
Computational Study on Temperature Driven Structure–Function Relationship of Polysaccharide Producing Bacterial Glycosyl Transferase Enzyme

 ,
,  , ,
, , 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Accession of Target Proteins
2.2. Primary Analysis of Physicochemical Parameters and Structure
2.3. Secondary Structure Assessment
2.4. Assessment of the Three Dimensional Structure, Modeling, and Validation
2.5. Functional Analyses of the Enzymes
2.6. Selection and Optimization of Ligand with the Target Proteins
2.7. Molecular Docking to Ensemble Protein Structure
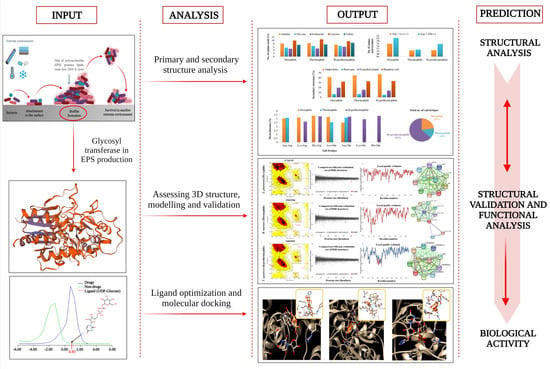
3. Results and Discussion
3.1. Accession of Target Proteins
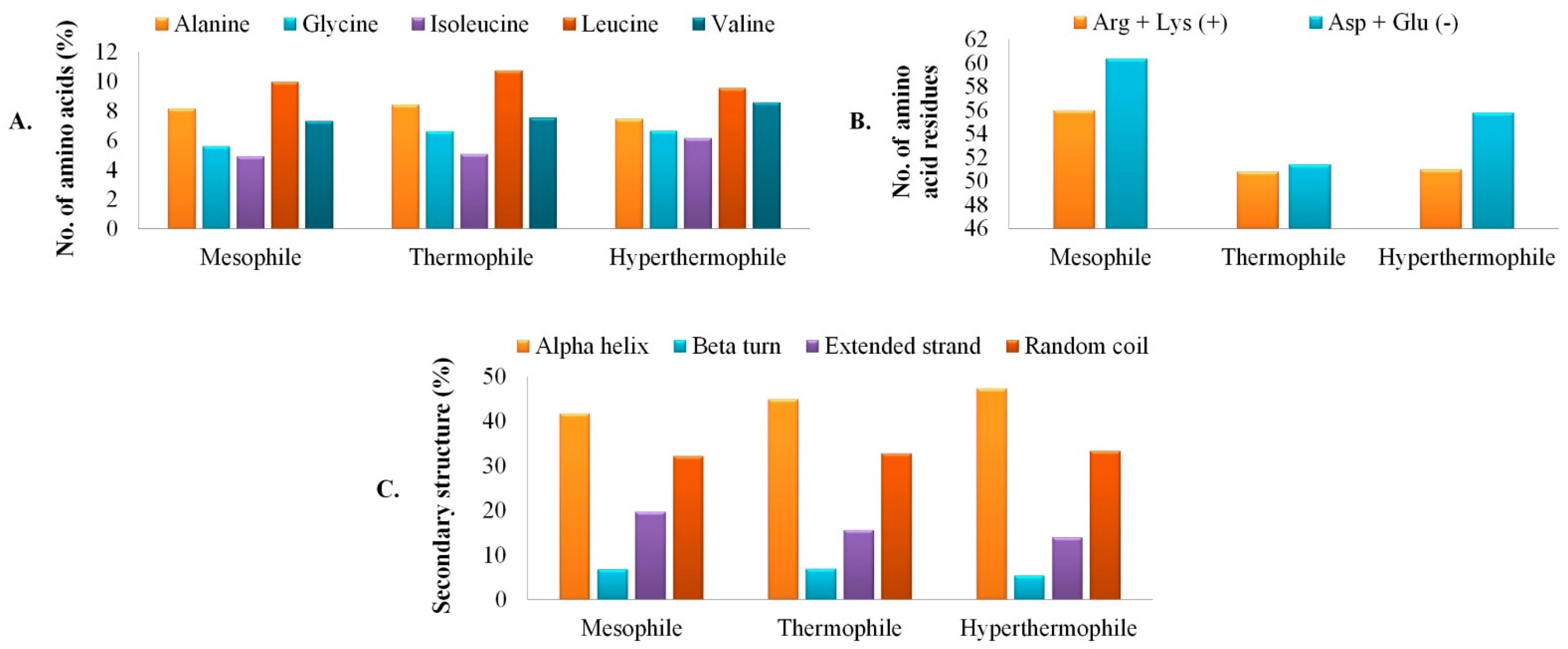
3.2. Primary Analysis of Physicochemical Parameters and Structure
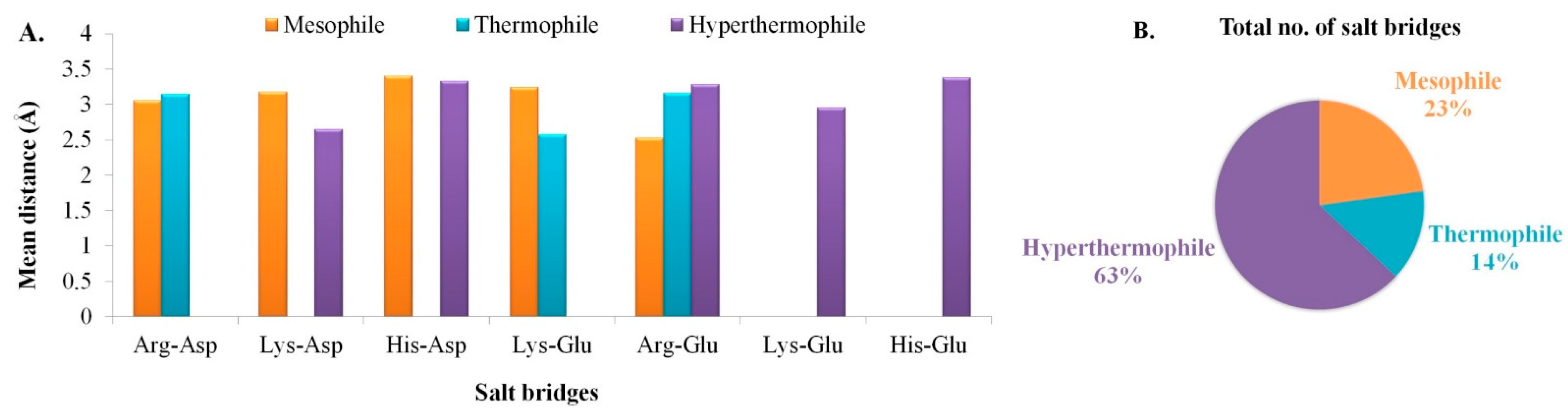
3.3. Secondary Structure Assessment
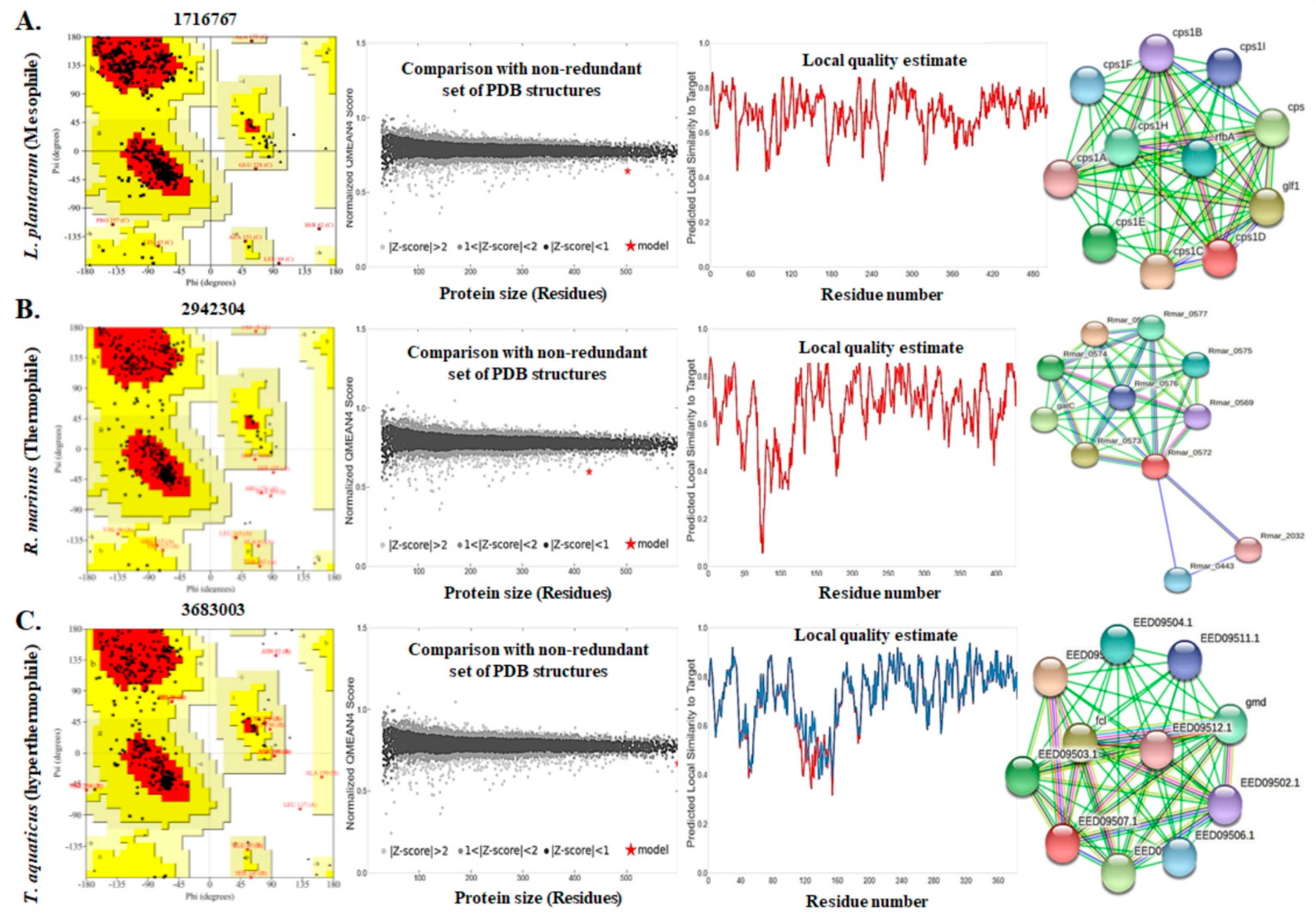
3.4. Assessment of the Three Dimensional Structure, Modeling, and Validation
3.5. Functional Analyses of the Enzymes
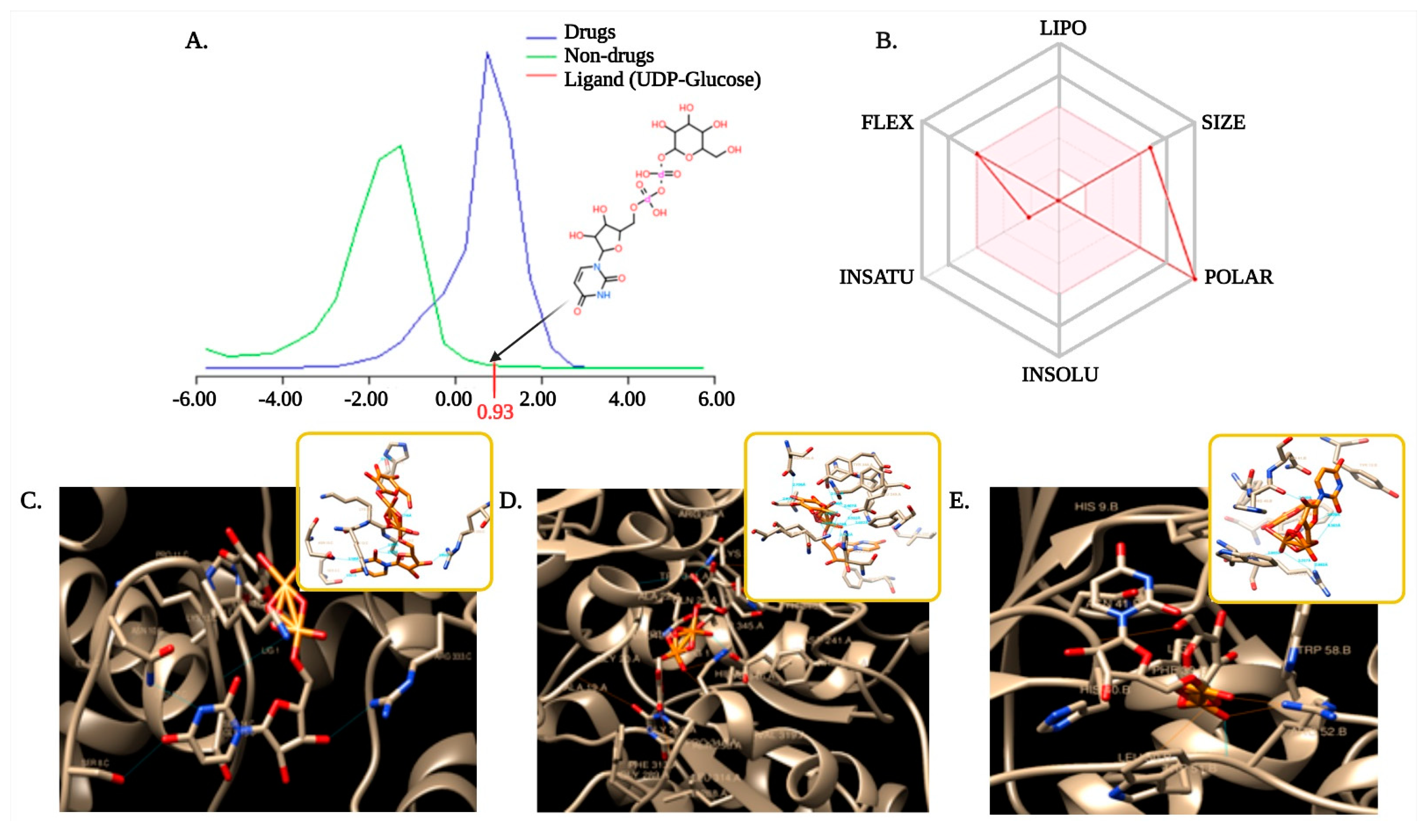
3.6. Selection and Optimization of Ligand with the Target Proteins
3.7. Molecular Docking to Ensemble Protein Structure
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yakovlieva, L.; Walvoort, M.T. Processivity in bacterial glycosyltransferases. ACS Chem. Biol. 2019, 15, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Das, D.; Rudra, S.G.; Mazumder, K.; Andler, R.; Bandopadhyay, R. Characterization of exopolysaccharide produced by Pseudomonas sp. PFAB4 for synthesis of EPS-coated AgNPs with antimicrobial properties. J. Polym. Environ. 2020, 28, 242–256. [Google Scholar] [CrossRef]
- Banerjee, A.; Rudra, S.G.; Mazumder, K.; Nigam, V.; Bandopadhyay, R. Structural and functional properties of exopolysaccharide excreted by a novel Bacillus anthracis (Strain PFAB2) of hot spring origin. Indian J. Microbiol. 2018, 58, 39–50. [Google Scholar] [CrossRef]
- Hinchliffe, J.D.; Madappura, A.P.; Syed Mohamed, S.M.D.; Roy, I. Biomedical applications of bacteria-derived polymers. Polymers 2021, 13, 1081. [Google Scholar] [CrossRef]
- Berillo, D.; Al-Jwaid, A.; Caplin, J. Polymeric Materials Used for Immobilisation of Bacteria for the Bioremediation of Contaminants in Water. Polymers 2021, 13, 1073. [Google Scholar] [CrossRef]
- Bilal, M.; Iqbal, H.M. Naturally-derived biopolymers: Potential platforms for enzyme immobilization. Int. J. Biol. Macromol. 2019, 130, 462–482. [Google Scholar] [CrossRef]
- Daba, G.M.; Elnahas, M.O.; Elkhateeb, W.A. Contributions of exopolysaccharides from lactic acid bacteria as biotechnological tools in food, pharmaceutical, and medical applications. Int. J. Biol. Macromol. 2021, 173, 79–89. [Google Scholar] [CrossRef]
- Caccamo, M.T.; Zammuto, V.; Gugliandolo, C.; Madeleine-Perdrillat, C.; Spanò, A.; Magazù, S. Thermal restraint of a bacterial exopolysaccharide of shallow vent origin. Int. J. Biol. Macromol. 2018, 114, 649–655. [Google Scholar] [CrossRef]
- Nešić, A.; Cabrera-Barjas, G.; Dimitrijević-Branković, S.; Davidović, S.; Radovanović, N.; Delattre, C. Prospect of polysaccharide-based materials as advanced food packaging. Molecules 2020, 25, 135. [Google Scholar] [CrossRef] [Green Version]
- Fukao, M.; Zendo, T.; Inoue, T.; Nakayama, J.; Suzuki, S.; Fukaya, T.; Yajima, N.; Sonomoto, K. Plasmid-encoded glycosyltransferase operon is responsible for exopolysaccharide production, cell aggregation, and bile resistance in a probiotic strain, Lactobacillus brevis KB290. J. Biosci. Bioeng. 2019, 128, 391–397. [Google Scholar] [CrossRef]
- Lairson, L.L.; Henrissat, B.; Davies, G.J.; Withers, S.G. Glycosyltransferases: Structures, functions, and mechanisms. Annu. Rev. Biochem. 2008, 77, 521–555. [Google Scholar] [CrossRef] [Green Version]
- Ardèvol, A.; Rovira, C. Reaction mechanisms in carbohydrate-active enzymes: Glycoside hydrolases and glycosyltransferases. Insights from ab initio quantum mechanics/molecular mechanics dynamic simulations. J. Am. Chem. Soc. 2015, 137, 7528–7547. [Google Scholar] [CrossRef]
- Schmid, J.; Heider, D.; Wendel, N.J.; Sperl, N.; Sieber, V. Bacterial glycosyltransferases: Challenges and opportunities of a highly diverse enzyme class toward tailoring natural products. Front. Microbiol. 2016, 7, 182. [Google Scholar] [CrossRef] [Green Version]
- Schmid, J. Recent insights in microbial exopolysaccharide biosynthesis and engineering strategies. Curr. Opin. Biotechnol. 2018, 53, 130–136. [Google Scholar] [CrossRef]
- Patel, A.K.; Singhania, R.R.; Pandey, A. Novel enzymatic processes applied to the food industry. Curr. Opin. Food Sci. 2016, 7, 64–72. [Google Scholar] [CrossRef]
- Chen, J.; Stites, W.E. Replacement of staphylococcal nuclease hydrophobic core residues with those from thermophilic homologues indicates packing is improved in some thermostable proteins. J. Mol. Biol. 2004, 344, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Greaves, R.B.; Warwicker, J. Mechanisms for stabilisation and the maintenance of solubility in proteins from thermophiles. BMC Struct. Biol. 2007, 7, 18. [Google Scholar] [CrossRef] [Green Version]
- Gromiha, M.M.; Pathak, M.C.; Saraboji, K.; Ortlund, E.A.; Gaucher, E.A. Hydrophobic environment is a key factor for the stability of thermophilic proteins. Proteins 2013, 81, 715–721. [Google Scholar] [CrossRef]
- Perl, D.; Welker, C.; Schindler, T.; Schroder, K.; Marahiel, M.A.; Jaenicke, R.; Schmid, F.X. Conservation of rapid two state folding in mesophilic, thermophilic and hyperthermophilic cold shock proteins. Nat. Struct. Biol. 1998, 5, 229–235. [Google Scholar] [CrossRef]
- Reeves, S.A.; Helman, L.J.; Allison, A.; Israel, M.A. Molecular cloning and primary structure of human glial fibrillary acidic protein. Proc. Natl. Acad. Sci. USA 1989, 86, 5178–5182. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [Green Version]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Rain, J.C.; Selig, L.; De Reuse, H.; Battaglia, V.; Reverdy, C.; Simon, S.; Chemama, Y. The protein–protein interaction map of Helicobacter pylori. Nature 2001, 409, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Hasan, R.; Rony, M.N.H.; Ahmed, R. In silico characterization and structural modeling of bacterial metalloprotease of family M4. J. Genet. Eng. Biotechnol. 2021, 19, 25. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [Green Version]
- Rost, B.; Yachdav, G.; Liu, J. The predictprotein server. Nucleic Acids Res. 2004, 32, W321–W326. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. Swissadme: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Wetzel, S.; Schuffenhauer, A.; Roggo, S.; Ertl, P.; Waldmann, H. Cheminformatic analysis of natural products and their chemical space. CHIMIA 2007, 61, 355–360. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1999, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Gamage, D.G.; Gunaratne, A.; Periyannan, G.R.; Russell, T.G. Applicability of instability index for in vitro protein stability prediction. Protein Pept. Lett. 2019, 26, 339–347. [Google Scholar] [CrossRef]
- Haki, G.D.; Rakshit, S.K. Developments in industrially important thermostable enzymes: A review. Bioresour. Technol. 2003, 89, 17–34. [Google Scholar] [CrossRef]
- Chang, K.Y.; Yang, J.R. Analysis and prediction of highly effective antiviral peptides based on random forests. PLoS ONE 2013, 8, e70166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, S.; Banerjee, A.; Chakraborty, N.; Soren, K.; Chakraborty, P.; Bandopadhyay, R. Structural-functional analyses of textile dye degrading azoreductase, laccase and peroxidase: A comparative in silico study. Electron. J. Biotechnol. 2020, 43, 48–54. [Google Scholar] [CrossRef]
- Miseta, A.; Csutora, P. Relationship between the occurrence of cysteine in proteins and the complexity of organisms. Mol. Biol. Evol. 2000, 17, 1232–1239. [Google Scholar] [CrossRef] [Green Version]
- Meruelo, A.D.; Han, S.K.; Kim, S.; Bowie, J.U. Structural differences between thermophilic and mesophilic membrane proteins. Protein Sci. 2012, 21, 1746–1753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiyagarajan, N.; Pham, T.T.; Stinson, B.; Sundriyal, A.; Tumbale, P.; Lizotte-Waniewski, M.; Acharya, K.R. Structure of a metal-independent bacterial glycosyltransferase that catalyzes the synthesis of histo-blood group A antigen. Sci. Rep. 2012, 2, 1–9. [Google Scholar] [CrossRef]
- Yu, H.; Yan, Y.; Zhang, C.; Dalby, P.A. Two strategies to engineer flexible loops for improved enzyme thermostability. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Marqusee, S.; Robbins, V.H.; Baldwin, R.L. Unusually stable helix formation in short alanine-based peptides. Proc. Natl. Acad. Sci. USA 1989, 86, 5286–5290. [Google Scholar] [CrossRef] [Green Version]
- Smith, L.J.; Fiebig, K.M.; Schwalbe, H.; Dobson, C.M. The concept of a random coil: Residual structure in peptides and denatured proteins. Fold. Des. 1996, 1, R95–R106. [Google Scholar] [CrossRef] [Green Version]
- Eswar, N.; Ramakrishnan, C.; Srinivasan, N. Stranded in isolation: Structural role of isolated extended strands in proteins. Protein Eng. 2003, 16, 331–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, R.; Rose, G.D. A physical basis for protein secondary structure. Proc. Natl. Acad. Sci. USA 1999, 96, 14258–14263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, C.; Shang, Y.; Xu, D. A deep dense inception network for protein beta-turn prediction. Proteins 2020, 88, 143–151. [Google Scholar] [CrossRef]
- Pace, C.N.; Fu, H.; Lee Fryar, K.; Landua, J.; Trevino, S.R.; Schell, D.; Thurlkill, R.L.; Imura, S.; Scholtz, J.M.; Gajiwala, K.; et al. Contribution of hydrogen bonds to protein stability. Protein Sci. 2014, 23, 652–661. [Google Scholar] [CrossRef]
- Kuhlman, B.; Bradley, P. Advances in protein structure prediction and design. Nat. Rev. Mol. Cell Biol. 2019, 20, 681–697. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Nussinov, R. Salt bridge stability in monomeric proteins. J. Mol. Biol. 1999, 293, 1241–1255. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Nussinov, R. Relationship between ion pair geometries and electrostatic strengths in proteins. Biophys. J. 2002, 83, 1595–1612. [Google Scholar] [CrossRef] [Green Version]
- Elcock, A.H. The stability of salt bridges at high temperatures: Implications for hyperthermophilic proteins. J. Mol. Biol. 1998, 284, 489–502. [Google Scholar] [CrossRef]
- Meuzelaar, H.; Vreede, J.; Woutersen, S. Influence of Glu/Arg, Asp/Arg, and Glu/Lys salt bridges on α-helical stability and folding kinetics. Biophys. J. 2016, 110, 2328–2341. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benkert, P.; Künzli, M.; Schwede. T. QMEAN server for protein model quality estimation. Nucleic Acids Res. 2009, 37, W510–W514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Mering, C.V. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.Y.; Li, S.T.; Li, Y. Identification and characterization of a new exopolysaccharide biosynthesis gene cluster from Streptomyces. FEMS Microbiol. Lett. 2003, 220, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L. Discovering novel sequence motifs with MEME. Curr. Protoc. Bioinform. 2003, 1, 2–4. [Google Scholar] [CrossRef]
- Behbahani, M.; Rabiei, P.; Mohabatkar, H. A Comparative Analysis of Allergen Proteins between Plants and Animals Using Several Computational Tools and Chou’s pseaac Concept. Int. Arch. Allergy Immunol. 2020, 181, 813–821. [Google Scholar] [CrossRef]
- Yachdav, G.; Kloppmann, E.; Kajan, L.; Hecht, M.; Goldberg, T.; Hamp, T.; Rost, B. Predictprotein—An open resource for online prediction of protein structural and functional features. Nucleic Acids Res. 2014, 42, W337–W343. [Google Scholar] [CrossRef] [Green Version]
- Chang, D.T.H.; Huang, H.Y.; Syu, Y.T.; Wu, C.P. Real value prediction of protein solvent accessibility using enhanced PSSM features. BMC Bioinform. 2008, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Gromiha, M.M. Protein Bioinformatics: From Sequence to Function; Academic Press: New Delhi, India, 2010. [Google Scholar]
- Lins, L.; Thomas, A.; Brasseur, R. Analysis of accessible surface of residues in proteins. Protein Sci. 2003, 12, 1406–1417. [Google Scholar] [CrossRef]
- Berezin, C.; Glaser, F.; Rosenberg, J.; Paz, I.; Pupko, T.; Fariselli, P.; Ben-Tal, N. Conseq: The identification of functionally and structurally important residues in protein sequences. Bioinformatics 2004, 20, 1322–1324. [Google Scholar] [CrossRef]
- Schlessinger, A.; Yachdav, G.; Rost, B. Profbval: Predict flexible and rigid residues in proteins. Bioinformatics 2006, 22, 891–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartuzi, D.; Kaczor, A.A.; Targowska-Duda, K.M.; Matosiuk, D. Recent advances and applications of molecular docking to G protein-coupled receptors. Molecules 2017, 22, 340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Shimshi, M.; Raz, K.; Nitoker Eliaz, N.; Mhashal, A.R.; Ansbacher, T.; Major, D.T. Enzydock: Protein–ligand docking of multiple reactive states along a reaction coordinate in enzymes. J. Chem. Theory Comput. 2019, 15, 5116–5134. [Google Scholar] [CrossRef] [PubMed]
- Ntie-Kang, F.; Nyongbela, K.D.; Ayimele, G.A.; Shekfeh, S. “Drug-likeness” properties of natural compounds. In Fundamental Concepts; De Gruyter: Berlin, Germany, 2020; pp. 81–102. [Google Scholar]
- Khan, T.; Dixit, S.; Ahmad, R.; Raza, S.; Azad, I.; Joshi, S.; Khan, A.R. Molecular docking, PASS analysis, bioactivity score prediction, synthesis, characterization and biological activity evaluation of a functionalized 2-butanone thiosemicarbazone ligand and its complexes. J. Chem. Biol. 2017, 10, 91–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kranenburg, R.V.; Marugg, J.D.; Van Swam, I.I.; Willem, N.J.; De Vos, W.M. Molecular characterization of the plasmid-encoded eps gene cluster essential for exopolysaccharide biosynthesis in Lactococcus lactis. Mol. Microbiol. 1997, 24, 387–397. [Google Scholar] [CrossRef]
- Ramírez, A.S.; Boilevin, J.; Mehdipour, A.R.; Hummer, G.; Darbre, T.; Reymond, J.L.; Locher, K.P. Structural basis of the molecular ruler mechanism of a bacterial glycosyltransferase. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Abdelhedi, O.; Nasri, R.; Mora, L.; Jridi, M.; Toldrá, F.; Nasri, M. In silico analysis and molecular docking study of angiotensin I-converting enzyme inhibitory peptides from smooth-hound viscera protein hydrolysates fractionated by ultrafiltration. Food Chem. 2018, 239, 453–463. [Google Scholar] [CrossRef]
- Gezegen, H.; Gürdere, M.B.; Dinçer, A.; Özbek, O.; Koçyiğit, Ü.M.; Taslimi, P.; Ceylan, M. Synthesis, molecular docking, and biological activities of new cyanopyridine derivatives containing phenylurea. Arch. Pharm. 2021, 354, 2000334. [Google Scholar] [CrossRef]
- Tao, X.; Huang, Y.; Wang, C.; Chen, F.; Yang, L.; Ling, L.; Chen, X. Recent developments in molecular docking technology applied in food science: A review. Int. J. Food Sci. Technol. 2020, 55, 33–45. [Google Scholar] [CrossRef]
- Goodsell, D.S. Computational docking of biomolecular complexes with autodock. Cold Spring Harb. Protoc. 2009, 5. [Google Scholar] [CrossRef]
- Zhao, H.; Huang, D. Hydrogen bonding penalty upon ligand binding. PLoS ONE 2011, 6, e19923. [Google Scholar] [CrossRef] [PubMed]
- Stroganov, O.V.; Novikov, F.N.; Stroylov, V.S.; Kulkov, V.; Chilov, G.G. Lead finder: An approach to improve accuracy of protein− ligand docking, binding energy estimation, and virtual screening. J. Chem. Inf. Model. 2008, 48, 2371–2385. [Google Scholar] [CrossRef] [PubMed]
- Desjarlais, R.L.; Sheridan, R.P.; Dixon, J.S.; Kuntz, I.D.; Venkataraghavan, R. Docking flexible ligands to macromolecular receptors by molecular shape. J. Med. Chem. 1986, 29, 2149–2153. [Google Scholar] [CrossRef]
- Dellafiora, L.; Oswald, I.P.; Dorne, J.L.; Galaverna, G.; Battilani, P.; Dall’Asta, C. An in silico structural approach to characterize human and rainbow trout estrogenicity of mycotoxins: Proof of concept study using zearalenone and alternariol. Food Chem. 2020, 312, 126088. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Nussinov, R. Different roles of electrostatics in heat and in cold: Adaptation by citrate synthase. Chembiochem 2004, 5, 280–290. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Serial No. | Bacterial Isolates | Physicochemical Characters | Quality Assessment Scores | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| No. of AA | Theoretical PI | MW (KDa) | II | AI | GRAVY | 3D-1D Score (%) | ERRAT Quality Factor | QMEAN Z- Score | AA in FR of Ramamchandran Plot (%) | ||
| 1 | Lactobacillus plantarum | 514 | 6.18 | 58.346 | 26.34 | 77.88 | −0.419 | 83.70 | 84.4898 | −4.01 | 89.7 |
| 2 | Rhodothermus marinus | 443 | 9.85 | 50.116 | 31.28 | 94.09 | −0.21 | 92.54 | 80.7786 | −4.98 | 83.4 |
| 3 | Thermus aquaticus | 396 | 9.04 | 44.256 | 40.53 | 88.21 | −0.223 | 87.53 | 85.0575 | −2.59 | 86.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Faune, P.; Sánchez-Arévalo, I.; Sarkar, S.; Majhi, K.; Bandopadhyay, R.; Cabrera-Barjas, G.; Gómez, A.; Banerjee, A. Computational Study on Temperature Driven Structure–Function Relationship of Polysaccharide Producing Bacterial Glycosyl Transferase Enzyme. Polymers 2021, 13, 1771. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13111771
González-Faune P, Sánchez-Arévalo I, Sarkar S, Majhi K, Bandopadhyay R, Cabrera-Barjas G, Gómez A, Banerjee A. Computational Study on Temperature Driven Structure–Function Relationship of Polysaccharide Producing Bacterial Glycosyl Transferase Enzyme. Polymers. 2021; 13(11):1771. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13111771
Chicago/Turabian StyleGonzález-Faune, Patricio, Ignacio Sánchez-Arévalo, Shrabana Sarkar, Krishnendu Majhi, Rajib Bandopadhyay, Gustavo Cabrera-Barjas, Aleydis Gómez, and Aparna Banerjee. 2021. "Computational Study on Temperature Driven Structure–Function Relationship of Polysaccharide Producing Bacterial Glycosyl Transferase Enzyme" Polymers 13, no. 11: 1771. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13111771