Controlled Synthesis of Linear Polyamidoamino Acids



Abstract
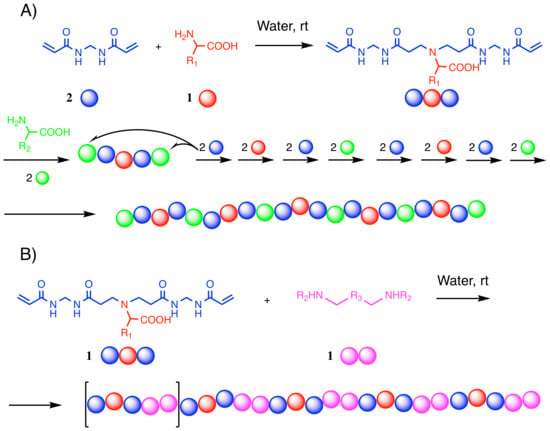
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Characterizations
2.3. Controlled Synthesis of PAACs
3. Results and Discussion
3.1. Rationale of the Synthetic Process
3.2. Stepwise Controlled Synthesis of PAAC Homo- and Copolymers
3.3. Use of Intermediates as Building Blocks for Preparing PAAC-PAA Copolymers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bauri, K.; Ghosh Roy, S.; De, P. Side-Chain Amino-Acid-Derived Cationic Chiral Polymers by Controlled Radical Polymerization. Macromol. Chem. Phys. 2016, 217, 365–379. [Google Scholar] [CrossRef]
- Sanda, F.; Nakamura, M.; Endo, T. Syntheses and Radical Copolymerization Behavior of Optically Active Methacrylamides Having l- and d-Leucine Moieties. Interaction between l- and d-Forms. Macromolecules 1996, 29, 8064–8068. [Google Scholar] [CrossRef]
- Casolaro, M.; Casolaro, I. Stimuli-Responsive Hydrogels Bearing α-Amino Acid Residues: A Potential Platform for Future Therapies. J. Biomed. Eng. Med. Device. 2016, 1, 100111. [Google Scholar] [CrossRef]
- Mori, H.; Kato, I.; Endo, T. Dual-Stimuli-Responsive Block Copolymers Derived from Proline Derivatives. Macromolecules 2009, 42, 4985–4992. [Google Scholar] [CrossRef]
- Maji, T.; Banerjee, S.; Bose, A.; Mandal, T.K. A Stimuli-Responsive Methionine-Based Zwitterionic Methacryloyl Sulfonium Sulfonate Monomer and The Corresponding Antifouling Polymer with Tunable Thermosensitivity. Polym. Chem. 2017, 8, 3164–3176. [Google Scholar] [CrossRef]
- Maji, T.; Banerjee, S.; Biswas, Y.; Mandal, T.K. Dual-Stimuli-Responsive l-Serine-Based Zwitterionic UCST-Type Polymer with Tunable Thermosensitivity. Macromolecules 2015, 48, 4597–4966. [Google Scholar] [CrossRef]
- Gao, G.; Sanda, F.; Masuda, T. Synthesis and Properties of Amino Acid-Based Polyacetylenes. Macromolecules 2003, 36, 3932–3937. [Google Scholar] [CrossRef]
- Cheuk, K.K.L.; Li, B.S.; Lam, J.W.Y.; Xie, Y.; Tang, B.Z. Synthesis, Chain Helicity, Assembling Structure, and Biological Compatibility of Poly(phenylacetylene)s Containing l-Alanine Moieties. Macromolecules 2008, 41, 5997–6005. [Google Scholar] [CrossRef]
- Hopkins, T.E.; Pawlow, J.H.; Koren, D.L.; Deters, K.S.; Solivan, S.M.; Davis, J.A.; Gómez, F.J.; Wagener, K.B. Chiral Polyolefins Bearing Amino Acids. Macromolecules 2001, 34, 7920–7922. [Google Scholar] [CrossRef]
- Sanda, S.; Endo, T. Synthesis and Cationic Polymerization of a Novel Optically Active Vinyl Ether with l-Proline Structure. Macromol. Chem. Phys. 1997, 198, 1209–1216. [Google Scholar] [CrossRef]
- Allcock, H.R.; Pucher, S.R.; Scopelianos, A.G. Poly[(amino acid ester)phosphazenes] as Substrates for the Controlled Release of Small Molecules. Biomaterials 1994, 15, 563–569. [Google Scholar] [CrossRef]
- Ferruti, P.; Mauro, N.; Falciola, L.; Pifferi, V.; Bartoli, C.; Gazzarri, M.; Chiellini, F.; Ranucci, E. Amphoteric, Prevailingly Cationic l-Arginine Polymers of Poly(amidoamino acid) Structure: Synthesis, Acid/Base Properties and Preliminary Cytocompatibility and Cell-Permeating Characterizations. Macromol. Biosci. 2014, 14, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, A.; Mauro, N.; Terenzi, A.; Alongi, J.; Lazzari, F.; Ganazzoli, F.; Raffaini, G.; Ranucci, E.; Ferruti, P. Self-Ordering Secondary Structure of d- and l-Arginine-Derived Polyamidoamino Acids. ACS Macro Lett. 2017, 6, 987–991. [Google Scholar] [CrossRef]
- Lazzari, F.; Manfredi, A.; Alongi, J.; Mendichi, R.; Ganazzoli, F.; Raffaini, G.; Ferruti, P.; Ranucci, E. Self-Structuring in Water of Polyamidoamino Acids with Hydrophobic Side Chains Deriving from Natural α-Amino Acids. Polymers 2018, 10, 1261. [Google Scholar] [CrossRef] [PubMed]
- Lazzari, F.; Manfredi, A.; Alongi, J.; Marinotto, D.; Ferruti, P.; Ranucci, E. d-, l- and d,l-Tryptophan-Based Polyamidoamino Acids: pH-Dependent Structuring and Fluorescent Properties. Polymers 2019, 11, 543. [Google Scholar] [CrossRef]
- Ferruti, P. Poly(amidoamine)s: Past, present, and perspectives. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 2319–2353. [Google Scholar] [CrossRef]
- Ranucci, E.; Manfredi, A. Polyamidoamines: Versatile Bioactive Polymers with Potential for Biotechnological Applications. Chem. Afr. 2019, 2, 167–193. [Google Scholar] [CrossRef] [Green Version]
- Green, D.W.; Lee, J.-M.; Kim, E.-J.; Lee, D.-J.; Jung, H.-S. Chiral Biomaterials: from Molecular Design to Regenerative Medicine. Adv. Mater. Interfaces 2016, 3, 1500411. [Google Scholar] [CrossRef]
- Hyland, L.L.; Twomey, J.D.; Vogel, S.; Hsieh, A.H.; Yu, Y.B. Enhancing Biocompatibility of d-Oligopeptide Hydrogels by Negative Charges. Biomacromolecules 2013, 14, 406–412. [Google Scholar] [CrossRef]
- Chen, J.; Wu, C.; Oupicky, D. Bioreducible Hyperbranched Poly(amido amine)s for Gene Delivery. Biomacromolecules 2009, 10, 2921–2927. [Google Scholar] [CrossRef] [Green Version]
- Elzes, M.R.; Akeroyd, N.; Engbersen, J.F.J.; Paulusse, J.M.J. Disulfide-functional Poly(amido amine)s with Tunable Degradability for Gene Delivery. J. Contr. Release 2016, 244, 357–365. [Google Scholar] [CrossRef]
- Parkhouse, M.S.; Garnett, M.C.; Chan, W.C. Targeting of Polyamidoamine-DNA Nanoparticles Using the Staudinger Ligation: Attachment of an RGD Motif Either Before or After Complexation. Bioorgan. Med. Chem. 2008, 16, 6641–6650. [Google Scholar] [CrossRef] [PubMed]
- Ferruti, P.; Franchini, J.; Bencini, M.; Ranucci, E.; Zara, G.P.; Serpe., L.; Primo, L.; Cavalli, R. Prevailingly Cationic Agmatine-Based Amphoteric Polyamidoamine as a Nontoxic, Nonhemolytic, and "Stealthlike" DNA Complexing Agent and Transfection Promoter. Biomacromolecules 2007, 8, 1498–1504. [Google Scholar] [CrossRef]
- Donghi, D.; Maggioni, D.; D’Alfonso, G.; Amigoni, A.; Ranucci, E.; Ferruti, P.; Manfredi, A.; Fenili, F.; Bisazza, A.; Cavalli, R. Tricarbonyl-Rhenium Complexes of a Thiol-Functionalized Amphoteric Poly(amidoamine). Biomacromolecules 2009, 10, 3273–3282. [Google Scholar] [CrossRef] [PubMed]
- Barshteyn, N.; Elfarra, A.A. Formation of Mono- and Bis-Michael Adducts by the Reaction of Nucleophilic Amino Acids with Hydroxymethylvinyl Ketone, a Reactive Metabolite of 1,3-Butadiene. Chem. Res. Toxicol. 2009, 22, 918–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferruti, F.; Alongi, J.; Manfredi, A.; Ranucci, E.; Ferruti, P. Controlled Synthesis of Linear Polyamidoamino Acids. Polymers 2019, 11, 1324. https://0-doi-org.brum.beds.ac.uk/10.3390/polym11081324
Ferruti F, Alongi J, Manfredi A, Ranucci E, Ferruti P. Controlled Synthesis of Linear Polyamidoamino Acids. Polymers. 2019; 11(8):1324. https://0-doi-org.brum.beds.ac.uk/10.3390/polym11081324
Chicago/Turabian StyleFerruti, Federica, Jenny Alongi, Amedea Manfredi, Elisabetta Ranucci, and Paolo Ferruti. 2019. "Controlled Synthesis of Linear Polyamidoamino Acids" Polymers 11, no. 8: 1324. https://0-doi-org.brum.beds.ac.uk/10.3390/polym11081324