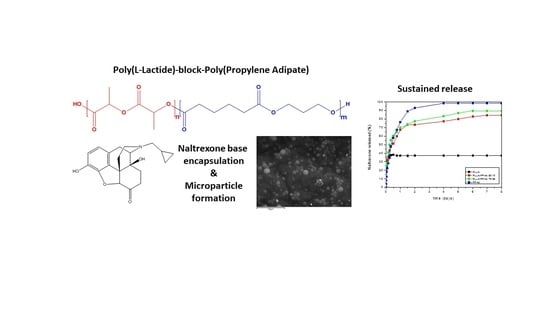
3.1. Synthesis and Characterization of Block Copolymers
PLLA-
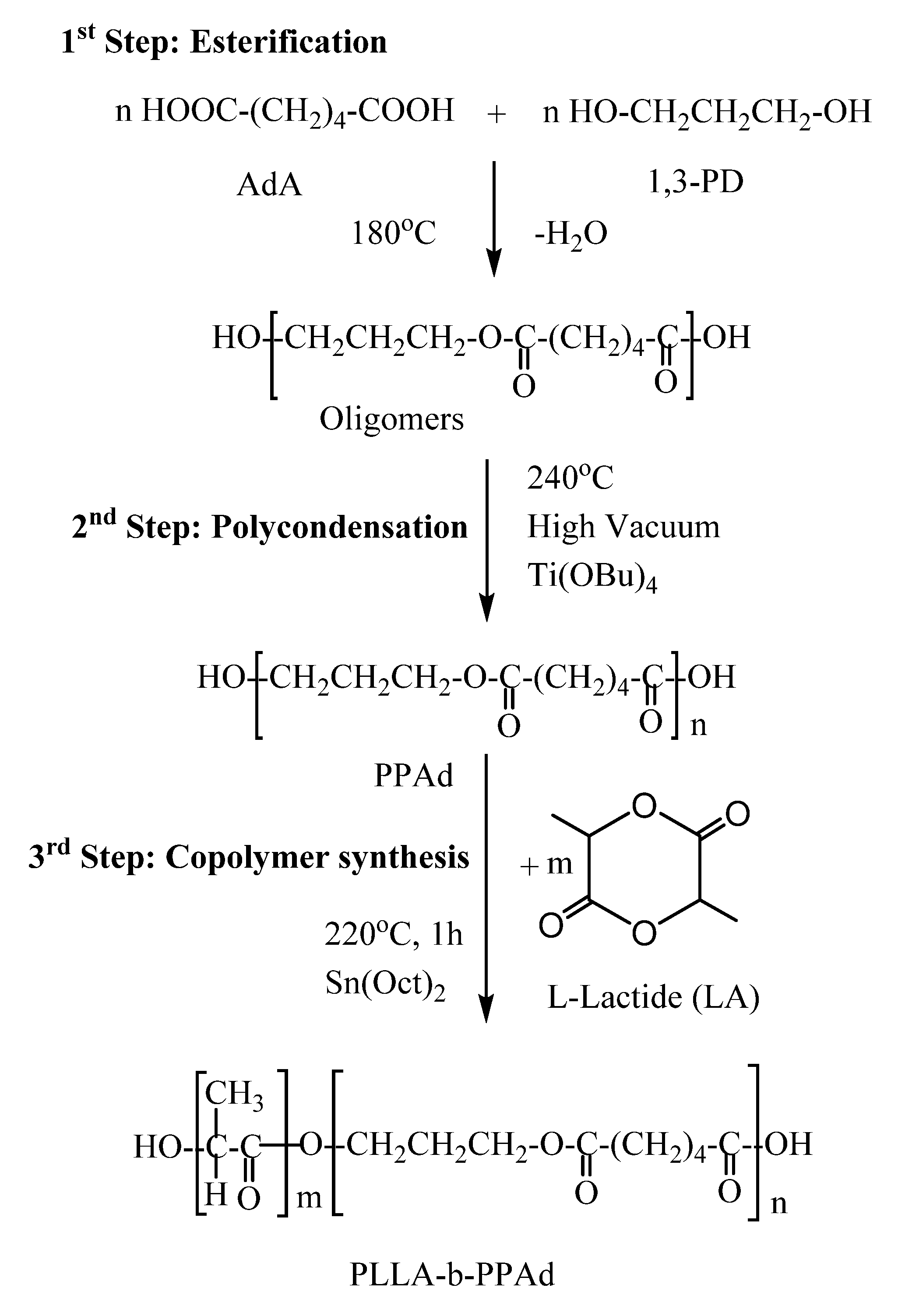
b-PPAd copolymers were synthesized by a combination of melt polycondensation and ring-opening polymerization, as described in
Figure 1. Initially, poly(propylene adipate) (PPAd) homopolymer was synthesized by a two-stage melt polycondensation method [
16,
17]. Briefly, during esterification (1st stage), PPAd oligomers were synthesized and water was removed as a byproduct. During polycondensation (2nd stage), the temperature was increased to 240 °C and reduced pressure was applied in order to build up the molecular weight of the polyester. Water and 1,3-propanediol byproducts were removed by distillation in a graduated cylinder. 1,3-propanediol was added in small excess ensuring that most of the prepared macromolecules acquired hydroxyl end groups. It is necessary to have hydroxyl-terminated macromolecules, as these will act as initiators for the ring-opening polymerization of lactide and carboxylic acid groups cannot initiate this polymerization.
The copolymerization between PLLA and PPAd was carried out at 220 °C in the presence of stannous octoate (Sn(Oct)
2), by ring-opening polymerization of LA, using the prepared PPAd homopolymer as a macroinitiator. After 1 h at atmospheric pressure, vacuum was applied (≈5 Pa) for 15 min to remove any unreacted LA. PLLA-
b-PPAd copolymers, containing 10 and 25 wt% of PPAd were thus synthesized (
Figure 1).
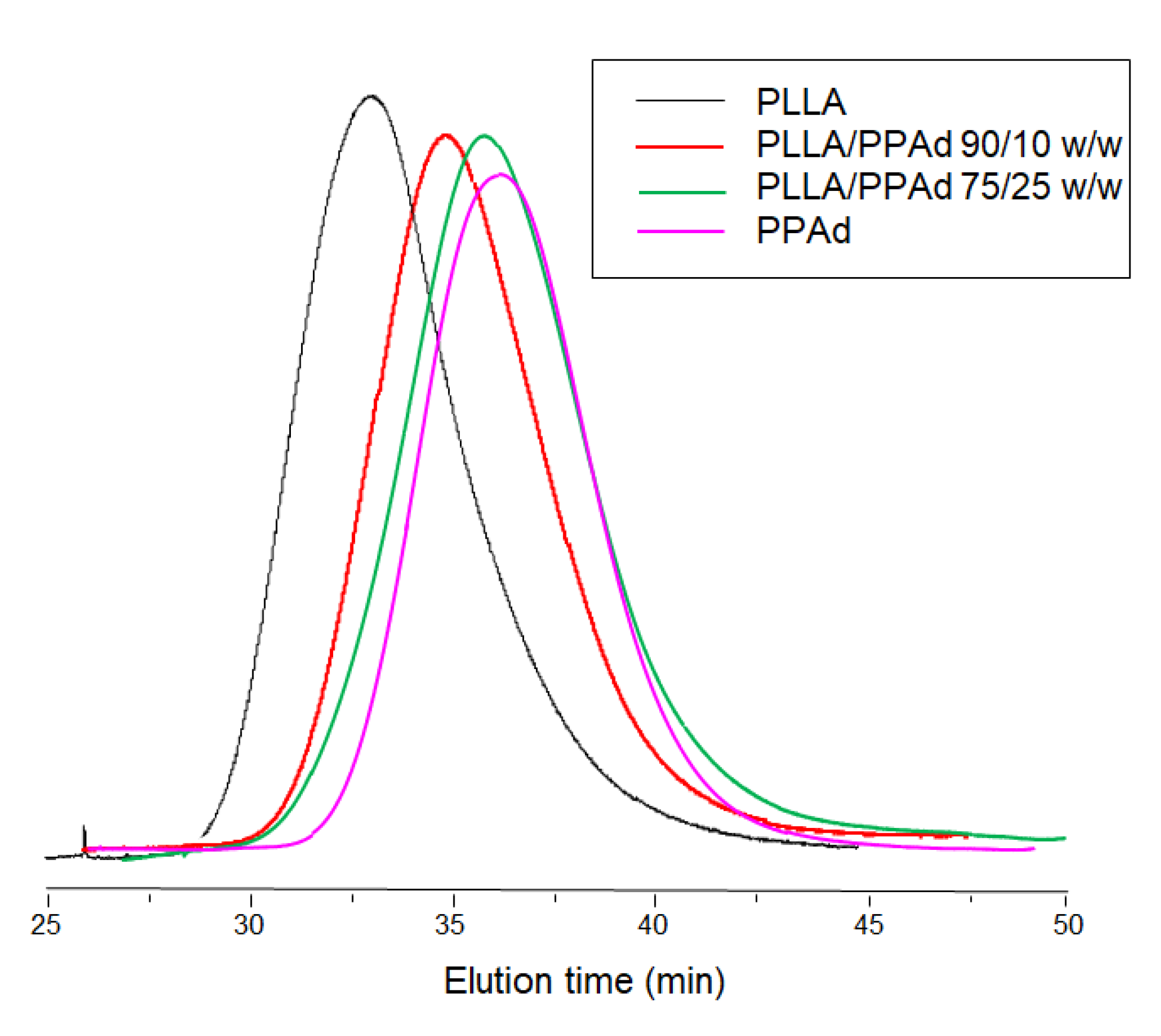
Characterization of the synthesized PPAd polyester by GPC measurements showed an average number molecular weight of 35,000 g/mol and an average weight molecular weight of 50,700 g/mol. The corresponding values for PLLA were 86,000 g/mol and 133,900 g/mol, respectively, while for the copolymers the recorded molecular weights ranged between 47,700–53,700 g/mol, as detailed in
Table 1. Intrinsic viscosities of PLLA and PPAd were found to be 0.99 and 0.38 dL/g respectively while the values for the copolymers ranged between 0.65–0.77 dL/g. It was observed that as the percentage of PPAd increased, the intrinsic viscosity decreased. In other words, the addition of PPAd led to a decrease in the molecular weight of the formed copolymers. This was expected since the LA amount for the preparation of copolymers was reduced and thus shorter chains were obtained. All samples exhibited unimodal distribution in GPC, i.e., only a single broad peak appeared (
Figure 2), with no oligomers being detected. In addition, the low polydispersity index (PDI) values indicate that no transesterification and/or backbiting reactions occurred during the copolymerization [
18].
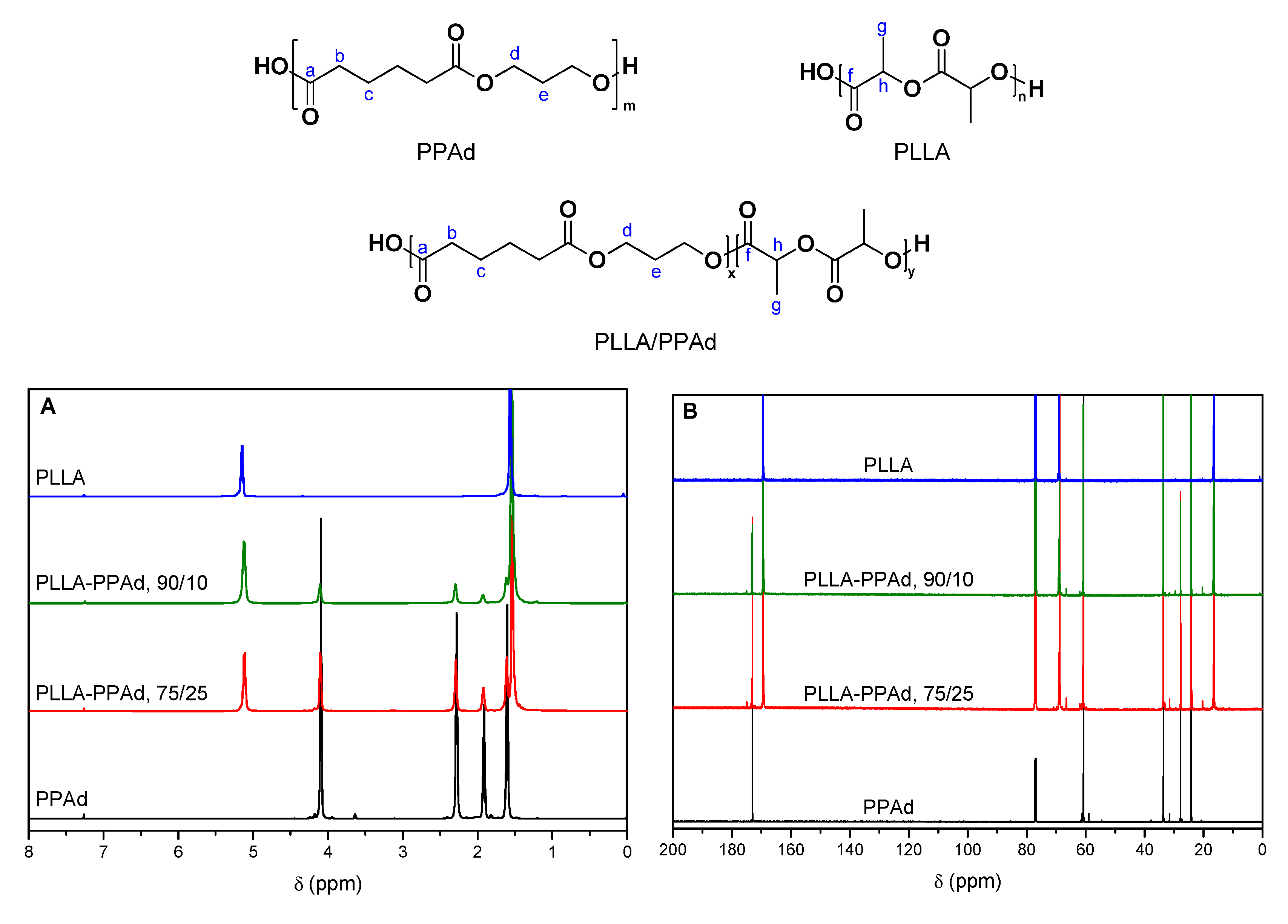
In order to evaluate the successful synthesis of the copolymers, NMR spectra were recorded. The
1H-NMR spectra of PLLA, PPAd, and PLLA-
b-PPAd copolymers are shown in
Figure 3A. The
1H-NMR spectrum of PPAd, exhibits a triple peak at 4.15 ppm corresponding to methylene group
d, a quintuple peak at 1.97 ppm corresponding to methylene group
e, a single peak at 2.33 ppm owing to methylene group
b and a multiple peak at 1.66 ppm attributed to methylene group
c. These peaks have also been documented in previous studies [
16,
17]. Typical resonance signals were obtained for PLLA, i.e., a quadruple peak at 5.17 ppm attributed to methine group
h, and a double peak at 1.6 ppm corresponding to methyl group
g. These peaks are in accordance with the literature [
19,
20].
1H-NMR spectra of copolymers were similar for all the copolymers synthesized, and exhibited the characteristic resonance signals of both PLLA and PPAd: methylene protons
b and
c at 2.29 and 1.62 ppm respectively, methylene protons
d and
e at 4.11 and 1.93 ppm respectively (PPAd segment) and methine and methyl protons
h and
g at 5.13 and 1.54 ppm respectively (PLLA segment). In the spectra of PPAd, the peak at 3.65 ppm is attributed to the protons
d′ which are adjacent to the hydroxyl end-group of PPAd chains. During the polymerization, PPAd chains initiate the ring-opening polymerization of lactide via the OH-terminated end. The disappearance of this peak in the spectra of the PLLA-PPAd copolymers indicates the successful polymerization and the formation of block copolymers. The molar composition of the copolymers was calculated by
1H-NMR spectra comparing the integrations of the CH(
h) group of the PLA unit and the CH
2(
e) group of PPAd. For both copolymers, a better incorporation of PPAd units in the copolymers was observed (15 and 31 wt% instead of 10 and 25 wt%). This could be attributed to some lactide sublimation, due to the high temperature of polymerization. All results are presented in
Table 1.
In the
13C spectrum (
Figure 3B), the carbonyl bond of the ester groups are observed at 169.5 ppm (C=O(
f), PLLA) and 173.1 ppm (C=O(
a), PPAd). Peaks at 24.2 (CH
2(
c)), 27.8 (CH
2(
e)), 33.7 (CH
2(
b)) and 60.8 ppm (CH
2(
d)) are attributed to the PPAd unit, while the peaks at 16.6 (CH
2(
g)) and 68.9 ppm (CH
2(
h)) are attributed to the PLLA unit. Similarly to the
1H-NMR spectra, the disappearance of the peak around 58.9 ppm indicates that PPAd chains react via the OH-terminated end. The absence of peaks between the two carbonyl
13C resonance signals further corroborates that block and not random copolymers were obtained.
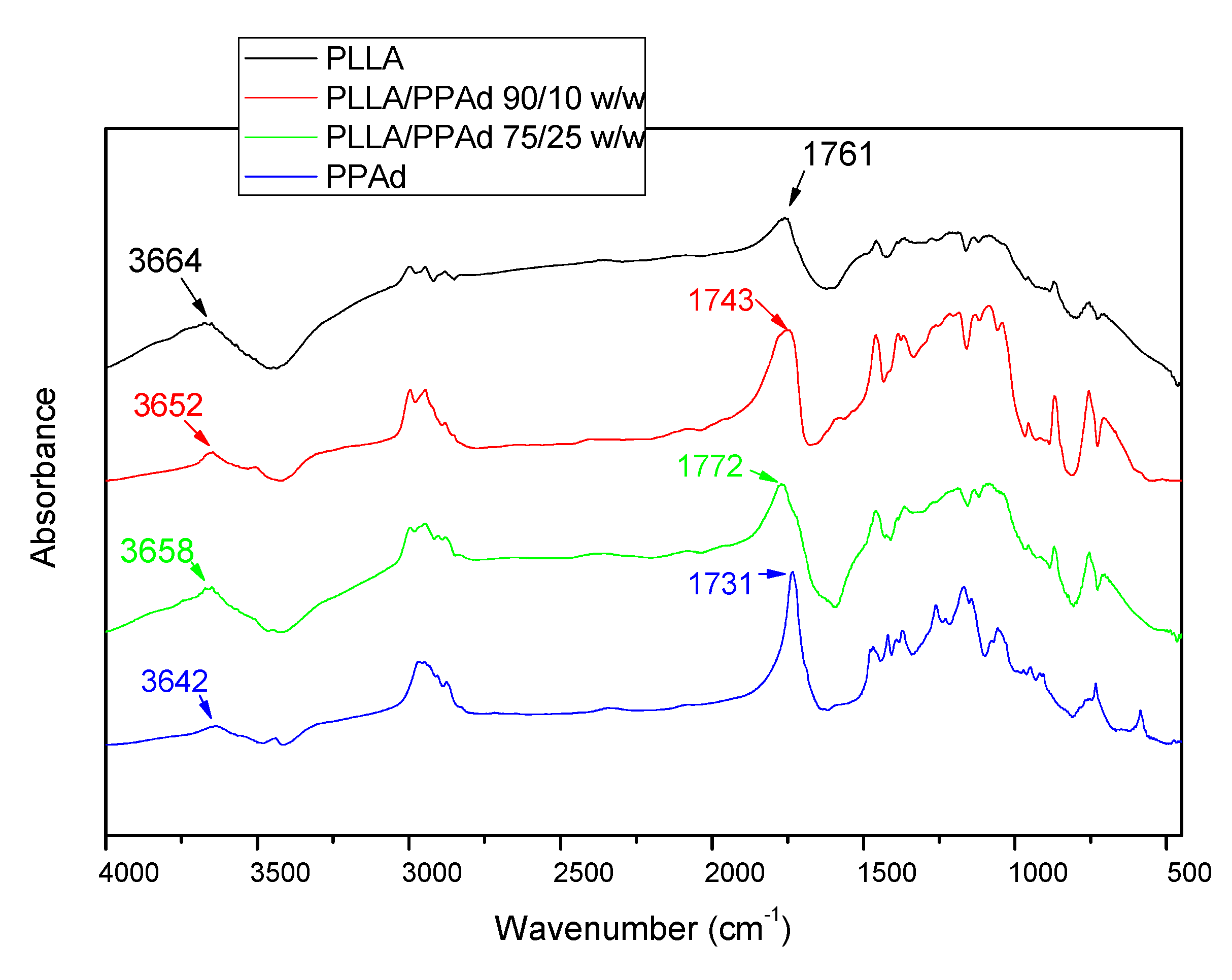
The formation of copolymers was also confirmed with FTIR spectroscopy.
Figure 4 shows the FTIR spectra of neat PLLA, PPAd and their copolymers. PLLA spectra exhibits a broad absorption peak in the range 3700–3600 cm
−1 which is assigned to ν(O–H) vibration modes. Hydrogen association can easily occur in hydroxyl groups, resulting in a shift of the corresponding absorption peak to lower wavenumbers and a gradual widening with the increase of the association degree [
21]. Furthermore, the two peaks at about 2951 and 3003 cm
−1 correspond to the ν(C–H) vibration modes of methyl groups. Besides, the δ(C–H) modes of methyl appear around 1458 and 1362 cm
−1. In the ν(C=O), ν(C–O) and ν(C–C) regions, PLLA exhibits sharp absorption peaks at 1760, 1081, and 1192 cm
−1 respectively. PPAd exhibits a peak at 3000–2900 cm
−1 corresponding to the ν(C–H) vibration modes of the methylene groups and sharp absorption peaks at 1731, 1254, and 1192 cm
−1 corresponding to ν(C=O), ν(C–O) and ν(C–C) respectively. No significant absorption was observed around 3400–3700 cm
-1, confirming a reasonably high polymerization degree. The main peaks appearing in the homopolymer spectra are retained in the copolymer spectra, without the appearance of any new peaks. These spectra exhibit sharp peaks between 1730 and 1775 cm
−1 due to vibration of the different ester bonds ν(C=O) and wider peaks at 1200–1100 cm
−1 attributed to the ν(C–O) bond, indicating that copolymer formation has taken place [
22].
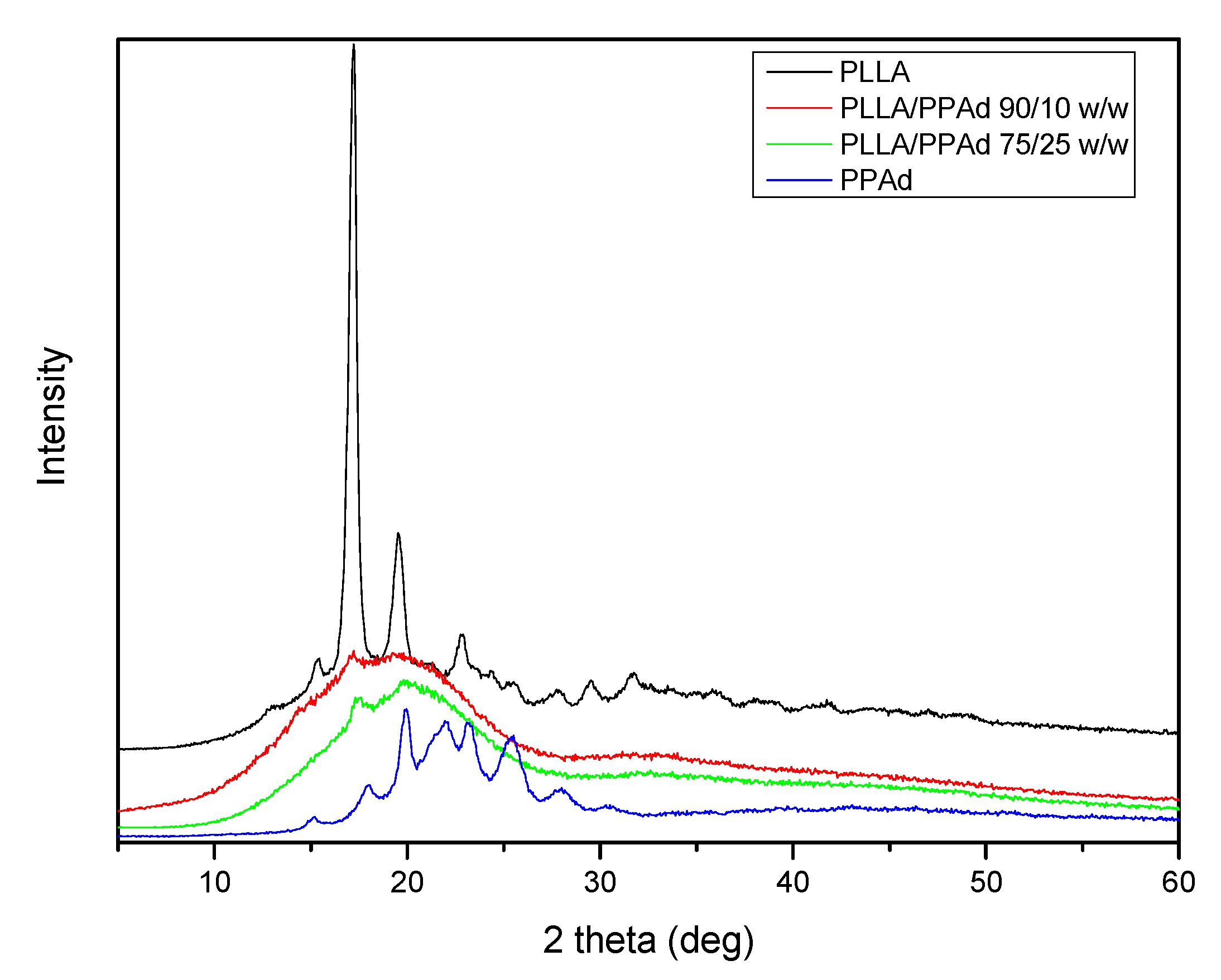
The physical state of the prepared copolymers is very crucial for drug release and was studied by XRD and DSC.
Figure 5 shows the X-ray diffraction patterns for the synthesized polyesters. Neat PLLA is a polymer with a high degree of crystallinity with characteristic peaks at 2θ = 17.20, 19.60, 22.81 and 31.60°. PPAd shows broad peaks at 2θ = 17.91, 19.90, 22.01, 23.20, 25.40 and 30.50° with low intensity, thus exhibiting a lower degree of crystallinity. Concerning the copolymers, only a small peak at 2θ = 17.20° appears, indicating that the synthesized copolymers have a reduced crystallinity compared to both homopolymers.
The degree of crystallinity of all samples was calculated by wide angle X-ray diffraction patterns using the relative areas under the crystalline peaks (
Ac) and the amorphous background (
Aam) using Equation (6), according to Lu and Hay [
23], and are summarized in
Table 1. As expected, PLLA showed the highest degree of crystallinity, with a value equal to 50.6%. In the copolymers the degree of crystallinity decreased with increasing PPAd content. This was expected since PPAd has a low degree of crystallinity (26%) and its incorporation into the macromolecular chains hinders the crystallization of PLLA. Consequently, the copolymer with the highest PPAd amount (25 wt%) has the lowest degree of crystallinity.
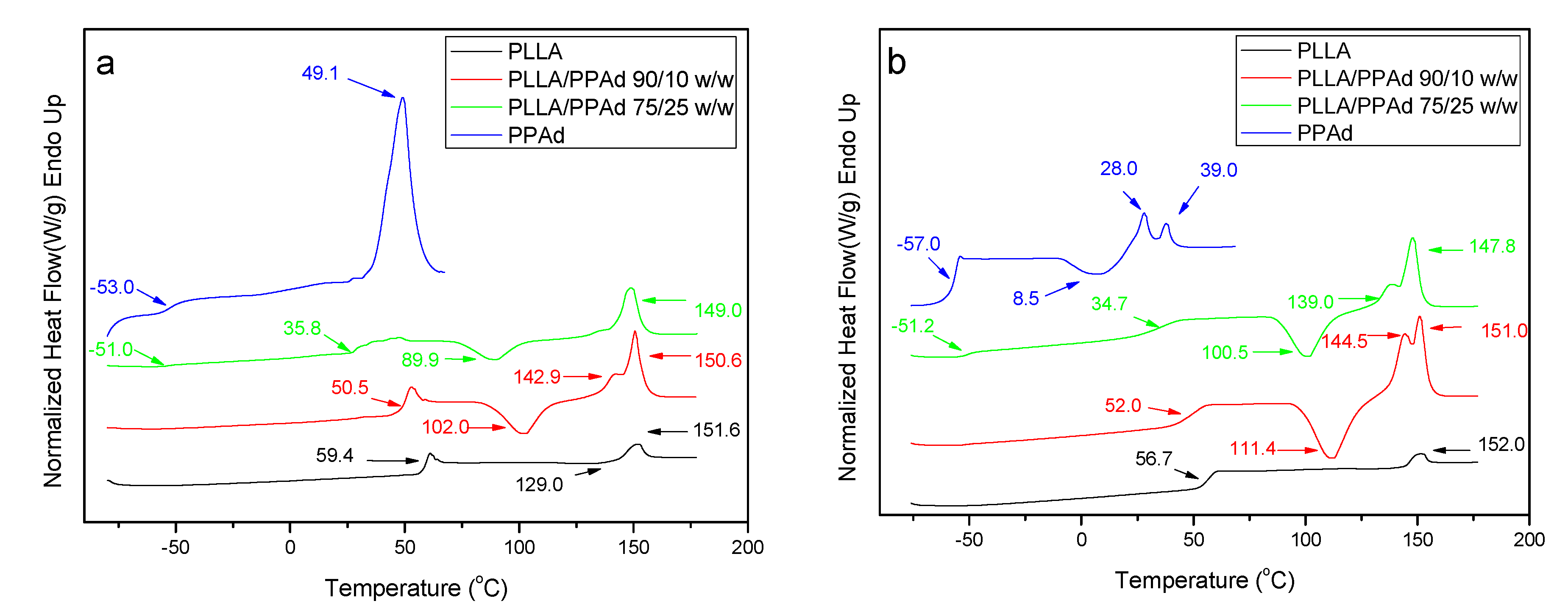
The thermal properties of all prepared polyesters were also studied. PPAd and PLLA are semicrystalline materials, which showed
Tg values at 59.4 and −53 °C, and
Tm values at 151.6 and 49.1 °C, respectively (
Figure 6A). The copolymer with 10 wt% PPAd exhibits also a single
Tg at 50.5 °C, which is very close to that of neat PLLA. On the other hand, copolymer PLLA/PPAd 75/25
w/
w has two glass transition temperatures at −51 °C and 35.8 °C (
Table 2). The former corresponds to the PPAd sequences and the latter to the PLLA sequences. This is in good agreement with the literature. Indeed, for a block copolymer consisting of two heterogeneous phases, two glass transition temperatures corresponding to those of the homopolymers are expected [
24]. Two
Tg values should also have been recorded for the copolymer with 10 wt% of PPAd. However, since the length of the PLLA segment is expected to be much higher than that of the PPAd segment, the
Tg of PPAd is difficult to be observed. The highly amorphous character of these copolymers, as already suggested from XRD studies, was also confirmed by the appearance of a cold crystallization peak at 102 °C and 89.9 °C for polyesters containing 10 and 25 wt% PPAd, respectively. The recorded
Tm values of PLLA sequence were at 150.6 and 149 °C.
Completely amorphous samples were obtained after melt quenching and their thermograms are presented in
Figure 6B. After the second heating process all synthesized copolymers exhibit a double peak at the melting point. The presence of the double peak is due either to the formation of two different crystals formed after the first melting or to the formation of a constituent copolymer where each peak corresponds to a different cluster [
16]. The
Tg and
Tcc values are very close to the ones recorded during the first heating with some small differences. As PLLA and PPAd are semicrystalline, the presence of both peaks is expected in the DSC thermographs of the copolymers [
25]. However, the
Tm of PPAd was not recorded, due to its low crystallization rate. This is also confirmed by the XRD of PLLA-
b-PPAd copolymers in which only some peaks of PLLA are recorded (
Figure 5).
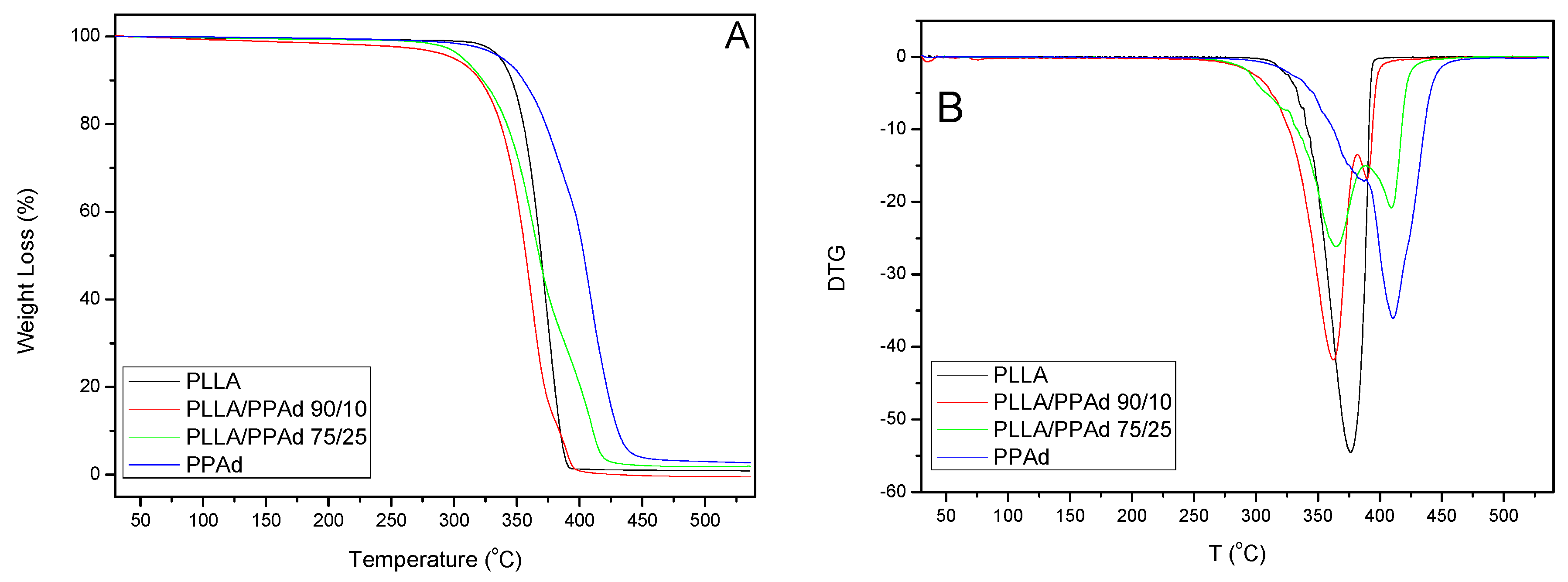
Thermal degradation of neat PLLA, PPAd and PLLA-
b-PPAd 90/10 and 75/25
w/
w copolymers was studied by determining their mass loss during heating, with thermogravimetric analysis (TGA). The mass loss (TG%) and the derivative mass loss (DTG) curves at a heating rate of 10 °C/min are presented in
Figure 7. With regard to thermal degradation mechanisms, PPAd is degraded in 2 steps with a maximum decomposition temperature of 410 °C. Aliphatic polyesters generally undergo β- and α- hydrogen bond scissions, yielding low molecular weight volatile products, such as CO
2, H
2O, aldehydes, ketones, and allylic products (H
2C=CH–CH
2R) [
26]. In the case of PLLA, only one degradation step was observed, with a maximum decomposition temperature of 376 °C. The main reaction pathway is an intramolecular transesterification, affording cyclic oligomers [
27]. In addition, acrylic acid from cis-elimination as well as carbon oxides and acetaldehyde from fragmentation reactions have also been observed. The onset of thermal degradation of neat PLLA, in spite of exhibiting a faster thermal degradation than the PLLA/PPAd copolymers, is observed at a higher temperature than for copolymers. PLLA/PPAd 90/10 and 75/25
w/
w copolymers follow a two-stage degradation. The lactide-rich sequences degrade first, and the PPAd-rich domains later. However, in neat PLLA the average sequence length of crystallizable lactide units is much larger, leading to a higher and more compact crystalline fraction than in copolymers, hence the onset of thermal degradation is delayed in neat PLLA because of the lower accessibility of lactide units when arranged in close compact crystallite aggregates [
28].
Table 3 shows the temperatures at which polyesters exhibited 2%, 50% and 98% mass loss, as well as temperatures with the maximum rate of mass loss.
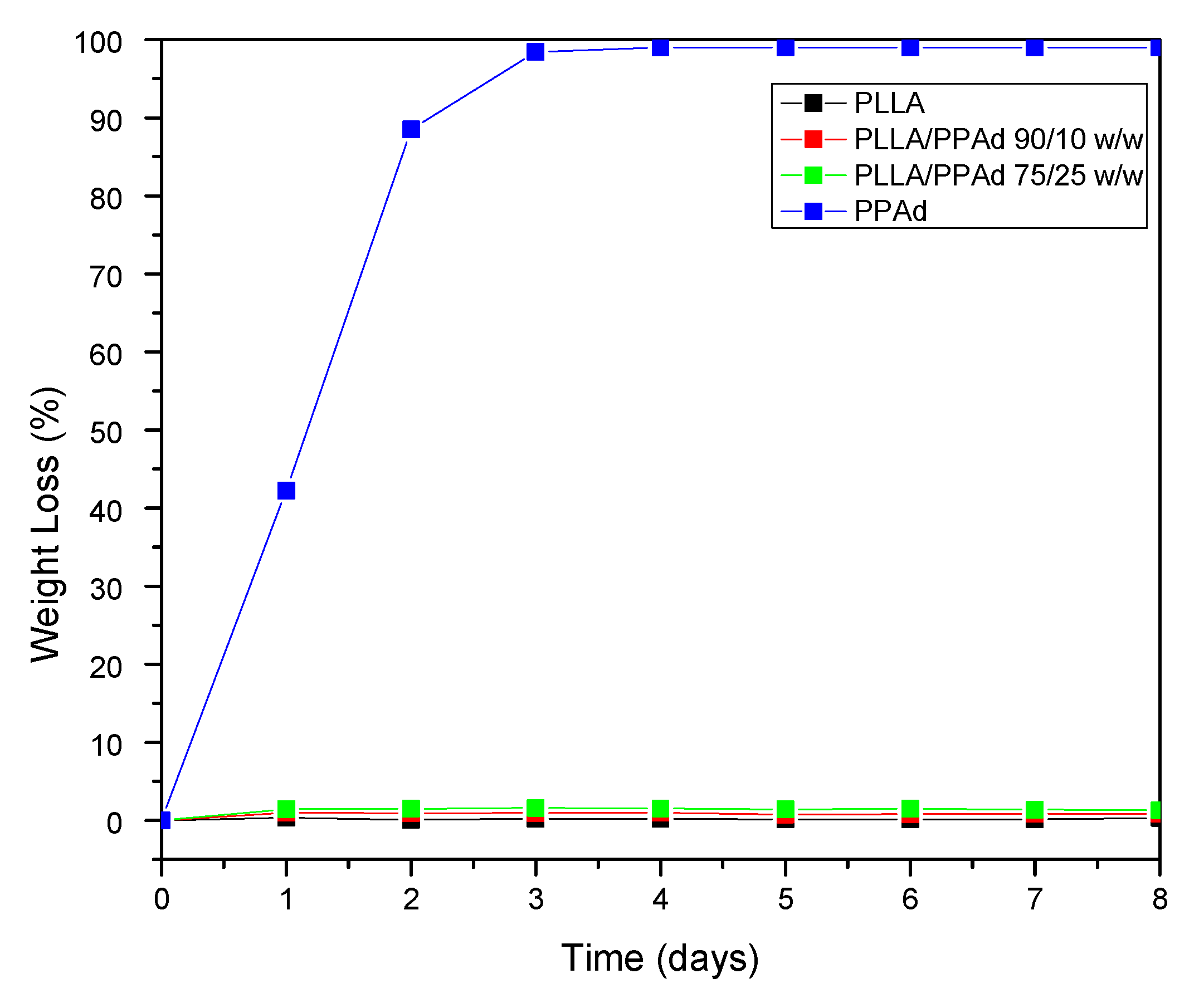
Polyester erosion via the hydrolysis of the ester bonds is one of the main mechanisms of drug release from such polymeric-based systems. Hence, the evaluation of hydrolysis is of crucial importance. As mentioned earlier, the in vivo hydrolysis of PLLA is slow and hence time-consuming [
29]. It is thus a common practice to evaluate the hydrolytic degradation in the presence of enzymes [
17]. More specifically, in the present work, the enzymatic hydrolysis of PLLA, PPAd and their copolymers was studied using a mixture of
Rhizopus oryzae and
Pseudomonas cepacia lipases. These lipases are activated by adsorption on hydrophobic surfaces and consequently are able to cleave ester bonds in the solid phase. As illustrated in
Figure 8, neat PLLA showed only limited mass loss during hydrolysis in the presence of enzymes, with approximately 0.3% of the initial mass being lost within the first eight days. No further changes in PLLA mass loss were recorded for up to 18 days (data not shown). On the contrary, PPAd was fully hydrolyzed within three days in the presence of enzymes, indicating that the prepared polyester is fully biodegradable in a short time. The different behavior of polyesters during hydrolysis can be attributed to several factors such as differences in molecular weight, hydrophilic/hydrophobic balance, degree of crystallinity, crystal morphology and melting point [
30]. With regard to PLLA/PPAd copolymers, the recorded mass loss rates during enzymatic hydrolysis were near the rates recorded for neat PLLA. Specifically, results in
Figure 8 show that as the PPAd content increased in the copolymers, the enzymatic hydrolysis rate slightly increased with approximately 0.9%, and 1.4% of mass loss being recorded within three days for copolymers having 10 and 25 wt% PPAd, respectively. Therefore, it can be safely assumed that the PPAd content of copolymers affected the enzymatic hydrolysis only to a limited extent. The molecular weight as well as the high
Tm, which are close to neat PLLA, had probably a more important impact.
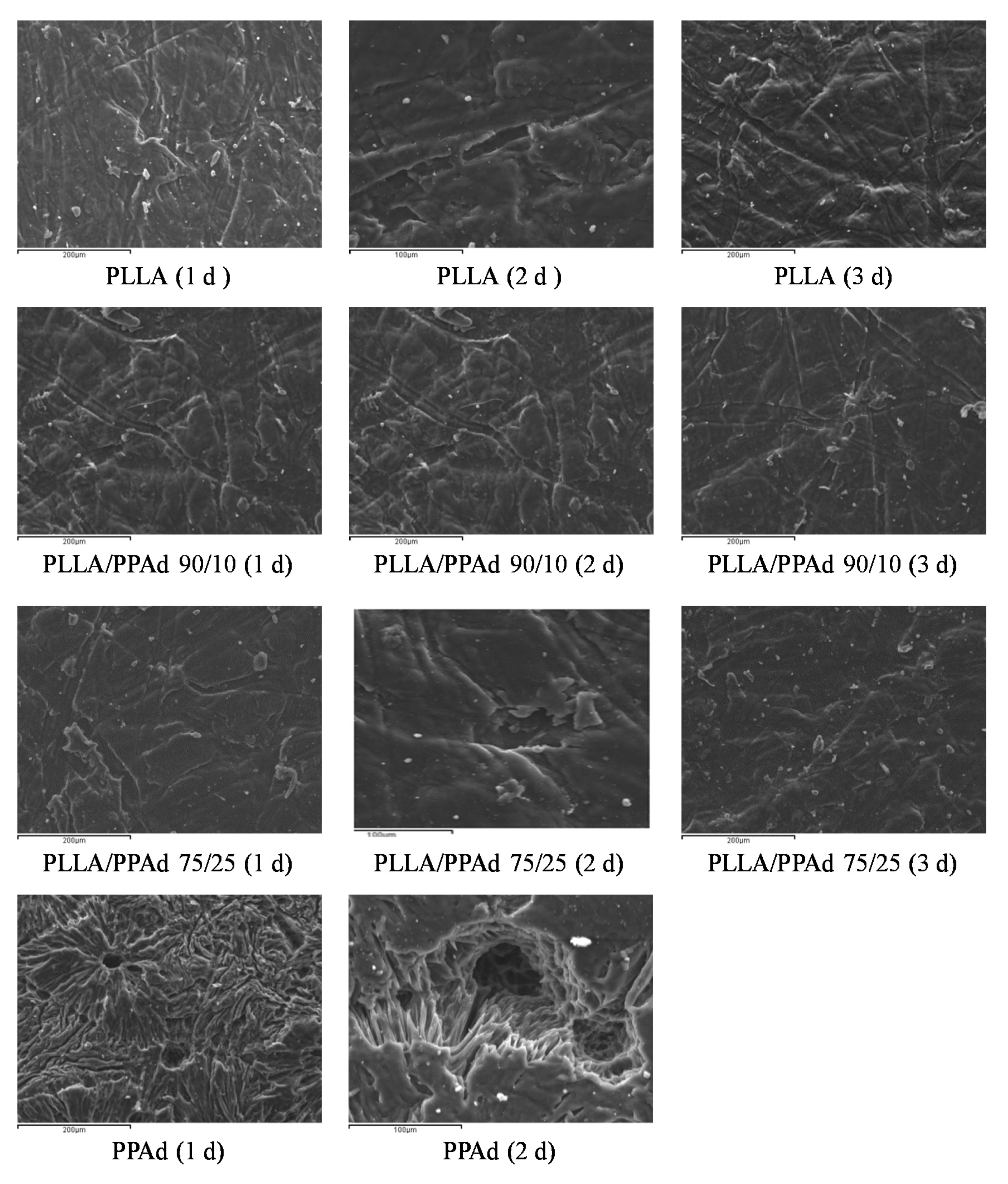
The morphology of enzymatically hydrolyzed polymers was further examined by SEM.
Figure 9 shows the photos taken during the first three days of enzymatic hydrolysis. As shown, the PLLA sample did not change significantly, an expected observation in accordance with the weight loss data. In the case of neat PPAd signs of enzymatic hydrolysis were obvious from the first day: the surface appears to have holes that are progressively growing. Furthermore, the surface became rough with the presence of cracks probably owing to the amorphous fragments of PPAd [
30]. With regard to PLLA/PPAd copolymers, structural deterioration on the surface texture of the films was not observed and an appearance close to neat PLLA is clear. The results of SEM micrographs are in full agreement with the mass loss measurements.
3.2. Microparticles Characterization
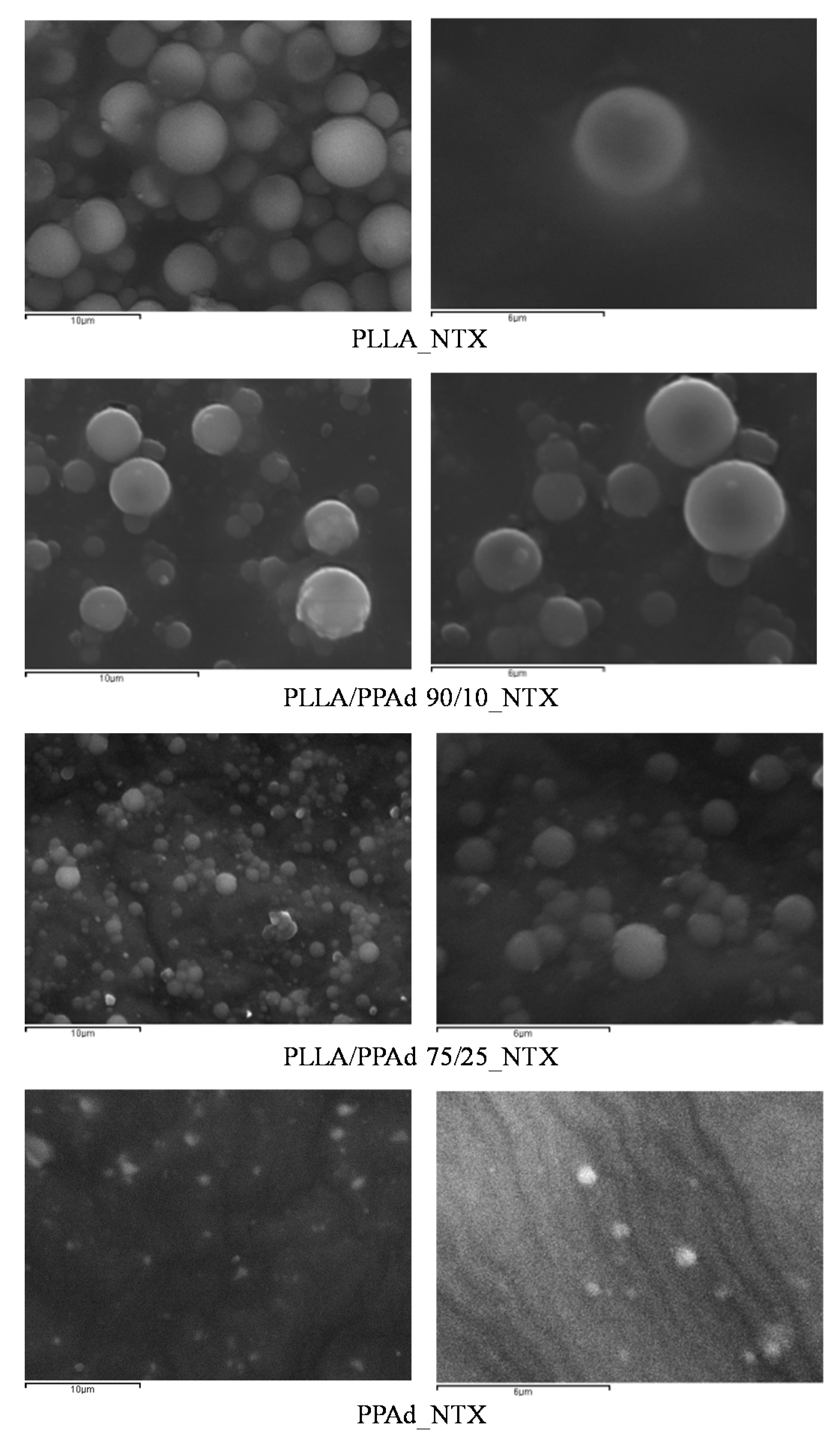
The prepared copolymers were used for the microencapsulation of NTX drug, applying the single emulsification method. Both the synthesized polyesters and NTX are soluble in DCM. Microparticles were obtained as evidenced by the SEM photos. As observed in
Figure 10 all the microparticles had a spherical morphology with smooth surfaces and without agglomeration. Microparticles prepared using PLLA had sizes ranging between 2.6 and 4.5 μm. The sizes for the other microspheres ranged between 2–4 μm for PLLA-
b-PPAd 90/10, 0.8–2 μm for PLLA-
b-PPAd 75/25 and 0.4–0.8 μm for PPAd. The size of the microparticles was affected by the PPAd content in the copolymer; i.e., as the content of PPAd increased, the size of the microparticles decreased. Concerning homopolymers this could be attributed to the low molecular weight of PPAd (lower than half the molecular weight of PLLA). Regarding the copolymers, the reduced size could be attributed either to the molecular weight compared with that of PLLA, either to the length of the block segments. Indeed, the length of the PLA segment in copolymer 90/10 is expected to be higher than in copolymer 75/25 and as the length of PLLA segment decreased, smaller particles were formed.
Table 4 presents the yields of microparticles, drug loading, and entrapment efficiency (EE). The yield of microparticles varied from 54.8% (in the case of block copolymer 75/25) to 62.5% (in the case of pure PLLA), indicating high process efficacy. Drug loading varied from 8.75% (in the case of PLLA) to 3.63% (in the case of pure PPAd) and % EE varied from 35.00% in the case of pure PLLA to 14.53% in the case of pure PPAd. Several factors may affect both drug loading and % EE, such as the affinity of the loaded drug with the polyesters, the hydrophobicity of the polymer matrix, the drug solubility in water, drug-drug interactions (i.e., its ability to self-aggregate), etc. It was observed that as the PPAd content increased, the drug loading and EE decreased. This may be attributed to the stereochemical inhibition of NTX and PPAd, as NTX has a relative rigid structure while PPAd is more flexible.
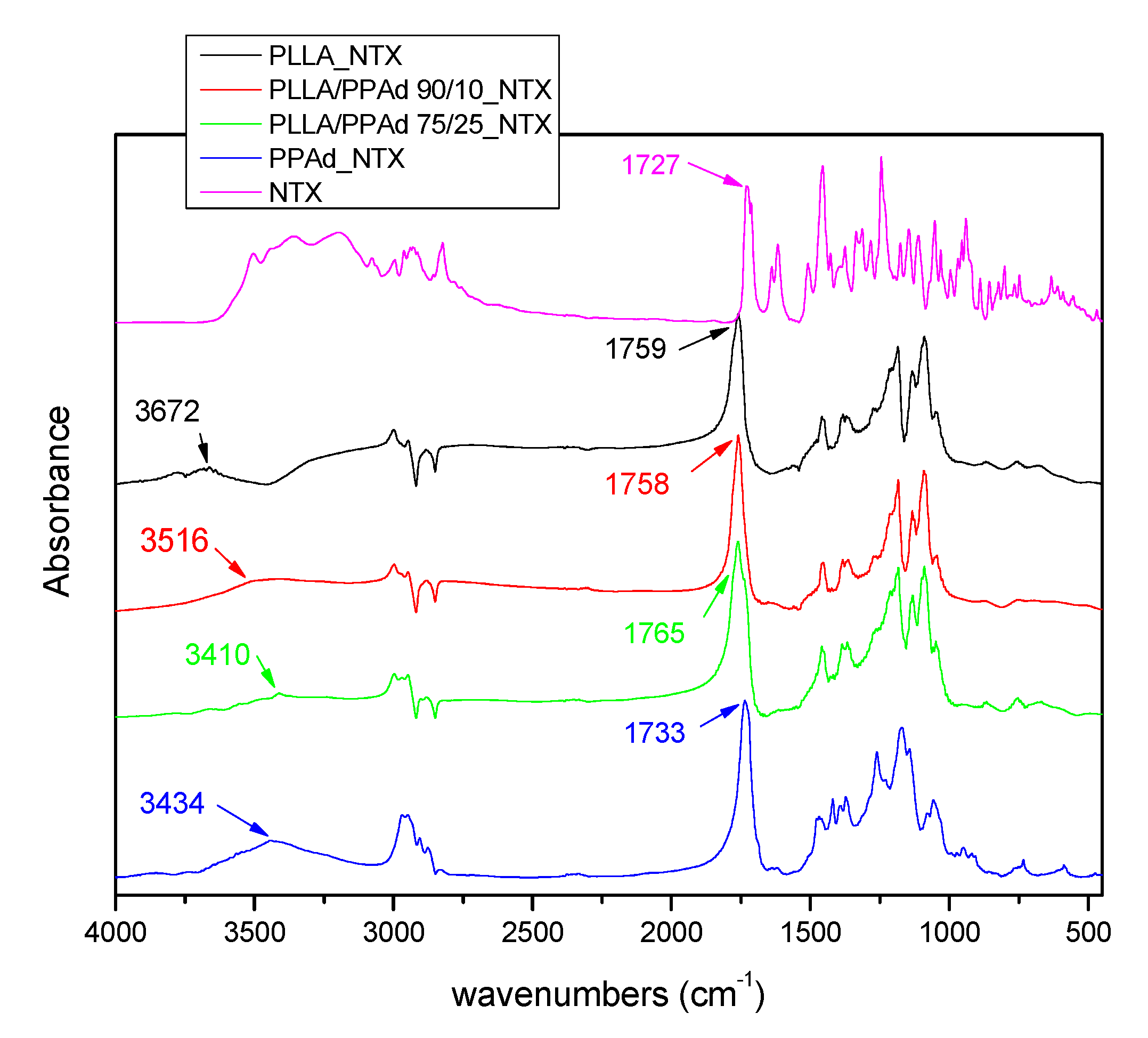
API solid state properties and physical state have an important role in the chemical and physical stability of the prepared formulations. FTIR was used in order to examine a possible bond formation between the drug and the polyesters in the prepared microparticles. FTIR spectra of NTX-loaded microparticles (
Figure 11) showed all the characteristic peaks of NTX and the copolymers. In the FTIR spectrum of NTX, the absorption band at 3000–3100 cm
−1 is assigned to the ν(C–H) vibrations of the benzene rings, peaks at 1508 and 1617 cm
−1 to the vibration of ν(C=C) bonds. In the ν(C–C) and ν(C–O) regions, NTX exhibits sharp absorption peaks at 1456 cm
−1 and 1239 cm
−1 respectively, a peak at 1727 cm
−1 corresponding to its ν(C=O) vibration modes, and a broad peak in the range 3200–3500 cm
−1 due to the existence of OH groups [
11]. After the incorporation of NTX, the FTIR spectra of microparticles showed shifts in the region of OH and C=O groups. For example, in PLLA_NTX, a shift was observed from 3664 to 3672 cm
−1, i.e., to higher wavenumbers, indicating that interactions took place with NTX. Additionally, shifts were also observed in the area of C=O, indicating hydrogen bond formation. Similar shifts were noticeable for all microparticles showing that strong interactions between the drug and the polymeric matrices took place in all cases.
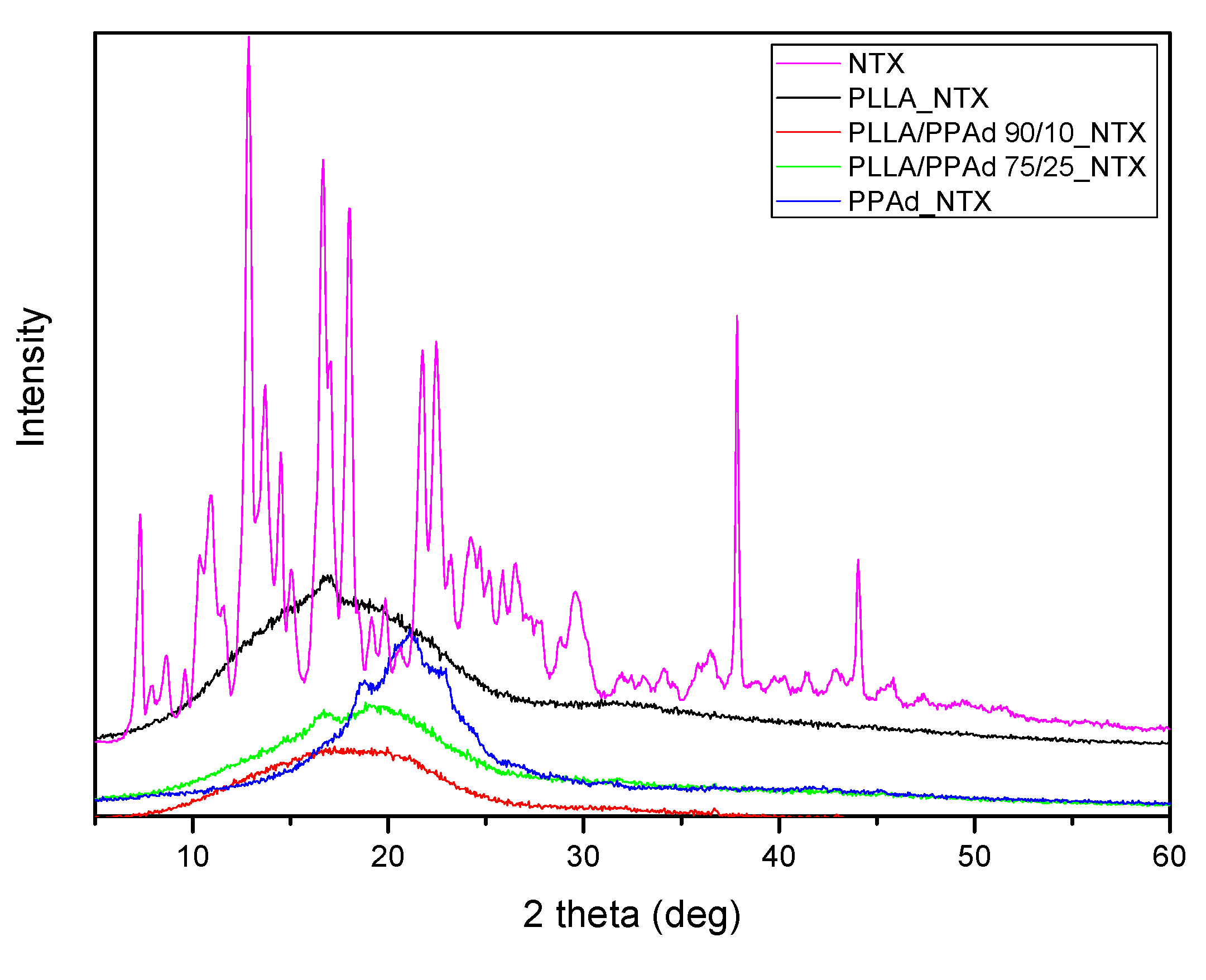
XRD was used in order to examine the crystallinity of NTX before and after its encapsulation. As it can be observed in
Figure 12, NTX is a highly crystalline drug with two sharp peaks at 2θ = 12.9° and 16.6°, three weaker ones at 21.6°, 22.5° and 37.7° and some much smaller ones [
31]. After encapsulation in microparticles no crystallinity of the drug was observed. An exception was the microparticles prepared with PLLA where a small peak at 16.7° was observed. This peak could be attributed either to NTX, meaning that the drug is in a crystalline state in microparticles, either to another type of crystalline formation of the polymer, due to the fact that PLLA has a sharp peak at 17.2°, and the presence of NTX could affect its structure. In microparticles prepared with PPAd it seems that a semicrystalline formulation was obtained mainly ought to the polymer rather than NTX. DSC was further used to confirm these observations.
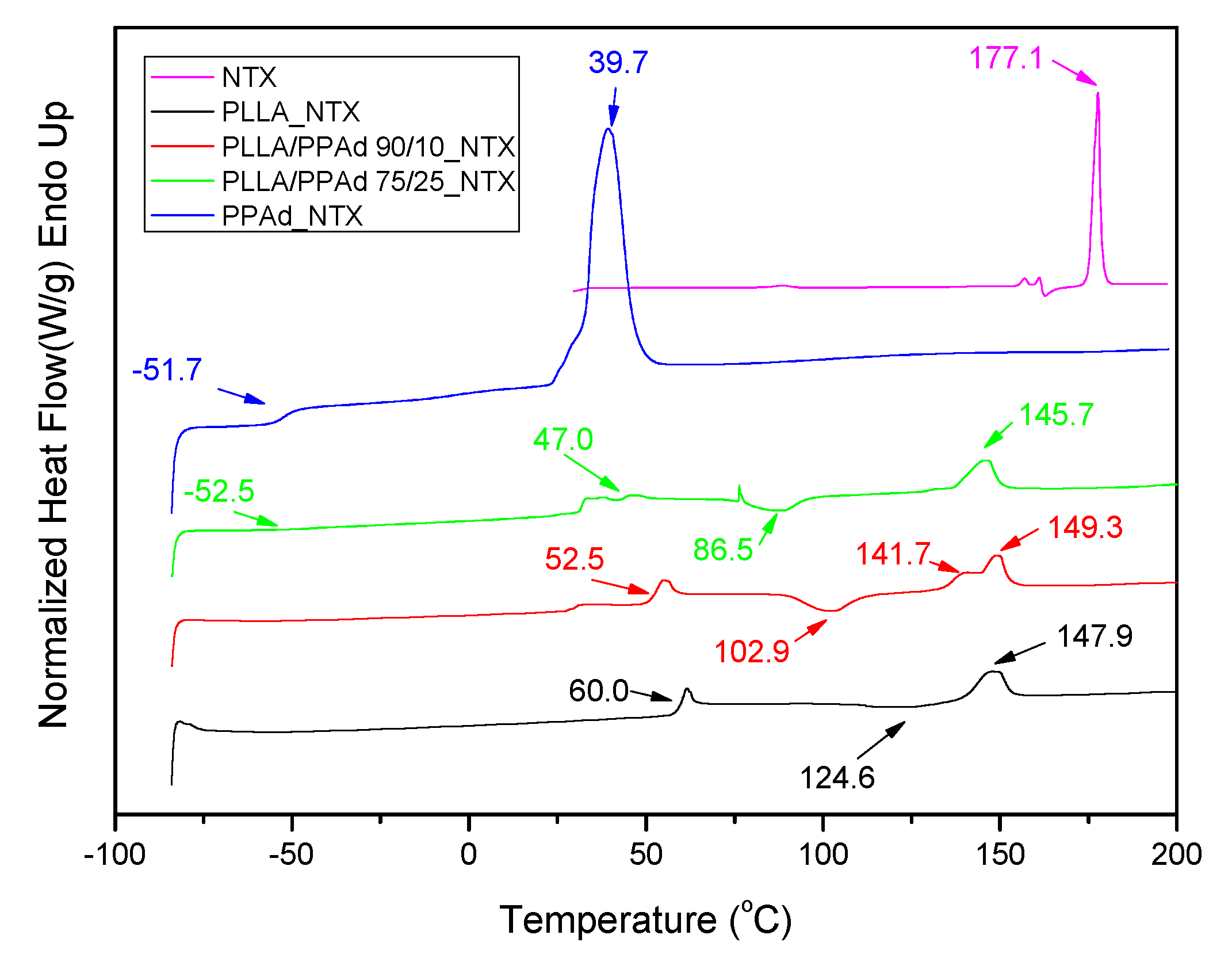
As shown in
Figure 13, a sharp endothermic peak is present for NTX at 177.1 °C [
11]. Concerning the DSC curves of the prepared microparticles, the peak of
Tm of NTX was not observed in any formulation indicating that the drug is present in an amorphous state in the microparticles, as was already suggested by XRD. Thermal properties of the polymers after NTX encapsulation, were also studied and some differences compared with neat polymers were observed. In PLLA microparticles, the
Tcc and
Tm values of PLLA changed from 129.0 and 151.6 to 124.6 and 147.9 °C, respectively, after NTX encapsulation. Similar changes in the
Tg and
Tm values of PPAd in microparticles were also observed.
Tg and
Tm of PPAd shifted from −53.0 and 49.1 °C to −51.7 and 39.7 °C, respectively, showing that the NTX also affected PPAd’s structure. For microparticles prepared by copolymers, changes in their characteristic temperatures were also observed;
Tg related to PLLA segment shifted from 50.5 to 52.5 °C, after NTX incorporation in PLA-b-PPAd 90/10
w/
w, and its value appeared at 47.0 °C in PLA-b-PPAd 75/25
w/
w. Concerning the T
m value due to the PPAd segment it shifted from 35.8 to 36.1 °C in PLA-b-PPAd 75/25
w/
w and appeared at 30.6 °C in PLA-b-PPAd 90/10
w/
w. From all these it is clear that drug loading affects slightly the thermal properties of the polymers.
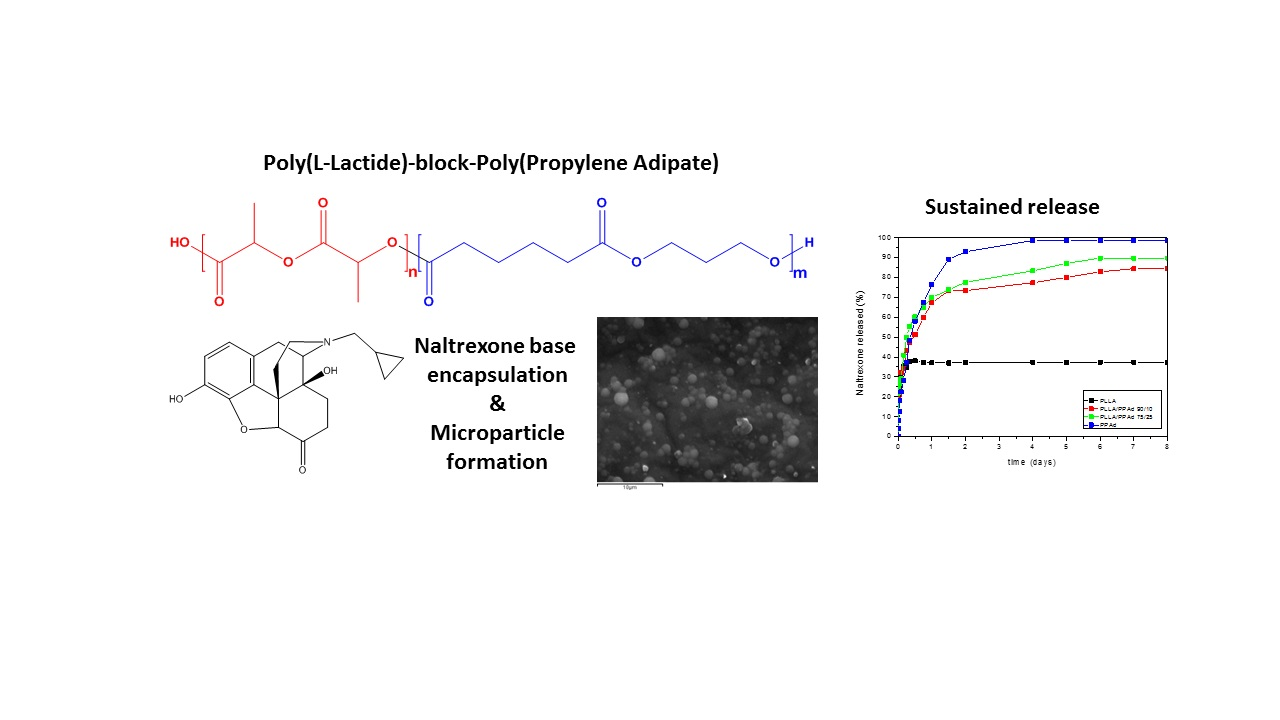
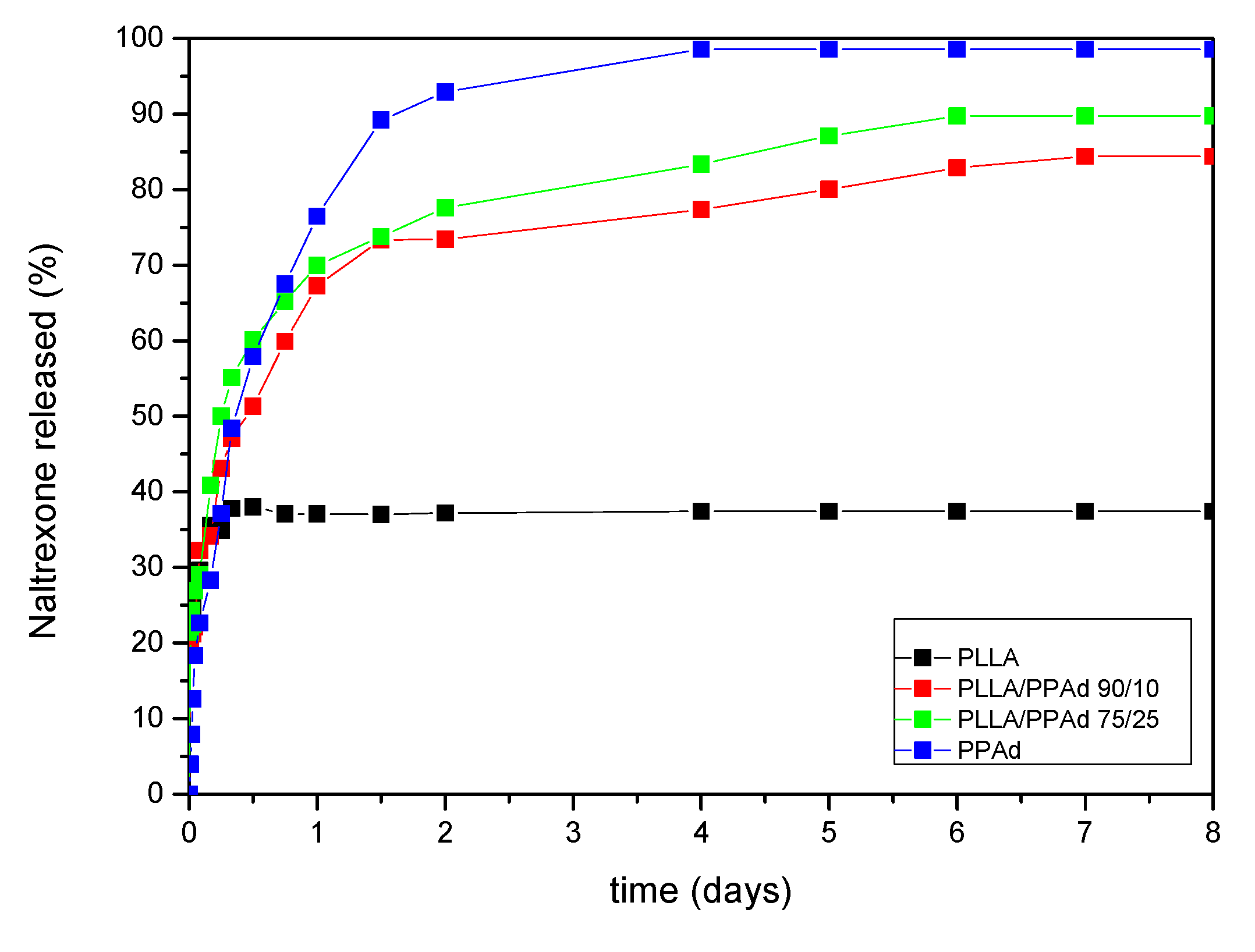
Naltrexone’s release profile from the microparticles was studied in phosphate buffered saline, at pH 7.4, containing Tween 20 (final concentration 0.2%
v/
v). The dissolution profiles are shown in
Figure 14. As it can be observed, there is an initial burst effect in all formulations in the first two hours reaching at about 30%. Thereafter each polymer showed a different behavior. Concerning PLLA, 36% of NTX was released from the microparticles, the release lasted half a day with no further changes after that time. PPAd exhibited a three-stage release. A burst release, as referred previously, with 30% release within the first two hours, followed by a second stage, with a reduced release rate. A total release of 93% was observed within two days. Finally, in the last stage, which lasted until the fourth day, a release of 98.5% was reached. The copolymers showed intermediate release rates, between neat polymers, with PLLA-
b-PPAd 90/10 and 75/25 release lasting six and five days in which 82% and 90% of the drug was released, respectively. According to these release profiles it is clear that the prepared copolymers have a better profile, compared with PLLA, and could be promising matrices for long-acting release formulations of naltrexone for at least one week.
Release profile is affected by many parameters such as the crystallinity of the drug, the microparticles size, diffusion and thermal properties of polymers. The drug was amorphous in all microparticles, a feature that enhanced its dissolution. The initial burst effect could be attributed to NTX located at the surface of the microparticles, since NTX is hydrophobic and could not be removed during washing steps. After that, diffusion of the drug took place leading to slower release rates [
16,
17]. Concerning PLLA, Cha and Pitt prepared microparticles of PLLA, PLGA and PCL of Naltrexone base by single emulsification method [
32]. It was found that release of NTX from PLLA microparticles was negligible, probably due to steric accessibility of the drug. This result is in agreement with the observation in our study where after the first two hours, no significant release was observed from PLLA microparticles. The same team also used PCL in microparticles preparation and found that NTX was completely released in two days. This was attributed to Fickian diffusion, due to the low
Tg of the polymer [
32]. In analogy to PCL, in the present work, NTX release from PPAd microparticles reached 98% in 4 days. This could be due to the low
Tm and
Tg of the microparticles (
Tg was found to be −51.7 °C, a value much lower than 37 °C where the dissolution study was conducted).
The diffusion of NTX through the amorphous polymeric matrix appears to be easier due to the higher mobility of the polyester macromolecules in the amorphous state, allowing an easier penetration of the dissolution medium and, as a result, a faster drug release. Moreover, the thermal properties of the polymer can also affect drug release. When the polymer is exposed to temperatures above the glass transition temperature (
Tg), an increase in free volume is observed, which allows for a greater segmental chain mobility and a faster drug delivery. According to the DSC, the
Tg of PPAd is −51.7 °C while PLLA-
b-PPAd 75/25
w/
w has also a similar
Tg, as well as one corresponding to the
Tg of PLLA (
Table 2). The copolymer has a lower degree of crystallinity, compared to neat PPAd (
Table 1), and thus a higher release rate was expected. However, this is not the case. As it can be seen in
Figure 14, PPAd has a higher release rate than PLLA-
b-PPAd copolymer. This behavior should be attributed to the existence of the PLLA segment in the copolymers, which has a lower mobility than PPAd. This was further confirmed with PLLA-
b-PPAd 90/10
w/
w copolymer, which, due to the higher PLLA amount, exhibits slower release than the 75/25
w/
w copolymer. The exposure of microparticles with neat PPAd and copolymers with higher PPAd amount at 37 °C, could explain the fast rate of drug release [
32,
33], indicating that NTX was probably released from microspheres by diffusion. So, it is clear that PLLA, due to its high
Tg, delays the release of NTX and its copolymerization with PPAd could enhance the release rate of NTX.
Figure 15 presents the morphology of the prepared microparticles after 8 days of dissolution, via SEM micrographs. It is clear that the surface of all the microspheres changed since the drug was dissolved. In all cases, aggregation of the microspheres was observed, their surface became rougher, and the normal spherical shape observed in the original microspheres was modified. PPAd microparticles exhibited extensive aggregation and their shape was destroyed. The presence of cavities in the internal morphology of the microspheres has not been confirmed, indicating that there is no erosion of the polymeric matrix. Thus, it is clear that diffusion was the main mechanism of drug release of these microparticles.
3.3. Release Data Modeling
According to the previous discussion and based on the experimental evidence, it appears that there is no polymer particle erosion, so the release is dominated by diffusion. This means that the drug release model must be a diffusion-based one. To set up the model the following assumptions are made: (i) the polymer particles are spherical, and they can be assumed to be monodispersed; (ii) the amount of the drug in the bath at the end of the release is much smaller than that corresponding to equilibrium. This is supported by the almost complete release for the case of PPAd. It appears that the non-released drug is trapped in the polymer matrix (maybe in an insoluble crystalline form); (iii) there is no mass transfer resistance to the bulk phase due to the agitation during release. There are approximate expressions for the diffusional release (in the form of power law in time functions) but they refer only to initial release stage [
34]. Here the interest is to reproduce the whole release curve so the partial differential equation of transient diffusion must be solved.
There are several levels of complication of the diffusion problem. The first level refers to the mode of diffusion (Fickian/non-Fickian). The second level refers to the possibility of non-uniformity in the polymer structure leading to different release paths with different diffusion coefficients. The third level is the possibility of non-uniform drug concentration along the radius of the polymer particle. Having experimental evidence for the third level and no evidence for the other two, Fickian diffusion with a single diffusion coefficient and non-uniform drug concentration is considered as the starting point to model the data.
The governing equation is:
where
r′ is the radial coordinate in the particle,
c is the local drug concentration in the particle and
D is the diffusion coefficient in the particle for the mobile fraction of the drug. The particle radius is denoted by
R and the boundary conditions for Equation (7) are:
c = 0 at
r′ =
R, d
c/d
r′ = 0 at
r′ = 0 and
c(
r′) =
co(
r′) at
t = 0. In order to facilitate the mathematical treatment of the problem the following normalization is made:
c =
Cavef(
r) where
Cave is the average concentration of mobile drug in the particles,
τ =
Dt/
R2 and
r =
r′/
R. The governing equation takes the form:
where the boundary conditions are
f = 0 at
r = 1, d
f/d
r = 0 at
r = 0 and
f(r) =
f0(
r) at
τ = 0. The function
f0(
r) denotes the initial drug distribution and by definition fulfills the relation
. According to the above model the release curve is computed as:
where
M∞ is the final % drug released. The mathematical problem is now completely defined. A semi-analytical solution is possible by transforming the spherical to planar geometry (by introducing the new function
g(
r) =
rf(
r)) and exploiting the solutions of the planar problem [
35]. The final result is:
It appears that for any profile of the initial drug distribution the functional form of the release curve is:
In the particular case of uniform initial profile, the coefficients
gi take the form
gi = 6/(
iπ)
2.
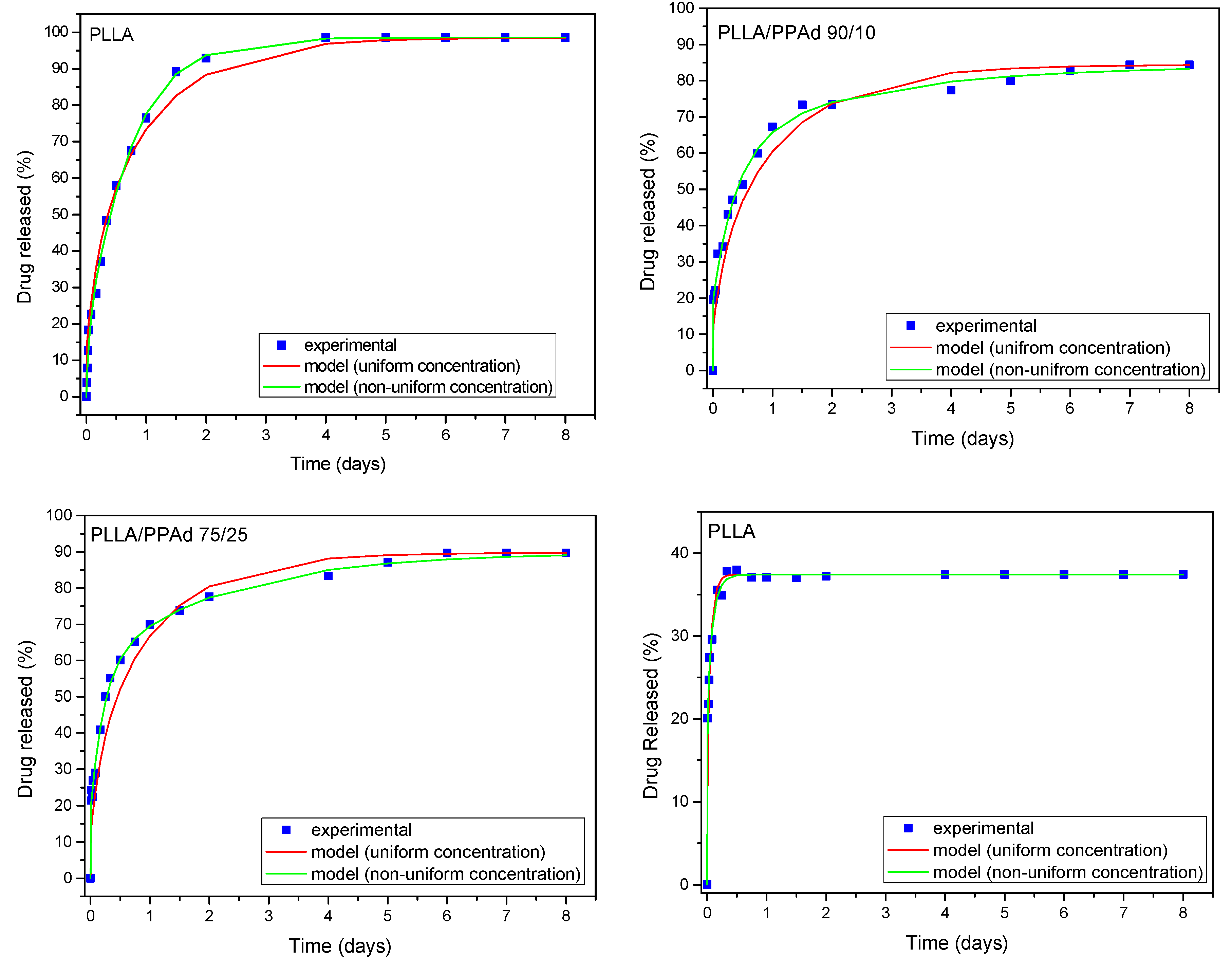
At first the possibility of fitting the experimental release curves assuming a uniform drug distribution will be examined. The fitting variable is the diffusion coefficient
D. The particle radius
R for each material is taken from the average diameter data presented in
Table 4. The fitting results appear in
Figure 16. As it is clear the fitting quality is acceptable only for the case of PLLA. In the other cases there is a systematic deviation between the data and the fitted curves. The next step is to consider non-uniform drug profile. No specific shape of the profile needs to be assumed. The fitting to the data is performed employing relation (11) with fitting parameters the diffusion coefficient
D and the coefficients
g1,
g2,
g3 (assuming
gi = 0 for
i > 3). The fitting curves are shown in
Figure 16 and a major improvement of the fitting quality (compared to that for uniform profile) is observed. The improvement appears to increase from PLLA to PPAd implying increasing drug profile non-uniformity in the particular order. It is denoted that in all cases the fittings are made using the least square criterion.
The success of the model to describe the data suggests that drug profile non-uniformity is enough to describe the experimental data and the other levels of complication do not have to be invoked. It is worth noticing that the profile of drug that fits the data is not determined directly by this approach. An additional complicated inverse problem must be set to go from the fitting parameters to the actual profile [
36]. The values of the diffusion coefficients that fit the data are: 8.67 × 10
−19 m
2/s for PPAd, 8.75 × 10
−20 m
2/s for PLLA/PPAd 75/25, 1.45 × 10
−20 m
2/s for PLLA/PPAd 90/10, 1.55 × 10
−19 m
2/s for PLLA. The diffusion coefficients are extremely small; almost ten order of magnitude smaller than those in liquids. It also appears that the diffusivity in neat polymers is higher than in polymer mixtures. The proposed model describes adequately the release data and it is compatible with the experimental observations of particle size and drug distribution non-uniformity.


 ,
, 


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}