos, A.; Lieb, K.P.; Uhrmacher, M.; Wiarda, D. Hyperfine interactions of 111Cd in TiO2 (Rutile) studied by perturbed angular correlations. Ann. Physik 1992, 504, 155–163. [Google Scholar] [CrossRef]
- Errico, L.A.; Fabricius, G.; Rentería, M.; de la Presa, P.; Forker, M. Anisotropic Relaxations Introduced by Cd Impurities in Rutile TiO2: First-Principles Calculations and Experimental Support. Phys. Rev. Lett. 2002, 89, 055503. [Google Scholar] [CrossRef] [Green Version]
- Ramos, J.M.; Carbonari, A.W.; Costa, M.S.; Saxena, R.N. Electric quadrupole interactions in nano-structured SnO2 as measured with PAC spectroscopy. Hyperfine Interact. 2010, 197, 239–243. [Google Scholar] [CrossRef]
- Schell, J.; Lupascu, D.C.; Carbonari, A.W.; Mansano, R.D.; Freitas, R.S.; Gonçalves, J.N.; Dang, T.T.; ISOLDE Collaboration; Vianden, R. Cd and In-doping in thin film SnO2. J. Appl. Phys. 2017, 121, 195303. [Google Scholar] [CrossRef]
- Schell, J.; Lupascu, D.C.; Carbonari, A.W.; Mansano, R.D.; Freitas, R.S.; Gonçalves, J.N.; Dang, T.T.; ISOLDE Collaboration; Vianden, R. Investigation of the local environment of SnO2 in an applied magnetic field. Phys. B 2020, 586, 412120. [Google Scholar] [CrossRef]
- Schell, J.; Dang, T.T.; Carbonari, A.W. Incorporation of Cd-Doping in SnO2. Crystals 2020, 10, 35. [Google Scholar] [CrossRef]
- Renteria, M.; Bibiloni, A.G.; Moreno, M.S.; Desimoni, J.; Mercader, R.C.; Bartos, A.; Uhrmacher, M.; Lieb, K.P. Hyperfine interactions of 111In-implanted tin oxide thin films. J. Phys. Condens. Matter 1991, 3, 3625. [Google Scholar] [CrossRef]
- Wolf, H.; Deubler, S.; Forkel-Wirth, D.; Foettinger, H.; Iwatschenko-Borho, M.; Meyer, F.; Renn, M.; Witthuhn, W.; Helbig, R. Acceptors and Donors in the Wide-Gap Semiconductors ZnO and SnO2. Mater. Sci. Forum 1986, 10–12, 863–868. [Google Scholar] [CrossRef]
- Muñoz, E.L.; Carbonari, A.W.; Errico, L.A.; Bibiloni, A.G.; Petrilli, H.M.; Rentería, M. TDPAC study of Cd-doped SnO. Hyperfine Interact. 2007, 178, 37. [Google Scholar] [CrossRef]
- Darriba, G.N.; Muñoz, E.L.; Richard, D.; Ayala, A.P.; Carbonari, A.W.; Petrilli, H.M.; Rentería, M. Insights into the aftereffects phenomenon in solids based on DFT and time-differential perturbed γ – γ angular correlation studies in 111In(→111Cd)-doped tin oxides. Phys. Rev. B 2022, 105, 195201. [Google Scholar] [CrossRef]
- Mercurio, M.E.; Carbonari, A.W.; Cordeiro, M.R.; Saxena, R.N.; D’Agostino, L.Z. Local investigation of hyperfine interactions in pure and Co-doped ZnO. J. Magn. Magn. Mater. 2010, 322, 1195–1197. [Google Scholar] [CrossRef]
- Mercurio, M.E.; Carbonari, A.W.; Cordeiro, M.R.; Saxena, R.N. Characterization of ZnO and Zn0.95Co0.05O prepared by sol-gel method using PAC spectroscopy. Hyperfine Interact. 2007, 178, 1–5. [Google Scholar] [CrossRef]
- Dogra, R.; Cordeiro, M.R.; Carbonari, A.W.; Saxena, R.N.; Costa, M.S. Absence of room temperature ferromagnetism in transition metal doped ZnO nanocrystalline powders from PAC spectroscopy. Hyperfine Interact. 2010, 197, 77. [Google Scholar] [CrossRef]
- Wang, R.; Gardner, J.A.; Evenson, W.E.; Sommers, J.A. Oxygen-vacancy complexes in cerium oxide studied by 111In time-differential perturbed-angular-correlation spectroscopy. Phys. Rev. B 1993, 47, 638. [Google Scholar] [CrossRef] [PubMed]
- Mowat, J.P.S.; Miller, S.R.; Griffin, J.M.; Seymour, V.R.; Ashbrook, S.E.; Thompson, S.P.; Fairen-Jimenez, D.; Banu, A.-M.; Düren, T.; Wright, P.A. Structural Chemistry, Monoclinic-to-Orthorhombic Phase Transition, and CO2 Adsorption Behavior of the Small Pore Scandium Terephthalate, Sc2(O2CC6H4CO2)3, and Its Nitro- And Amino-Functionalized Derivatives. Inorg. Chem. 2011, 50, 10844–10858. [Google Scholar] [CrossRef] [PubMed]
- Baldinozzi, G.; Bérar, J.F.; Calvarin-Amiri, G. Rietveld Refinement of Two-Phase Zr-Doped Y2O3. Mater. Sci. Forum 1998, 278–281, 680–685. [Google Scholar] [CrossRef]
- Marezio, M. Refinement of the Crystal Structure of In2O3 at two Wavelengths. Acta Cryst. 1966, 20, 723–728. [Google Scholar] [CrossRef]
- Haas, H.; Röder, J.; Correia, J.G.; Schell, J.; Fenta, A.S.; Vianden, R.; Larsen, E.M.H.; Aggelund, P.A.; Fromsejer, R.; Hemmingsen, L.B.S.; et al. Free Molecule Studies by Perturbed γ-γ Angular Correlation: A New Path to Accurate Nuclear Quadrupole Moments. Phys. Rev. Lett. 2021, 126, 103001. [Google Scholar] [CrossRef]
- Butz, T.; Lerf, A. Comment on “Mössbauer studies of the 6.2 keV γ-rays of 181Ta in Ta-dichalcogenides”. Phys. Lett. A 1983, 97A, 217–218. [Google Scholar] [CrossRef]
- Ryu, S.; Das, S.K.; Butz, T. Nuclear quadrupole interaction at 44Sc in the anatase and rutile modifications of TiO2: Time-differential perturbed angular correlation measurements and ab initio calculations. Phys. Rev. B 2008, 77, 094124. [Google Scholar] [CrossRef]
- Budzinsky, M.; Velichkov, A.I.; Karaivanov, D.V.; Kochetov, O.I.; Salamatin, A.V.; Filosofov, D.V. Use of 44Ti in the Time-Differential γγ Perturbed-Angular-Correlation Method for Studying Condensed Matter. Instrum. Exp. Tech. 2017, 60, 775–781. [Google Scholar] [CrossRef]
- Rentería, M.; Bibiloni, A.G.; Requejo, F.G.; Pasquevich, A.F.; Shitu, J.; Errico, L.A.; Freitag, K. Impurity Cationic-Site Population and Electric-Field Gradient Dependence on Ionic Size in Bixbyites Sesquioxides Implanted With 181Hf →181Ta. Mod. Phys. Lett. B 1998, 12, 819–827. [Google Scholar] [CrossRef]
- Richard, D.; Rentería, M.; Carbonari, A.W.; Romero, M.; Faccio, R. Preparation of In-doped Y2O3 ceramics through a sol-gel process: Effects on the structural and electronic properties. Ceram. Int. 2020, 46, 16088–16095. [Google Scholar] [CrossRef]
- Sorokin, A.A.; Shirani, E.N.; Shpinkova, L.G.; Akselrod, Z.Z.; Komissarova, B.A.; Ryasny, G.K.; Semyonov, S.I.; Denisenko, G.A.; Zibrov, I.P.; Buev, A.R. PAC studies of the electric quadrupole interaction of 181Ta in yttrium and copper oxides and in super conducting ceramics YBa2Cu3O7-δ. Hyperfine Interact. 1992, 73, 337–346. [Google Scholar] [CrossRef]
- Pasquevich, A.A.; Bibiloni, A.G.; Massolo, C.P.; Rentería, M.; Vercesi, J.A.; Freitag, K. Electric-field gradients at the 181Ta impurity site in Yb, Y, and Dy sesquioxides. Phys. Rev. B 1994, 49, 14331–14336. [Google Scholar] [CrossRef] [PubMed]
- Uhrmacher, M.; Bolse, W. TDPAC study of indium oxide. Hyperfine Interact. 1983, 15/16, 445–448. [Google Scholar] [CrossRef]
- Desimoni, J.; Bibiloni, A.G.; Mendoza-Zélis, L.; Pasquevich, A.F.; Sánchez, F.H.; López-García, A. Kinetics studies and oxide characterization in the internal oxidation of AgIn alloys. Phys. Rev. B 1983, 28, 5739. [Google Scholar] [CrossRef]
- Bibiloni, A.G.; Desimoni, J.; Massolo, C.P.; Mendoza-Zélis, L.; Pasquevich, A.F.; Sánchez, F.H.; López-García, A. Temperature dependence of electron-capture aftereffects in the semiconductor In2O3. Phys. Rev. B 1984, 29, 1109–1111. [Google Scholar] [CrossRef]
- Bibiloni, A.G.; Massolo, C.P.; Desimoni, J.; Mendoza-Zélis, L.A.; Sánchez, F.H.; Pasquevich, A.F.; Damonte, L.; López-García, A.R. Time-differential perturbed-angular-correlation study of pure and Sn-doped In2O3 semiconductors. Phys. Rev. B 1985, 32, 2393–2400. [Google Scholar] [CrossRef]
- Massolo, C.P.; Desimoni, J.; Bibiloni, A.G.; Mendoza-Zélis, L.A.; Sánchez, F.H.; Pasquevich, A.F.; López-García, A.R. TDPAC studies of after-effects in In2O3 precipitates in a silver matrix. Hyperfine Interact. 1986, 30, 1–8. [Google Scholar] [CrossRef]
- Bolse, W.; Uhrmacher, M.; Lieb, K.P. Perturbed angular-correlation experiments on 111In in oxidized fcc metals and their oxides. Phys. Rev. B 1987, 36, 1818–1830. [Google Scholar] [CrossRef]
- Habenicht, S. PAC-studies of Sn-doped In2O3: Electronic defect relaxation following the 111In(EC)111Cd-decay. Z. Phys. B 1996, 101, 187–196. [Google Scholar] [CrossRef]
- Ramallo-Lopez, J.M.; Rentería, M.; Miró, E.E.; Requejo, F.G.; Traverse, A. Structure of Extremely Nanosized and Confined In–O Species in Ordered Porous Materials. Z. Phys. B 2003, 91, 108304. [Google Scholar] [CrossRef] [PubMed]
- Sena, C.; Costa, M.S.; Cabrera-Pasca, G.A.; Saxena, R.N.; Carbonari, A.W. TDPAC measurements in pure and Fe-doped In2O3. Hyperfine Interact. 2013, 221, 105–110. [Google Scholar] [CrossRef]
- Vercesi, J.A.; Bibiloni, A.G.; Massolo, C.P.; Moreno, M.S.; Pasquevich, A.F.; Freitag, K. Hyperfine characterization of 181Ta in In2O3. Phys. Rev. B 1993, 41, 490–492. [Google Scholar] [CrossRef] [PubMed]
- Renteria, M.; Requejo, F.G.; Bibiloni, A.G.; Pasquevich, A.F.; Shitu, J.; Freitag, K. Perturbed-angular-correlation study of the electric-field gradient in 181Hf-doped and implanted indium sesquioxide. Phys. Rev. B 1997, 55, 14200–14207. [Google Scholar] [CrossRef]
- Atkins, P.W.; Overton, T.L.; Rourke, J.P.; Weller, M.T.; Armstrong, F.A. Shriver and Atkins’ Inorganic Chemistry, 5th ed.; Oxford University Press: Oxford, UK, 2010; pp. 783–784. [Google Scholar]
- Aleshina, L.A.; Loginova, S.V. Rietveld analysis of X-ray diffraction pattern from β-Ta2O5 oxide. Crystallogr. Rep. 2002, 47, 415–419. [Google Scholar] [CrossRef]
- Stephenson, N.C.; Roth, R.S. The Crystal Structure of the High Temperature Form of Ta2O5. J. Solid State Chem. 1971, 3, 145–153. [Google Scholar] [CrossRef]
- Autschbach, J.; Zheng, S.; Schurko, R.W. Analysis of Electric Field Gradient Tensors at Quadrupolar Nuclei in Common Structural Motifs. Inc. Concepts Magn. Reson. Part A 2010, 36A, 84–126. [Google Scholar] [CrossRef]
- Blaha, P.; Schwarz, K.; Dederichs, P.H. First-principles calculation of the electric-field gradient in hcp metals. Phys. Rev. B 1988, 37, 2792–2797. [Google Scholar] [CrossRef]



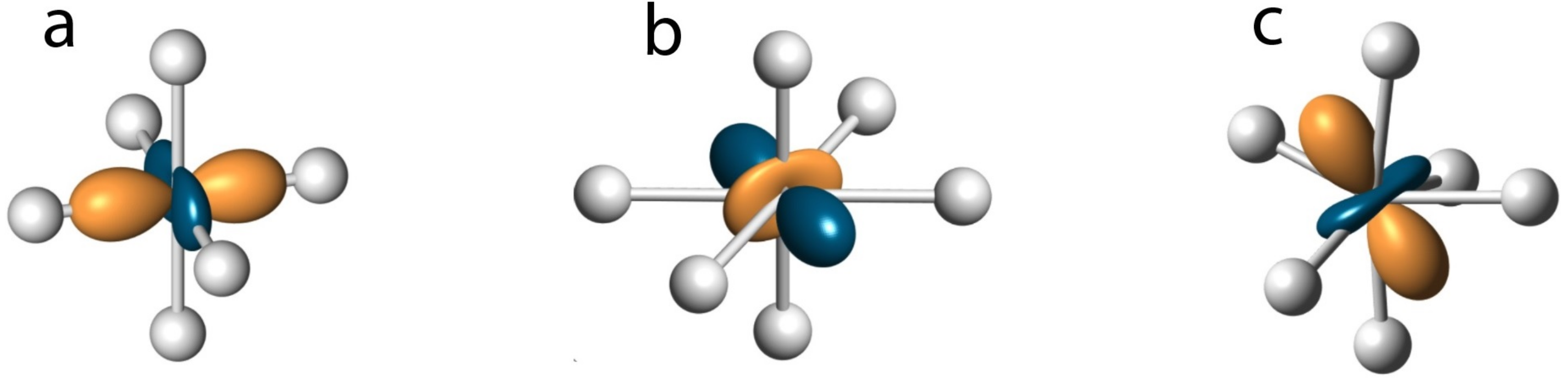{kind=link}
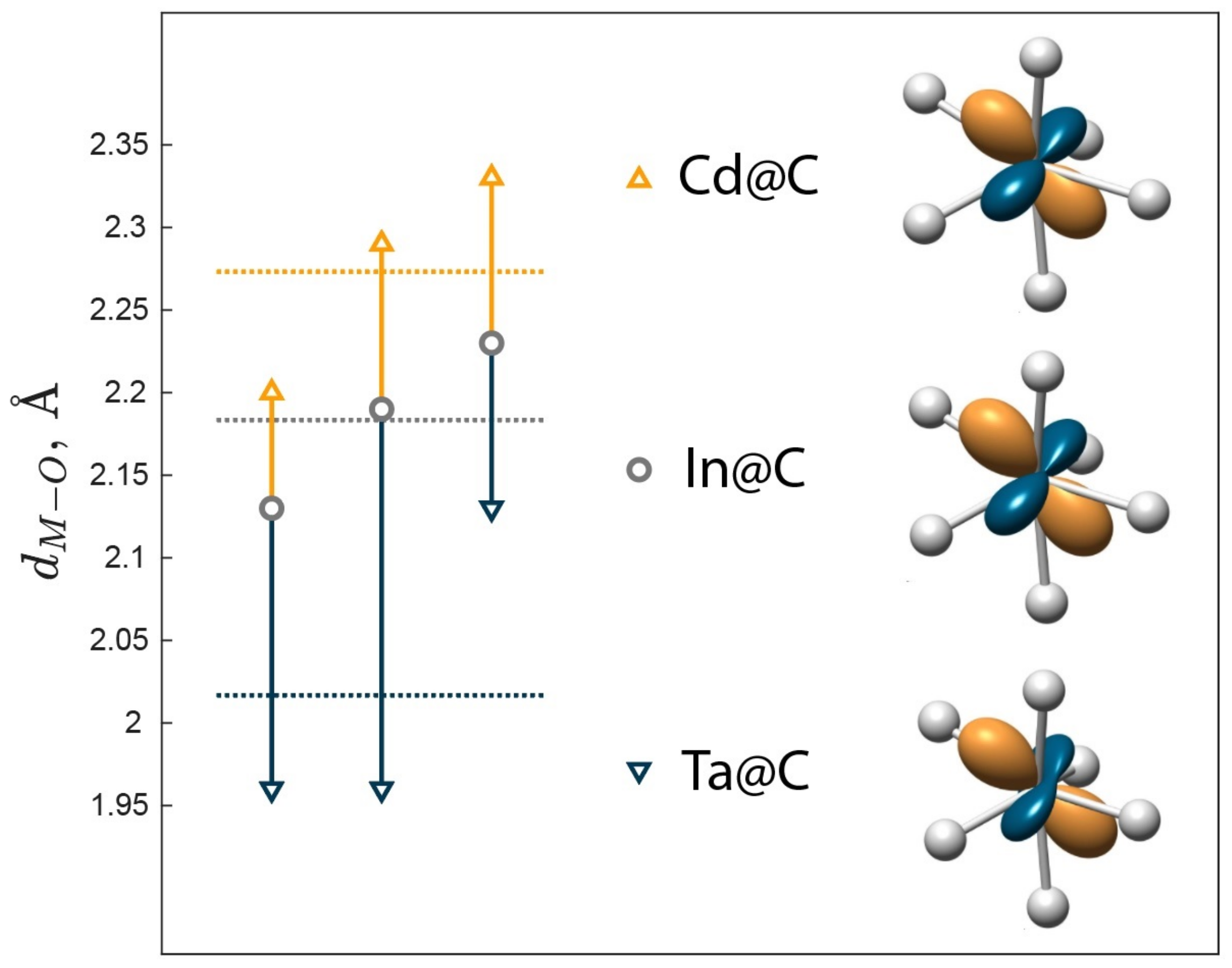{kind=link}
| Oxide | Experimental | Theoretical | Crystalline Structure | Ref. | |||||
|---|---|---|---|---|---|---|---|---|---|
| , V/Å | , V/Å | Charge State | Space Group | Lattice Parameters | , Å | ||||
| TiO | 50.7 | 0.18 | 45.5 | 0.26 | [Cd] | Cd–O1 = 2.18 | [20] | ||
| 55.74 | 0.12 | Cd–O2 = 2.11 | [21] | ||||||
| 52.3 | 0.18 | [22] | |||||||
| [23] | |||||||||
| SnO | 56.8 e | 0.13 e | 54.3 e | 0.14 e | [Cd] | Cd–O1 = 2.16 | e [11] | ||
| 57.3 | 0.1 | 51.1 e | 0.36 e | [Cd] | Cd–O2 = 2.20 | [24] | |||
| 48.2 | 0.15 | [25] | |||||||
| 45.0 | 0.37 | [26] | |||||||
| 56–63 | 0.42–0.18 | [27] | |||||||
| 58.3 | 0.18 | [28] | |||||||
| 57.8 | 0.1 | [29] | |||||||
| SnO | 57.0 | 0.2 | 62.0 | 0.0 | [Cd] | Cd–O = 2.26 | [30] | ||
| ∼53.0 | ∼0.2 | 53.0 | 0.0 | [Cd] | [2] | ||||
| o [31] | |||||||||
| ZnO | 15.5 | 0.0 | 16.8 | 0.0 | [Cd] | Cd–O = 2.21 | [32] | ||
| 15.7 | 0.0 | [33] | |||||||
| 16.0 | 0.09 | [34] | |||||||
| [3] | |||||||||
| CeO, | 26.4 | 0.1 | 27.6 | 0.0 | [Cd] | Cd–O = 2.36 | [12] | ||
| monovacancy | 26.5 | 0.0 | [35] | ||||||
| CeO, | 30.4 | 0.4 | −31.5 | 0.37 | [Cd] | ||||
| divacancy | 29.6 | 0.35 | |||||||
| Compound | Impurity | , Å | , Å | , V/Å | , V/Å | Ref. | ||
|---|---|---|---|---|---|---|---|---|
| ScO | – | 0 | [19] | |||||
| Sc | 0.63 | 0 | [19,42] | |||||
| Cd | 0 | [1,6] | ||||||
| [Cd] | 0 | [5] | ||||||
| [Cd] | 0 | |||||||
| Ta | 141.2 | 1 | 178.6 | 0.16 | [43] | |||
| [Ta] | 127.2 | 0.82 | 90 | 0 | [7] | |||
| [Ta] | −167.5 | 0.45 | 165 | 0 | ||||
| [Ta] | −183 | 0.18 | 273 | 0 | ||||
| YO | Cd | 0 | [1,44] | |||||
| Ta | 0.54 | 0.07 | [4,45,46] | |||||
| [Ta] | −115.4 | 0.65 | −99.6 | 0 | [4] | |||
| [Ta] | −189 | 0.44 | −27.8 | 0 | ||||
| [Ta] | −151.3 | 0.66 | 236.2 | 0 | ||||
| [Ta] | 57.2 | 0.65 | 50.3 | 0 | ||||
| [Ta] | −76.5 | 0.71 | 147.1 | 0 | ||||
| [Ta] | −137.1 | 0.60 | 259.4 | 0 | ||||
| InO | – | 54 | 0.98 | 82 | 0 | [8] | ||
| Cd | [1,18,47,48,49,50,51,52,53,54,55] | |||||||
| [Cd] | relaxed | −30.8 | 0.01 | 71.5 | 0 | [18] | ||
| [Cd] | 48 | 0.96 | 78 | 0 | [8] | |||
| [Cd] | 56 | 0.68 | 76 | 0 | ||||
| Ta | [10,56,57] * | |||||||
| [Ta] | 1.95/1.95/2.12 | 2.02 | −174 | 0.13 | 199 | 0 | [10] | |
| [Ta] | 1.95/1.96/2.13 | 2.00 | −175 | 0.12 | 209 | 0 | ||
| [Ta] | 1.96/1.96/2.13 | 2.01 | −174 | 0.10 | 208 | 0 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burimova, A.; Carbonari, A.W.; de Lima, N.P.; Miranda Filho, A.A.; dos Santos Souza, A.P.; da Silva Nascimento Sales, T.; Ferreira, W.L.; Pereira, L.F.D.; Correa, B.S.; Saxena, R.N. Local Crystalline Structure of Doped Semiconductor Oxides Characterized by Perturbed Angular Correlations: Experimental and Theoretical Insights. Crystals 2022, 12, 1204. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12091204
Burimova A, Carbonari AW, de Lima NP, Miranda Filho AA, dos Santos Souza AP, da Silva Nascimento Sales T, Ferreira WL, Pereira LFD, Correa BS, Saxena RN. Local Crystalline Structure of Doped Semiconductor Oxides Characterized by Perturbed Angular Correlations: Experimental and Theoretical Insights. Crystals. 2022; 12(9):1204. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12091204
Chicago/Turabian StyleBurimova, Anastasia, Artur Wilson Carbonari, Nicole Pereira de Lima, Arnaldo Alves Miranda Filho, Alexandre Pinho dos Santos Souza, Tatiane da Silva Nascimento Sales, Wanderson Lobato Ferreira, Luciano Fabricio Dias Pereira, Bruno Santos Correa, and Rajendra Narain Saxena. 2022. "Local Crystalline Structure of Doped Semiconductor Oxides Characterized by Perturbed Angular Correlations: Experimental and Theoretical Insights" Crystals 12, no. 9: 1204. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12091204