Crystallization of Poly(ethylene)s with Regular Phosphoester Defects Studied at the Air–Water Interface


Abstract
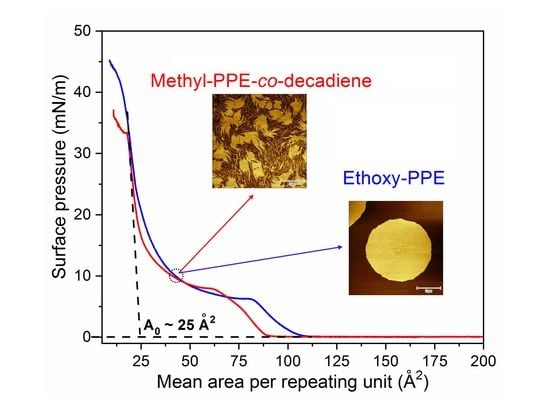
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Langmuir Isotherms Measurement
2.3. Microscopic Studies on Langmuir Films
2.4. Langmuir–Blodgett (LB) Film Transfer, AFM, and GI-WAXS
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Reiter, G. Dewetting of thin polymer films. Phys. Rev. Lett. 1992, 68, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Factor, B.J.; Russell, T.P.; Toney, M.F. Grazing incidence X-ray scattering studies of thin films of an aromatic polyimide. Macromolecules 1993, 26, 2847–2859. [Google Scholar] [CrossRef]
- Keddie, J.L.; Jones, R.A.L.; Cory, R.A. Interface and surface effects on the glass-transition temperature in thin polymer films. Faraday Discuss. 1994, 98, 219–230. [Google Scholar] [CrossRef]
- Hobbs, J.K.; Humphris, A.D.L.; Miles, M.J. In-situ atomic force microscopy of polyethylene crystallization. 1. crystallization from an oriented backbone. Macromolecules 2001, 34, 5508–5519. [Google Scholar] [CrossRef]
- Si, L.; Massa, M.V.; Dalnoki-Veress, K.; Brown, H.R.; Jones, R.A.L. Chain entanglement in thin freestanding polymer films. Phys. Rev. Lett. 2005, 94, 127801–127804. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Hu, W.; Reiter, G. Lamellar crystal orientations biased by crystallization kinetics in polymer thin films. Macromolecules 2006, 39, 5159–5164. [Google Scholar] [CrossRef]
- Kossack, W.; Seidlitz, A.; Thurn-Albrecht, T.; Kremer, F. Interface and confinement induced order and orientation in thin films of poly(ϵ-caprolactone). Macromolecules 2016, 49, 3442–3451. [Google Scholar] [CrossRef]
- Flieger, A.-K.; Schulz, M.; Thurn-Albrecht, T. Interface-induced crystallization of polycaprolactone on graphite via first-order prewetting of the crystalline phase. Macromolecules 2018, 51, 189–194. [Google Scholar] [CrossRef] [Green Version]
- Polymer Thin Films; Hashim, A.A. (Ed.) In-Tech: Vukovar, Croatia, 2010; pp. 1–309. [Google Scholar]
- Nagaraja, A.; Jalageri, M.D.; Puttaiahgowda, Y.M. A thirst for polymeric antimicrobial surfaces/coatings for diverse applications. In Engineered Antimicrobial Surfaces. Materials Horizons: From Nature to Nanomaterials; Snigdha, S., Thomas, S., Radhakrishnan, E.K.N., Eds.; Springer: Singapore, 2020; pp. 13–31. [Google Scholar]
- Reiter, G. Dewetting as a probe of polymer mobility in thin films. Macromolecules 1994, 27, 3046–3052. [Google Scholar] [CrossRef]
- Tseng, K.C.; Turro, N.J.; Durning, C.J. Molecular mobility in polymer thin films. Phys. Rev. E 2000, 61, 1800–1811. [Google Scholar] [CrossRef] [Green Version]
- Bunn, C.W. The crystal structure of long-chain normal paraffin hydrocarbons. The “shape” of the CH2 group. Trans. Faraday Soc. 1939, 35, 482–491. [Google Scholar] [CrossRef]
- Forrest, J.A.; Dalnoki-Veress, K.; Dutcher, J.R. Interface and chain confinement effects on the glass transition temperature of thin polymer films. Phys. Rev. E 1997, 56, 5705–5716. [Google Scholar] [CrossRef] [Green Version]
- De Gennes, P.G. Glass transitions in thin polymer films. Eur. Phys. J. E 2000, 2, 201–205. [Google Scholar]
- Tsui, O.K.C.; Zhang, H.F. Effects of chain ends and chain entanglement on the glass transition temperature of polymer thin films. Macromolecules 2001, 34, 9139–9142. [Google Scholar] [CrossRef]
- Roth, C.B.; Dutcher, J.R. Glass transition and chain mobility in thin polymer films. J. Electroanal. Chem. 2005, 584, 13–22. [Google Scholar] [CrossRef]
- Inoue, R.; Kanaya, T.; Nishida, K.; Tsukushi, I.; Telling, M.T.F.; Gabrys, B.J.; Tyagi, M.; Soles, C.; Wu, W.-L. Glass transition and molecular mobility in polymer thin films. Phys. Rev. E 2009, 80, 031802. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.L.; Kumar, S.K.; Ho, D.L.; Briber, R.M.; Russell, T.P. Chain conformation in ultrathin polymer films. Nature 1999, 400, 146–149. [Google Scholar] [CrossRef]
- Kraus, J.; Müller-Buschbaum, P.; Kuhlmann, T.; Schubert, D.W.; Stamm, M. Confinement effects on the chain conformation in thin polymer films. Europhys. Lett. 2000, 49, 210–216. [Google Scholar] [CrossRef]
- Hall, D.B.; Underhill, P.; Torkelson, J.M. Spin coating of thin and ultrathin polymer films. Polym. Eng. Sci. 1998, 38, 2039–2045. [Google Scholar] [CrossRef]
- Crisp, D.J. Surface films of polymers. Part, I. Films of the fluid type. J. Colloid Sci. 1946, 1, 49–70. [Google Scholar] [CrossRef]
- Crisp, D.J. Surface films of polymers. Part II. Films of the coherent and semi-crystalline type. J. Colloid Sci. 1946, 1, 161–184. [Google Scholar] [CrossRef]
- Petty, M.C. Film Deposition; Cambridge University Press: Cambridge, UK, 1996; pp. 12–64. [Google Scholar]
- Busse, K.; Fuchs, C.; Hasan, N.; Pulst, M.; Kressler, J. Crystallization of poly(ethylene oxide) on the surface of aqueous salt solutions studied by grazing incidence wide-angle X-ray scattering. Langmuir 2018, 34, 12759–12763. [Google Scholar] [CrossRef] [PubMed]
- Hasan, N.; Fuchs, C.; Schwieger, C.; Busse, K.; Dolynchuk, O.; Kressler, J. Crystallization of poly(ε-caprolactone) at the air-water interface studied by IRRAS and GI-WAXS. Polymer 2020, 196, 122468. [Google Scholar] [CrossRef]
- Kumaki, J.; Kawauchi, T.; Yashima, E. Two-dimensional folded chain crystals of a synthetic polymer in a Langmuir−Blodgett film. J. Am. Chem. Soc. 2005, 127, 5788–5789. [Google Scholar] [CrossRef]
- Kumaki, J. Observation of polymer chain structures in two-dimensional films by atomic force microscopy. Polym. J. 2016, 48, 3–14. [Google Scholar] [CrossRef]
- Gaines, G.L. Monolayers of polymers. Langmuir 1991, 7, 834–839. [Google Scholar] [CrossRef]
- Watanabe, K.; Kumaki, J. Extended-chain crystallization and stereocomplex formation of polylactides in a Langmuir monolayer. Polym. J. 2020, 52, 601–613. [Google Scholar] [CrossRef]
- Fuchs, C.; Busse, K.; Flieger, A.-K.; Kressler, J. Polymer crystallization on the surface of water or aqueous salt solution. Chem. Eng. Technol. 2016, 39, 1333–1340. [Google Scholar] [CrossRef]
- Sorokin, A.V.; Bai, M.; Ducharme, S.; Poulsen, M. Langmuir—Blodgett films of polyethylene. J. Appl. Phys. 2002, 92, 5977–5981. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, K.; Suzuki, H.; Katoh, T.; Imai, S.; Nakayama, Y.; Miki, H. Deposition of polyethylene thin films using synchrotron radiation ablation. Jpn. J. Appl. Phys. 1999, 38, 863–867. [Google Scholar] [CrossRef]
- Chung, T.C.; Lu, H.L.; Li, C.L. Synthesis and functionalization of unsaturated polyethylene: Poly(ethylene-co-1,4-hexadiene). Macromolecules 1994, 27, 7533–7537. [Google Scholar] [CrossRef]
- Hong, M.; Liu, J.-Y.; Li, B.-X.; Li, Y.-S. Facile functionalization of polyethylene via click chemistry. Macromolecules 2011, 44, 5659–5665. [Google Scholar] [CrossRef]
- Boffa, L.S.; Novak, B.M. Copolymerization of polar monomers with olefins using transition-metal complexes. Chem. Rev. 2000, 100, 1479–1494. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, C.; Mecking, S.; Jian, Z. Ultrahigh branching of main-chain-functionalized polyethylenes by inverted insertion selectivity. Angew. Chem. 2020, 132, 14402–14408. [Google Scholar] [CrossRef]
- Long, B.K.; Eagan, J.M.; Mulzer, M.; Coates, G.W. Semi-crystalline polar polyethylene: Ester-functionalized linear polyolefins enabled by a functional-group-tolerant, cationic nickel catalyst. Angew. Chem. Int. Ed. 2016, 55, 7106–7110. [Google Scholar] [CrossRef] [PubMed]
- Wagener, K.B.; Boncella, J.M.; Nel, J.G. Acyclic diene metathesis (ADMET) polymerization. Macromolecules 1991, 24, 2649–2657. [Google Scholar] [CrossRef]
- Few, C.S.; Wagener, K.B.; Thompson, D.L. Systematic studies of morphological changes of precision polyethylene. Macromol. Rapid Commun. 2014, 35, 123–132. [Google Scholar] [CrossRef]
- Zheng, Y.-R.; Tee, H.T.; Wei, Y.; Wu, X.-L.; Mezger, M.; Yan, S.; Landfester, K.; Wagener, K.; Wurm, F.R.; Lieberwirth, I. Morphology and thermal properties of precision polymers: The crystallization of butyl branched polyethylene and polyphosphoesters. Macromolecules 2016, 49, 1321–1330. [Google Scholar] [CrossRef]
- Haider, T.; Suraeva, O.; O’Duill, M.L.; Mars, J.; Mezger, M.; Lieberwirth, I.; Wurm, F.R. Controlling the crystal structure of precisely spaced polyethylene-like polyphosphoesters. Polym. Chem. 2020, 11, 3404–3415. [Google Scholar] [CrossRef]
- Hasan, N.; Schwieger, C.; Tee, H.T.; Wurm, F.R.; Busse, K.; Kressler, J. Crystallization of a polyphosphoester at the air-water interface. Eur. Polym. J. 2018, 101, 350–357. [Google Scholar] [CrossRef]
- Stumm, W.W.; Morgan, J.J. Aquatic Chemistry: Chemical Equilibria and Rates in Natural Waters; John Wiley & Sons: Hoboken, NJ, USA, 1996; p. 528. [Google Scholar]
- Kobayashi, Y.; Amano, T.; Taga, K.; Yamamoto, Y.; Shervani, Z.; Yamamoto, M. Surface properties of novel surfactant, dihexadecyl gemini phosphate, monolayers on water surface by dropping method. J. Biophys. Chem. 2017, 08, 39–50. [Google Scholar] [CrossRef] [Green Version]
- Kaganer, V.M.; Möhwald, H.; Dutta, P. Structure and phase transitions in Langmuir monolayers. Rev. Mod. Phys. 1999, 71, 779–819. [Google Scholar] [CrossRef] [Green Version]
- Amado, E.; Kerth, A.; Blume, A.; Kressler, J. Infrared reflection absorption spectroscopy coupled with Brewster angle microscopy for studying interactions of amphiphilic triblock copolymers with phospholipid monolayers. Langmuir 2008, 24, 10041–10053. [Google Scholar] [CrossRef] [PubMed]
- Pulst, M.; Schneemann, C.; Ruda, P.; Golitsyn, Y.; Grefe, A.-K.; Stühn, B.; Busse, K.; Reichert, D.; Kressler, J. Chain tilt and crystallization of ethylene oxide oligomers with midchain defects. ACS Macro Lett. 2017, 6, 1207–1211. [Google Scholar] [CrossRef]
- Fritzsching, K.J.; Mao, K.; Schmidt-Rohr, K. Avoidance of density anomalies as a structural principle for semicrystalline polymers: The importance of chain ends and chain tilt. Macromolecules 2017, 50, 1521–1540. [Google Scholar] [CrossRef]
- Elzein, T.; Nasser-Eddine, M.; Delaite, C.; Bistac, S.; Dumas, P. FTIR study of polycaprolactone chain organization at interfaces. J. Colloid Interface Sci. 2004, 273, 381–387. [Google Scholar] [CrossRef]
- Kraack, H.; Tamam, L.; Sloutskin, E.; Deutsch, M.; Ocko, B.M. Alkyl-thiol Langmuir films on the surface of liquid mercury. Langmuir 2007, 23, 7571–7582. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, R.; Mao, G.; Flach, C.R. Infrared reflection–absorption spectroscopy: Principles and applications to lipid—protein interaction in Langmuir films. Biochim. Biophys. Acta-Biomembr. 2010, 1798, 788–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki, A.; Fukayama, S. Organic thin film solar cell composed of hetero-deposited Langmuir-Blodgett films. Electrochemistry 2010, 78, 178–180. [Google Scholar] [CrossRef]
- Li, B.; Wu, Y.; Liu, M.; Esker, A.R. Brewster angle microscopy study of poly(ε-caprolactone) crystal growth in Langmuir films at the air/water interface. Langmuir 2006, 22, 4902–4905. [Google Scholar] [CrossRef]
- Lösche, M.; Möhwald, H. Fluorescence microscope to observe dynamical processes in monomolecular layers at the air/water interface. Rev. Sci. Instrum. 1984, 55, 1968–1972. [Google Scholar] [CrossRef]
- Lösche, M.; Rabe, J.; Fischer, A.; Rucha, B.U.; Knoll, W.; Möhwald, H. Microscopically observed preparation of Langmuir-Blodgett films. Thin Solid Films 1984, 117, 269–280. [Google Scholar] [CrossRef]
- Lösche, M.; Möhwald, H. Impurity controlled phase transitions of phospholipid monolayers. Eur. Biophys. J. 1984, 11, 35–42. [Google Scholar] [CrossRef]
- Helm, C.A.; Möhwald, H.; Kjaer, K.; Als-Nielsen, J. Phospholipid monolayers between fluid and solid states. Biophys. J. 1987, 52, 381–390. [Google Scholar] [CrossRef]
- Kajiyama, T.; Oishi, Y. Novel concepts of aggregation structure of fatty acid monolayers on the water surface. In New Developments in Construction and Functions of Organic Thin Films; Kajiyama, T., Aizawa, M., Eds.; Elsevier: Amsterdam, The Netherlands, 1996; pp. 15–16. [Google Scholar]
- Organ, S.J.; Keller, A. Solution crystallization of polyethylene at high temperatures. J. Mater. Sci. 1985, 20, 1571–1585. [Google Scholar] [CrossRef]
- Gedde, U.W.; Hedenqvist, M.S. Fundamental Polymer Science; Springer Nature: Cham, Switzerland, 2019; pp. 251–321. [Google Scholar]
- Weber, C.H.M.; Chiche, A.; Krausch, G.; Rosenfeldt, S.; Ballauff, M.; Harnau, L.; Göttker-Schnetmann, I.; Tong, Q.; Mecking, S. Single lamella nanoparticles of polyethylene. Nano Lett. 2007, 7, 2024–2029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misra, R.D.K.; Depan, D.; Shah, J. The effect of dimensionality of nanostructured carbon on the architecture of organic–inorganic hybrid materials. Phys. Chem. Chem. Phys. 2013, 15, 12988. [Google Scholar] [CrossRef]
- Li, L.; Li, C.Y.; Ni, C. Polymer crystallization-driven, periodic patterning on carbon nanotubes. J. Am. Chem. Soc. 2006, 128, 1692–1699. [Google Scholar] [CrossRef]
- Gedde, U.W.; Jansson, J.-F. Molecular fractionation in melt-crystallized polyethylene: 3. Microscopy of solvent-treated samples. Polymer 1984, 25, 1263–1267. [Google Scholar] [CrossRef]
- Geuchies, J.J.; Soligno, G.; Geraffy, E.; Hendrikx, C.P.; van Overbeek, C.; Montanarella, F.; Slot, M.R.; Konovalov, O.V.; Petukhov, A.V.; Vanmaekelbergh, D. Unravelling three-dimensional adsorption geometries of PbSe nanocrystal monolayers at a liquid-air interface. Commun. Chem. 2020, 3, 28. [Google Scholar] [CrossRef]
- Denicolò, I.; Doucet, J.; Craievich, A.F. X-ray study of the rotator phase of paraffins (III): Even-numbered paraffins C18H38, C20H42, C22H46, C24H50, and C26H54. J. Chem. Phys. 1983, 78, 1465–1469. [Google Scholar] [CrossRef]
- Wentzel, N.; Milner, S.T. Crystal and rotator phases of n-alkanes: A molecular dynamics study. J. Chem. Phys. 2010, 132, 044901. [Google Scholar] [CrossRef] [PubMed]
- Weiss, V.M.; Naolou, T.; Amado, E.; Busse, K.; Mäder, K.; Kressler, J. Formation of structured polygonal nanoparticles by phase-separated comb-like polymers. Macromol. Rapid Commun. 2012, 33, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Bera, P.K.; Kandar, A.K.; Krishnaswamy, R.; Fontaine, P.; Impéror-Clerc, M.; Pansu, B.; Constantin, D.; Maiti, S.; Sanyal, M.K.; Sood, A.K. Grazing incidence X-ray diffraction studies of lipid–peptide mixed monolayers during shear flow. ACS Omega 2020, 5, 14555–14563. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Sworen, J.; Pyda, M.; Nowak-Pyda, E.; Habenschuss, A.; Wagener, K.B.; Wunderlich, B. Effect of the precise branching of polyethylene at Each 21st CH2 group on its phase transitions, crystal structure, and morphology. Macromolecules 2006, 39, 204–217. [Google Scholar] [CrossRef]
- Cankaya, A.; Steinmann, M.; Bülbül, Y.; Lieberwirth, I.; Wurm, F.R. Side-chain poly(phosphoramidate)s via acyclic diene metathesis polycondensation. Polym. Chem. 2016, 7, 5004–5010. [Google Scholar] [CrossRef] [Green Version]
- Smilgies, D.-M. Scherrer grain-size analysis adapted to grazing-incidence scattering with area detectors. J. Appl. Crystallogr. 2009, 42, 1030–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, H.C.; Lee, C.H.; Lin, M.Z.; Huang, T.W. A comparison between X-ray reflectivity and atomic force microscopy on the characterization of a surface roughness. Chin. J. Phys. 2012, 50, 291–300. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
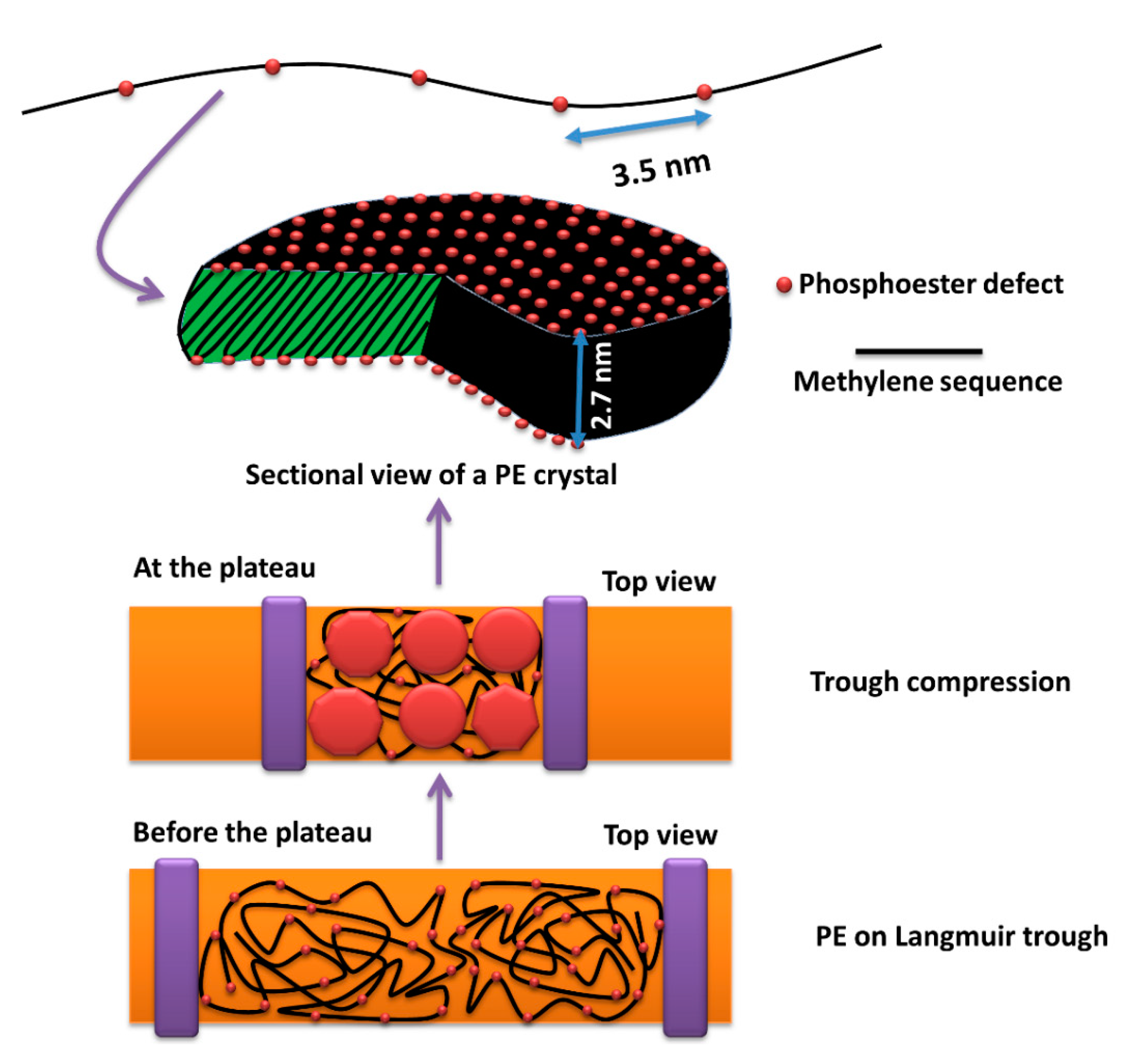{kind=link}
{kind=link}
| Polymer Name and Chemical Structure | Abbreviatedas | Mn* (g/mol) | Mw/Mn* |
|---|---|---|---|
 | Ethoxy-PPE | 9900 | 2.30 |
 | Methyl-PPE-co-decadiene | 8500 | 2.08 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasan, N.; Busse, K.; Haider, T.; Wurm, F.R.; Kressler, J. Crystallization of Poly(ethylene)s with Regular Phosphoester Defects Studied at the Air–Water Interface. Polymers 2020, 12, 2408. https://0-doi-org.brum.beds.ac.uk/10.3390/polym12102408
Hasan N, Busse K, Haider T, Wurm FR, Kressler J. Crystallization of Poly(ethylene)s with Regular Phosphoester Defects Studied at the Air–Water Interface. Polymers. 2020; 12(10):2408. https://0-doi-org.brum.beds.ac.uk/10.3390/polym12102408
Chicago/Turabian StyleHasan, Nazmul, Karsten Busse, Tobias Haider, Frederik R. Wurm, and Jörg Kressler. 2020. "Crystallization of Poly(ethylene)s with Regular Phosphoester Defects Studied at the Air–Water Interface" Polymers 12, no. 10: 2408. https://0-doi-org.brum.beds.ac.uk/10.3390/polym12102408