
Evaluation of Composition Effects on the Physicochemical and Biological Properties of Polypeptide-Based Hydrogels for Potential Application in Wound Healing

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthetic Procedures
2.2.1. Nε-tert-Butyloxycarbonyl l-lysine (H-Lys(Boc)-OH)
2.2.2. Synthesis of N-carboxyanhydride (NCA) of Nε-tert-butyloxycarbonyl l-lysine (NCA-Lys(Boc))
2.2.3. Synthesis of N-carboxyanhydride (NCA) of l-alanine (NCA-Ala)
2.2.4. Random NCA Copolymerization
2.2.5. Deprotection of Polypeptides
2.3. Preparation of Polypeptide-Based Hydrogels
2.4. Hydrogel Characterization
2.4.1. Gelation and Rheometry
2.4.2. Amine Quantification
2.4.3. Pore Morphology
2.4.4. Swelling Behavior
2.4.5. Degradation Time
2.5. In Vitro Cell Studies
2.5.1. Cytotoxicity
2.5.2. Cell Adhesion
2.6. Statistical Evaluation
3. Results and Discussion
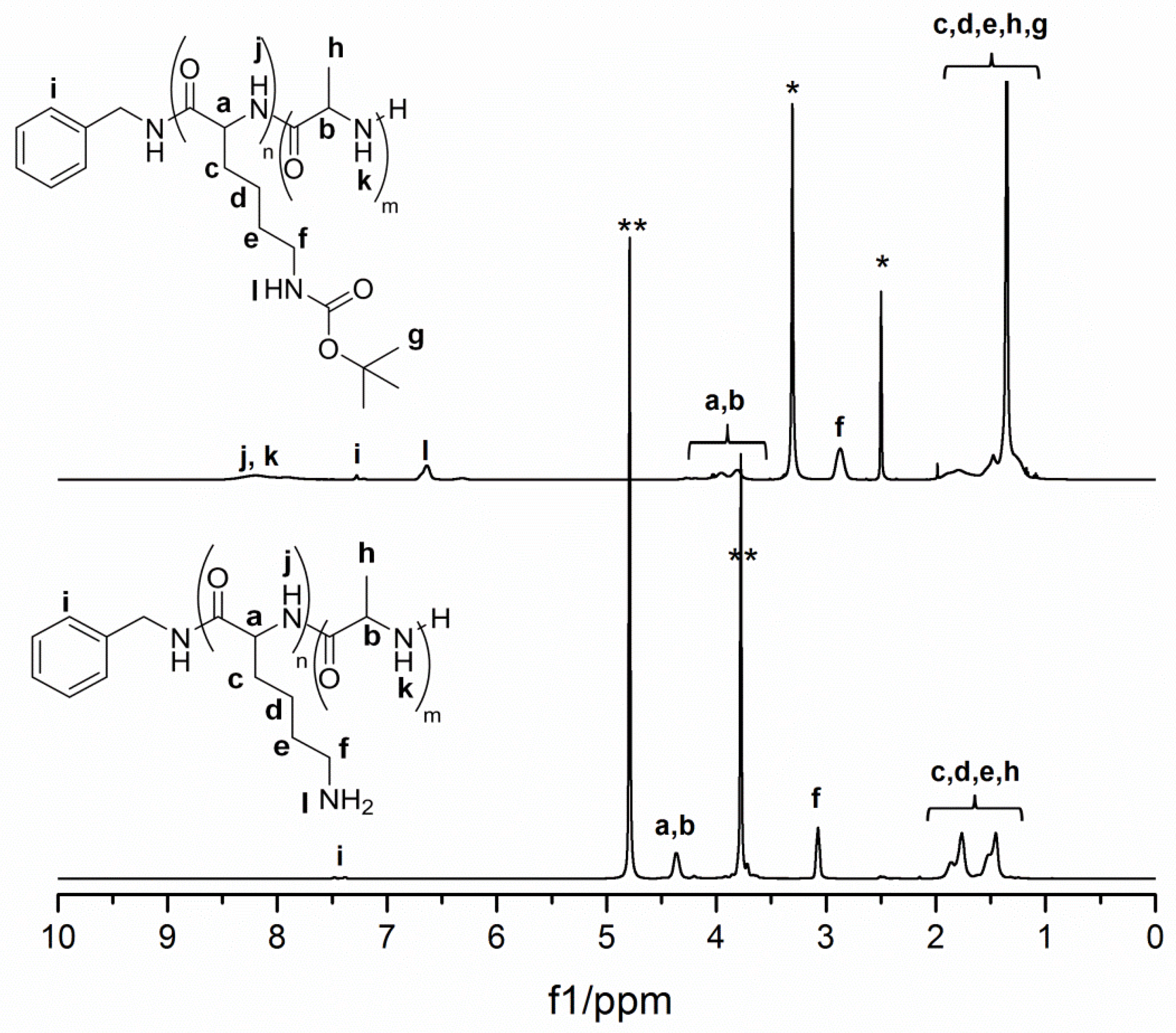
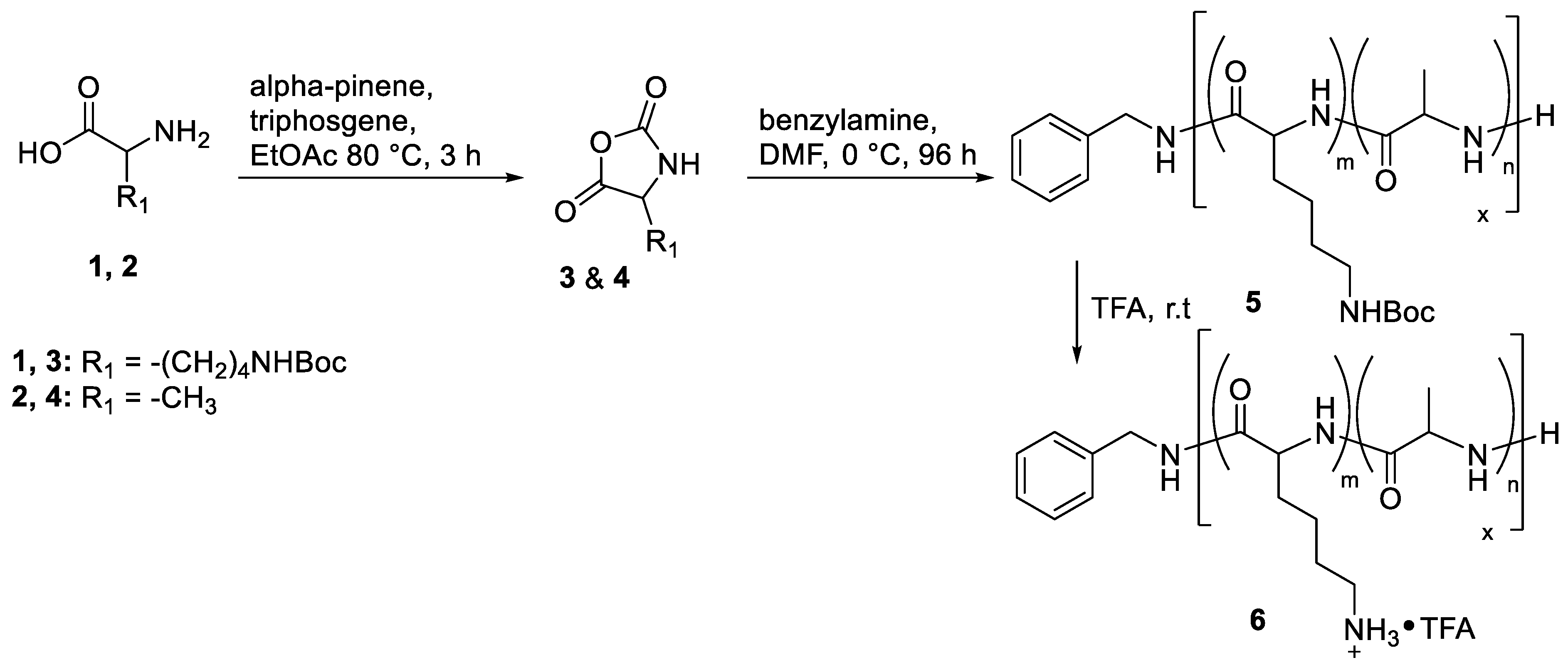
3.1. Preparation of Controlled Polypeptides
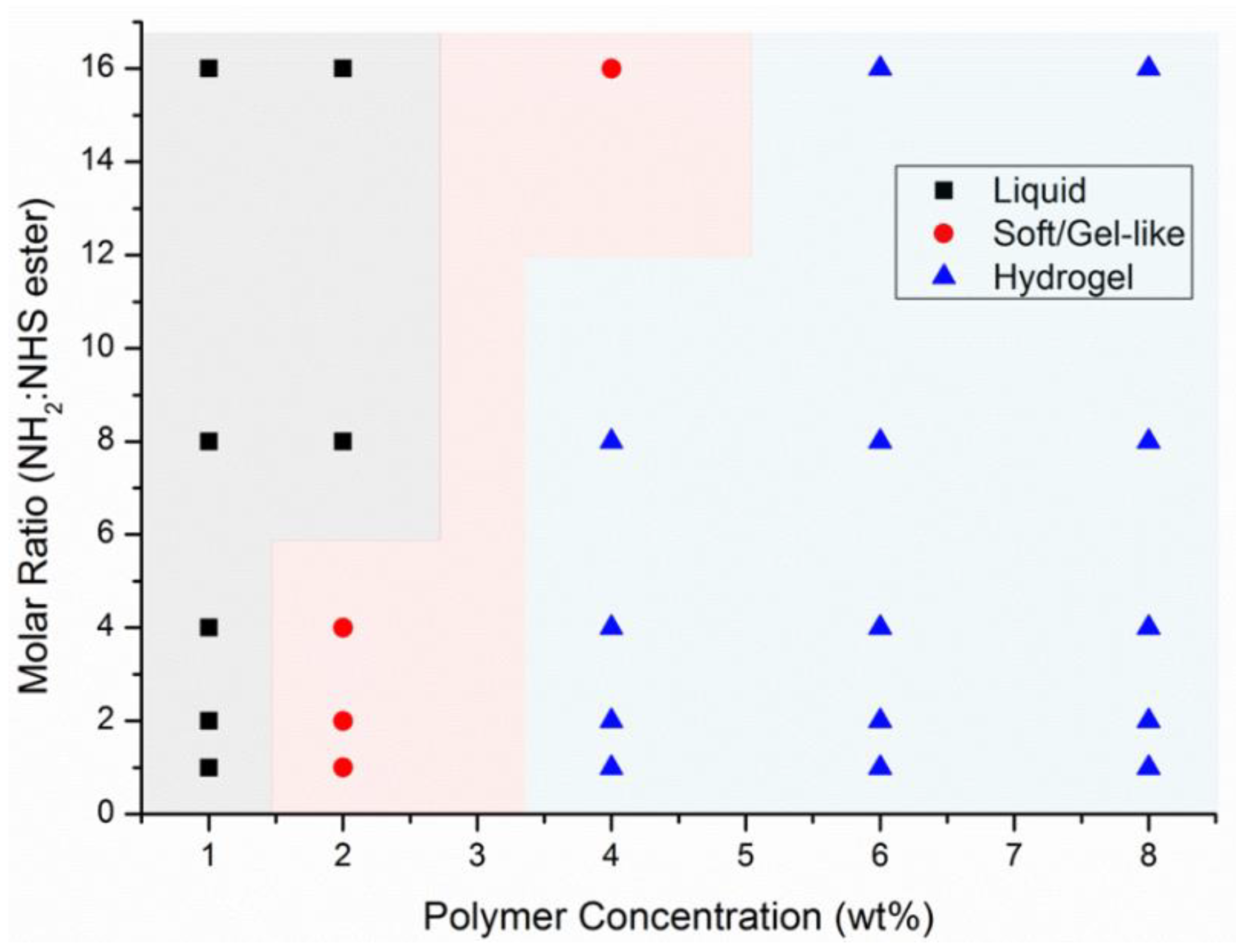
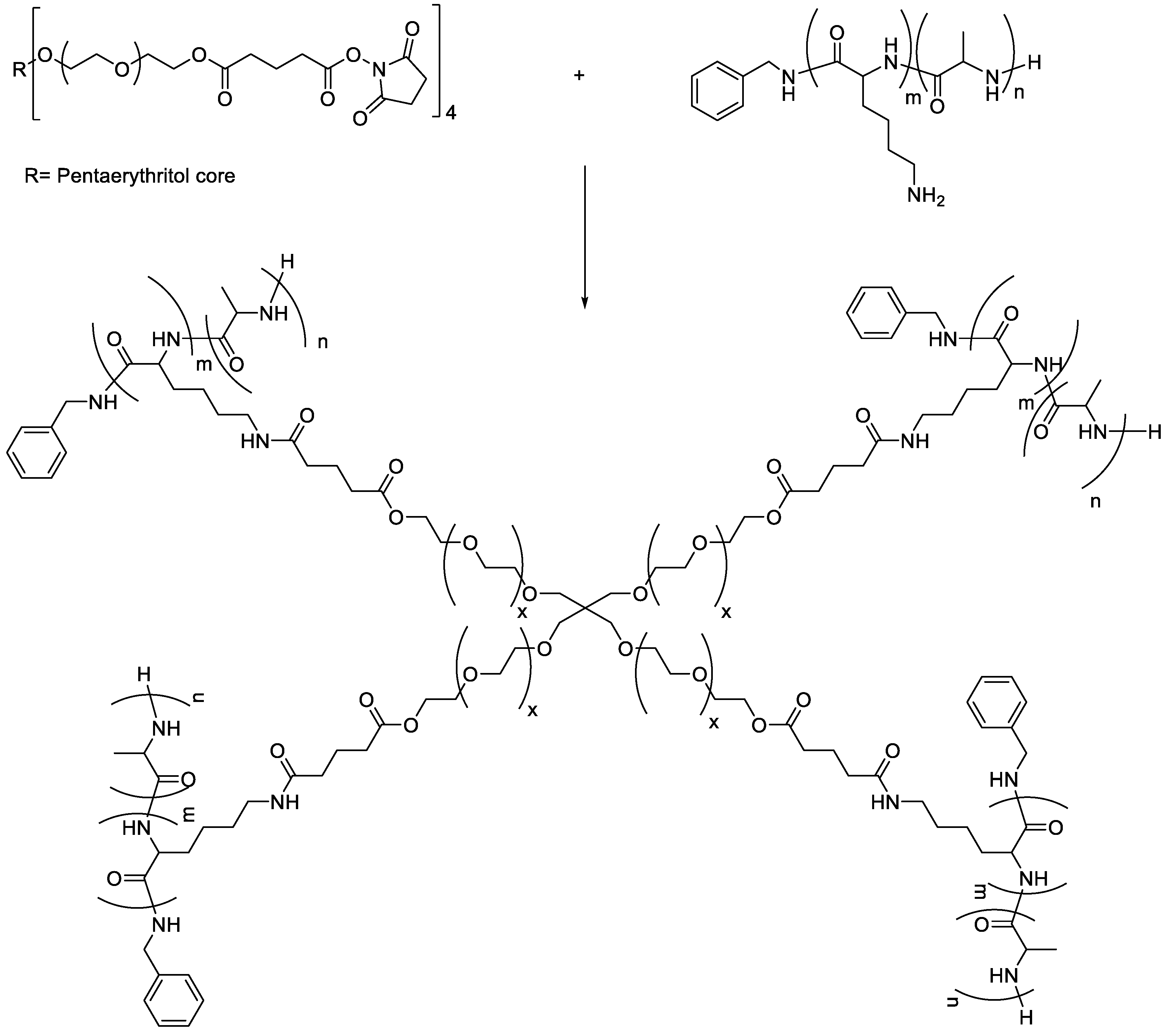
3.2. Preparation of a Library of Hydrogels
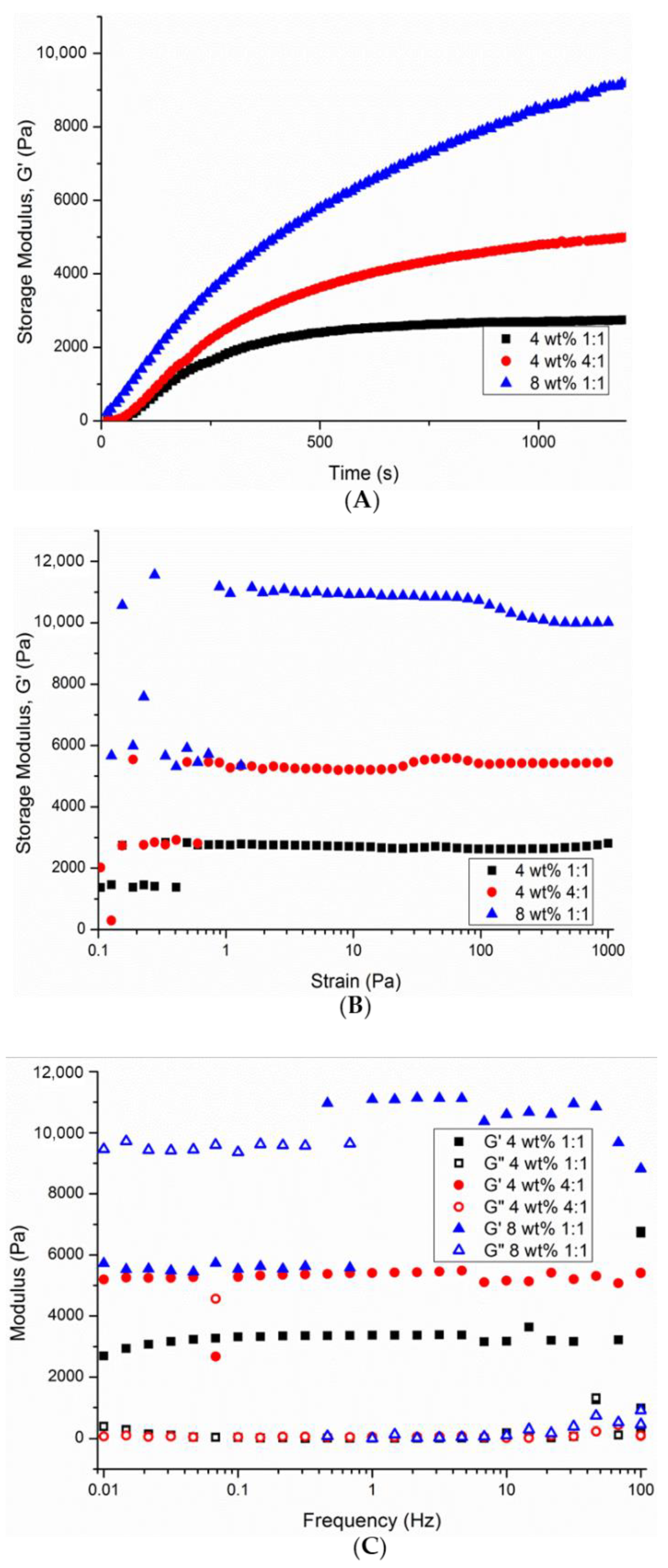
3.3. Assessment of the Rheological Nature of Selected P(KA)/4-PEG Hydrogels
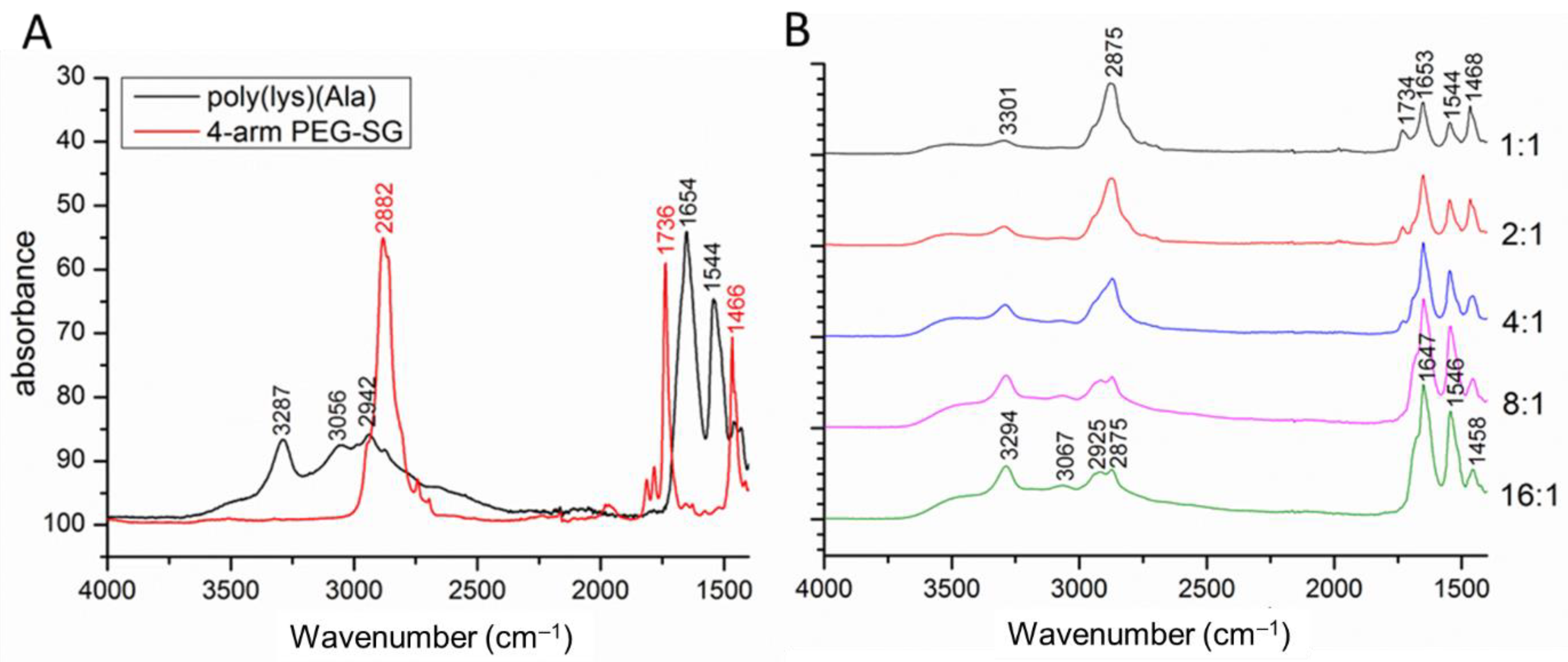
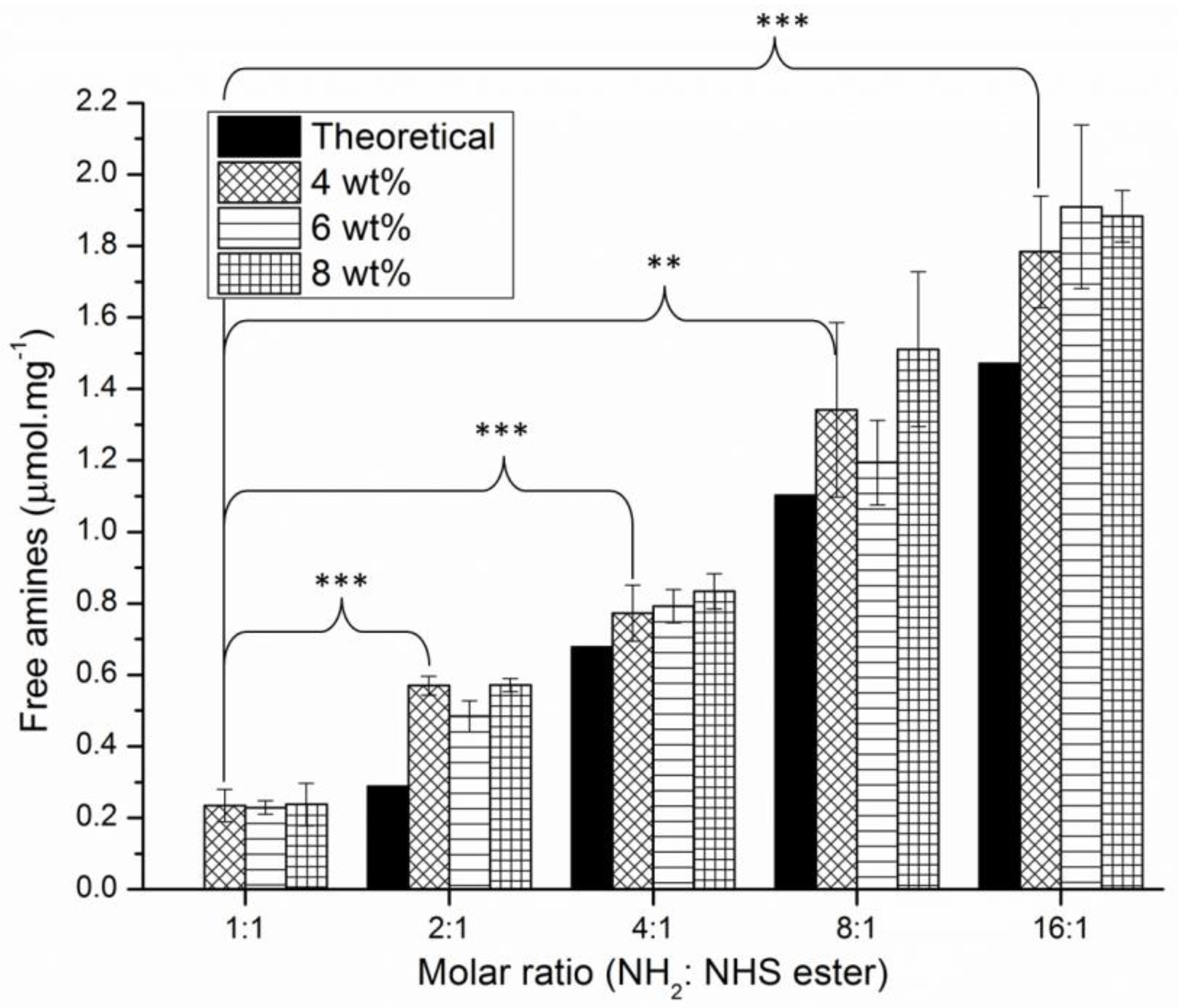
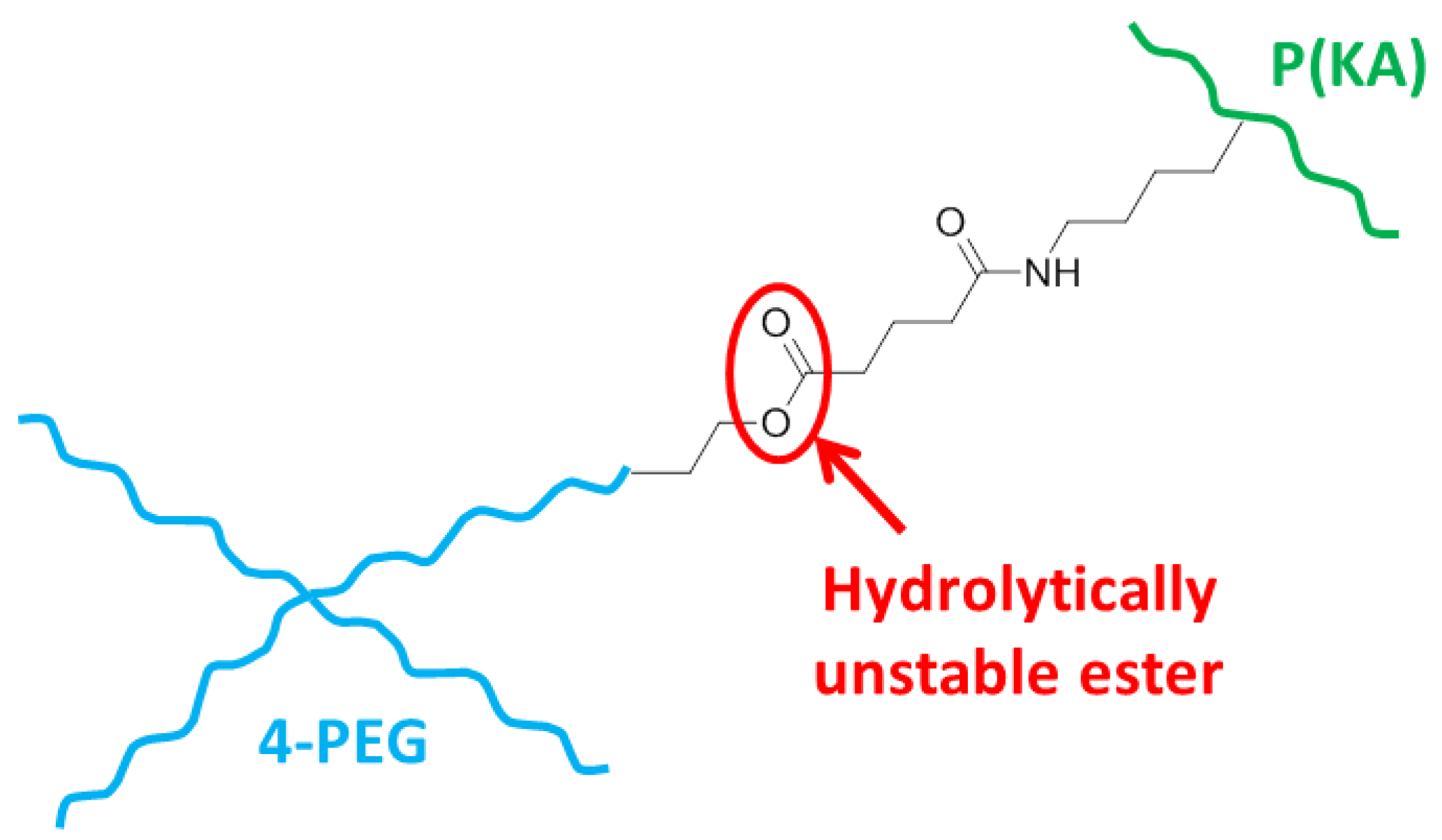
3.4. Chemical Analysis of the Hydrogel Compositions
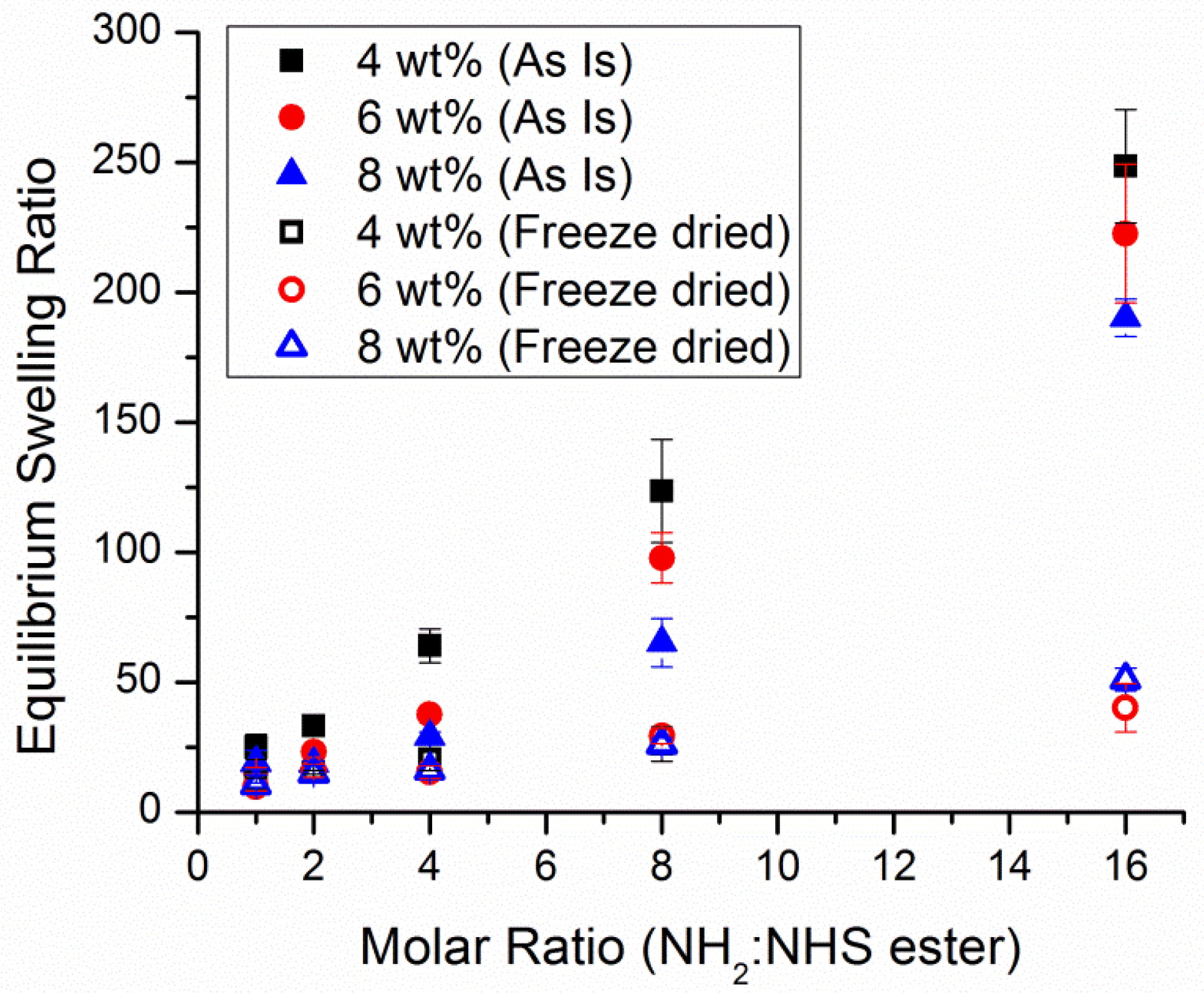
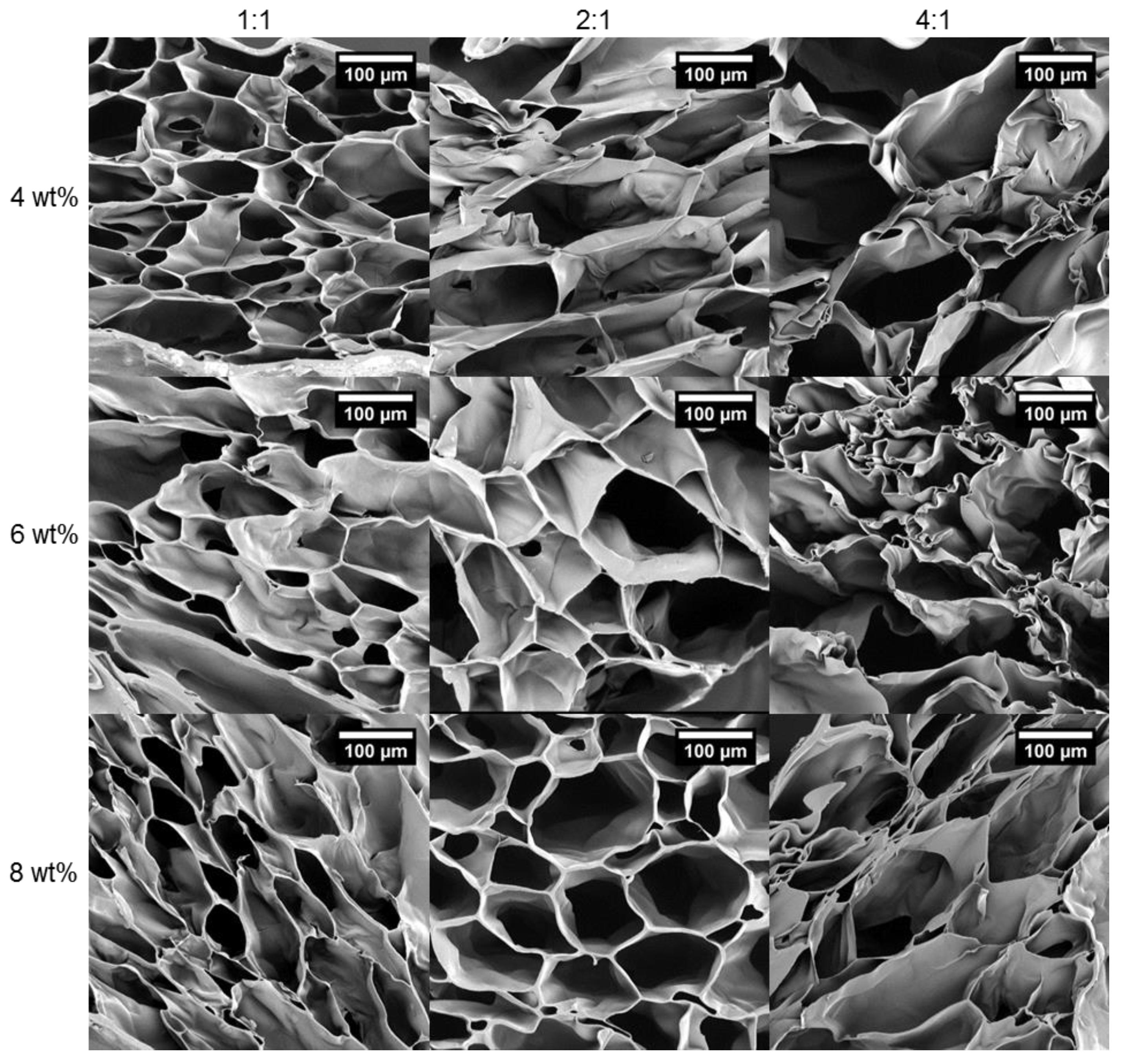
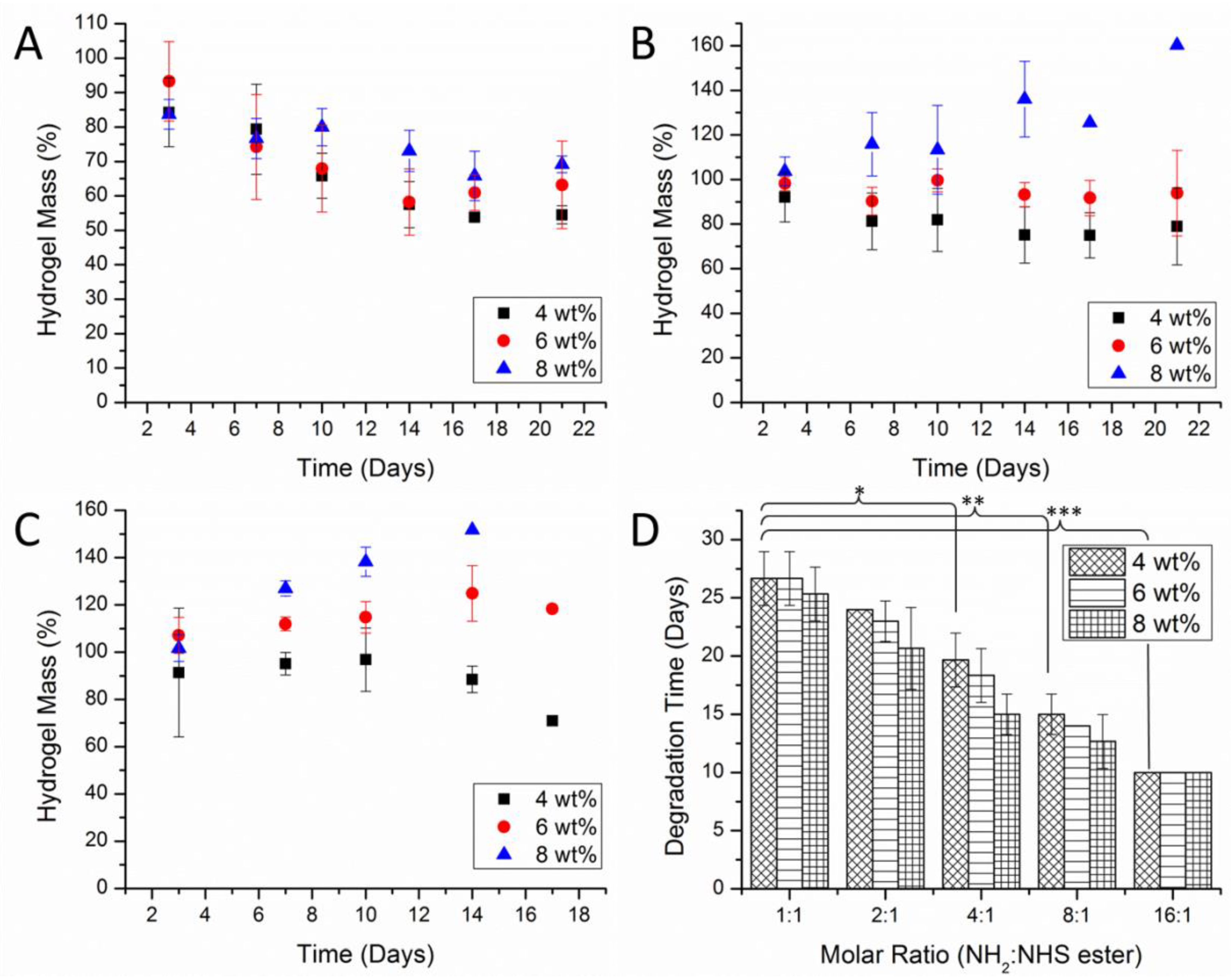
3.5. The Effect of Crosslinking and Concentration on the Hydrogel Network
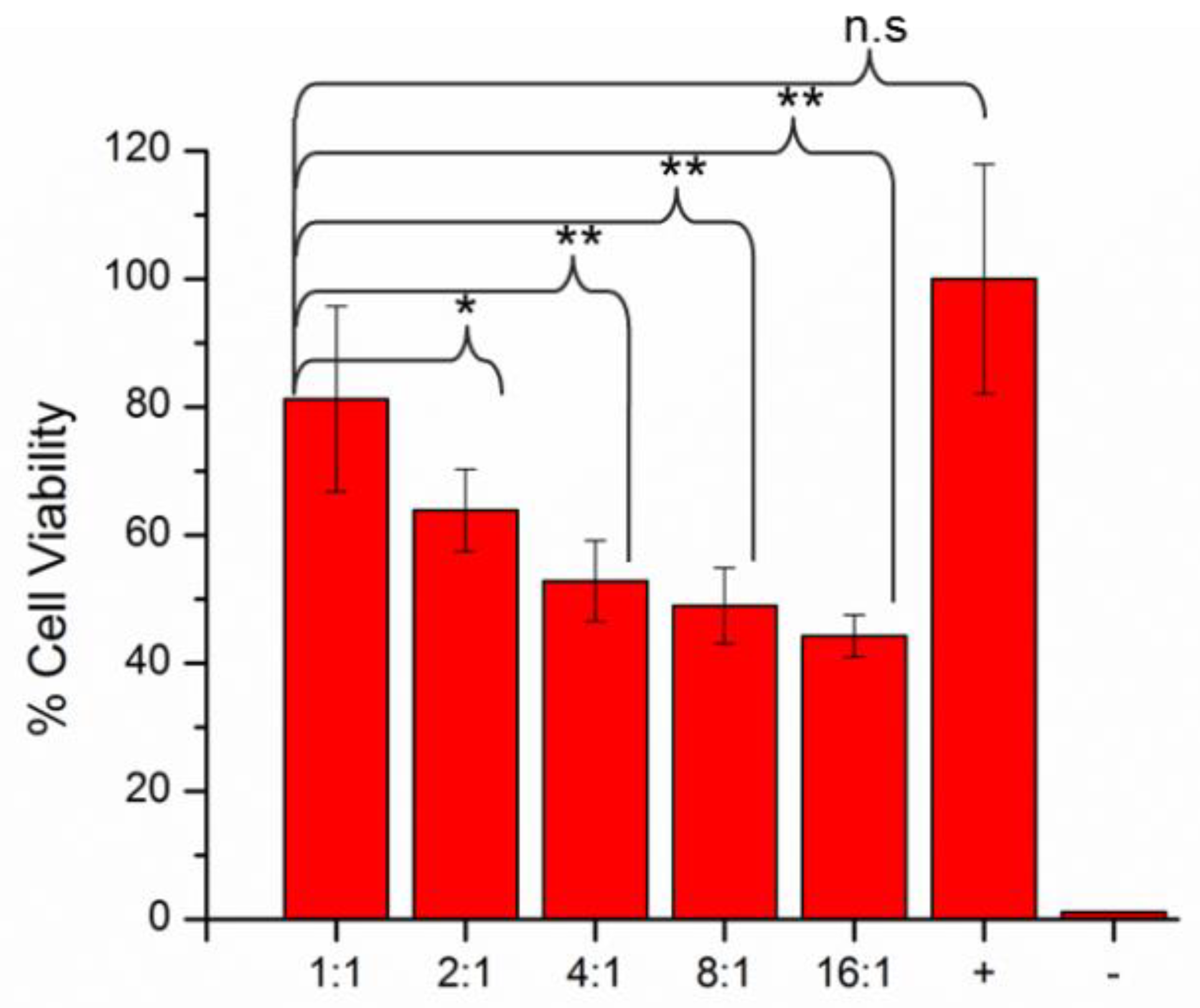
3.6. Biocompatibility Assessment of the Hydrogels
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singer, A.J.; Clark, R.A.F. Cutaneous Wound Healing. N. Engl. J. Med. 1999, 341, 738. [Google Scholar] [CrossRef] [PubMed]
- Werner, S.; Grose, R. Regulation of Wound Healing by Growth Factors and Cytokines. Physiol. Rev. 2003, 83, 835–870. [Google Scholar] [CrossRef]
- Broughton, G.; Janis, J.E.; Attinger, C.E. The Basic Science of Wound Healing. Plast. Reconstr. Surg. 2006, 117 (Suppl. 7), 12S–34S. [Google Scholar] [CrossRef]
- Barrientos, S.; Stojadinovic, O.; Golinko, M.S.; Brem, H.; Tomic-Canic, M. Growth factors and cytokines in wound healing. Wound Repair Regen. 2008, 16, 585–601. [Google Scholar] [CrossRef]
- Boateng, J.; Matthews, K.; Stevens, H.N.; Eccleston, G.M. Wound Healing Dressings and Drug Delivery Systems: A Review. J. Pharm. Sci. 2008, 97, 2892–2923. [Google Scholar] [CrossRef]
- Shahrokhi, S.; Arno, A.; Jeschke, M.G. The use of dermal substitutes in burn surgery: Acute phase. Wound Repair Regen. 2014, 22, 14–22. [Google Scholar] [CrossRef]
- Stone, R.I.; Natesan, S.; Kowalczewski, C.J.; Mangum, L.H.; Clay, N.E.; Clohessy, R.M.; Carlsson, A.H.; Tassin, D.H.; Chan, R.K.; Rizzo, J.A.; et al. Advancements in Regenerative Strategies Through the Continuum of Burn Care. Front. Pharmacol. 2018, 9, 672. [Google Scholar] [CrossRef] [PubMed]
- Selig, H.F.; Lumenta, D.B.; Giretzlehner, M.; Jeschke, M.G.; Upton, D.; Kamolz, L.P. The properties of an “ideal” burn wound dressing—What do we need in daily clinical practice? Results of a worldwide online survey among burn care specialists. Burns 2012, 38, 960–966. [Google Scholar] [CrossRef] [PubMed]
- Zarrintaj, P.; Moghaddam, A.S.; Manouchehri, S.; Atoufi, Z.; Amiri, A.; Amirkhani, M.A.; Nilforoushzadeh, M.A.; Saeb, M.R.; Hamblin, M.R.; Mozafari, M. Can regenerative medicine and nanotechnology combine to heal wounds? The search for the ideal wound dressing. Nanomedicine 2017, 12, 2403–2422. [Google Scholar] [CrossRef] [PubMed]
- Haddad, A.G.; Giatsidis, G.; Orgill, D.P.; Halvorson, E.G. Skin Substitutes and Bioscaffolds:Temporary and Permanent Coverage. Clin. Plast. Surg. 2018, 44, 627–634. [Google Scholar] [CrossRef]
- Koehler, J.; Brandl, F.P.; Goepferich, A.M. Hydrogel wound dressings for bioactive treatment of acute and chronic wounds. Eur. Polym. J. 2018, 100, 1–11. [Google Scholar] [CrossRef]
- Mayet, N.; Choonara, Y.E.; Kumar, P.; Tomar, L.K.; Tyagi, C.; Du Toit, L.C.; Pillay, V. A Comprehensive Review of Advanced Biopolymeric Wound Healing Systems. J. Pharm. Sci. 2014, 103, 2211–2230. [Google Scholar] [CrossRef]
- Jhon, M.S.; Andrade, J.D. Water and hydrogels. J. Biomed. Mater. Res. 1973, 7, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Mooney, D.J. Hydrogels for Tissue Engineering. Chem. Rev. 2001, 101, 203–225. [Google Scholar] [CrossRef] [PubMed]
- El-Sherbiny, I.M.; Yacoub, M.H. Hydrogel scaffolds for tissue engineering: Progress and challenges. Glob. Cardiol. Sci. Pract. 2013, 2013, 316–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Wang, J.; Song, Z.; Yin, L.; Zhang, Y.; Tang, H.; Tu, C.; Lin, Y.; Cheng, J. Recent advances in amino acid N-carboxyanhydrides and synthetic polypeptides: Chemistry, self-assembly and biological applications. Chem. Commun. 2014, 50, 139–155. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Li, Z. Advances and Biomedical Applications of Polypeptide Hydrogels Derived from α-Amino Acid N -Carboxyanhydride (NCA) Polymerizations. Adv. Healthc. Mater. 2018, 7, 1800020. [Google Scholar] [CrossRef] [PubMed]
- González-Henríquez, C.M.; Sarabia-Vallejos, M.A.; Rodríguez-Hernández, J. Strategies to Fabricate Polypep-tide-Based Structures via Ring-Opening Polymerization of N-Carboxyanhydrides. Polymers 2017, 9, 551. [Google Scholar] [CrossRef] [Green Version]
- Penczek, S. Models of Biopolymers by Ring-Opening Polymerization of Cyclic Phosphorus Containing Compounds; Elsevier BV: Amsterdam, The Netherlands, 1980; pp. 133–143. [Google Scholar]
- Deng, C.; Wu, J.; Cheng, R.; Meng, F.; Klok, H.-A.; Zhong, Z. Functional polypeptide and hybrid materials: Precision synthesis via α-amino acid N-carboxyanhydride polymerization and emerging biomedical applications. Prog. Polym. Sci. 2014, 39, 330–364. [Google Scholar] [CrossRef]
- Li, S.; Dong, S.; Xu, W.; Tu, S.; Yan, L.; Zhao, C.; Ding, J.; Chen, X. Antibacterial Hydrogels. Adv. Sci. 2018, 5, 1700527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowak, A.P.; Breedveld, V.; Pakstis, L.; Ozbas, B.; Pine, D.J.; Pochan, D.; Deming, T. Rapidly recovering hydrogel scaffolds from self-assembling diblock copolypeptide amphiphiles. Nat. Cell Biol. 2002, 417, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Deming, T.J. Polypeptide hydrogels via a unique assembly mechanism. Soft Matter 2005, 1, 28–35. [Google Scholar] [CrossRef]
- Fischer, D.; Youxin, L.; Barbara, A.; Josef, K.; Thomas, K. In vitro cytotoxicity testing of polycations: Influence of polymer structure on cell viability and hemolysis. Biomaterials 2003, 24, 1121–1131. [Google Scholar] [CrossRef]
- Kadlecova, Z.; Baldi, L.; Hacker, D.; Wurm, F.M.; Klok, H.-A. Comparative Study on the In Vitro Cytotoxicity of Linear, Dendritic, and Hyperbranched Polylysine Analogues. Biomacromolecules 2012, 13, 3127–3137. [Google Scholar] [CrossRef] [PubMed]
- Pakstis, L.M.; Ozbas, B.; Hales, K.D.; Nowak, A.P.; Deming, T.J.; Pochan, D. Effect of Chemistry and Morphology on the Biofunctionality of Self-Assembling Diblock Copolypeptide Hydrogels. Biomacromolecules 2004, 5, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Hynes, S.R.; Rauch, M.F.; Bertram, J.P.; Lavik, E.B. A library of tunable poly(ethylene glycol)/poly(L-lysine) hydrogels to investigate the material cues that influence neural stem cell differentiation. J. Biomed. Mater. Res. Part A 2008, 89, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Song, A.; Rane, A.A.; Christman, K.L. Antibacterial and cell-adhesive polypeptide and poly(ethylene glycol) hydrogel as a potential scaffold for wound healing. Acta Biomater. 2012, 8, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Ambekar, R.S.; Kandasubramanian, B. Progress in the Advancement of Porous Biopolymer Scaffold: Tissue Engineering Application. Ind. Eng. Chem. Res. 2019, 58, 6163–6194. [Google Scholar] [CrossRef]
- Höck, S.; Marti, R.; Riedl, R.; Simeunovic, M. Thermal Cleavage of the Fmoc Protection Group. Chim. Int. J. Chem. 2010, 64, 200–202. [Google Scholar] [CrossRef]
- Habraken, G.J.M.; Wilsens, K.H.R.M.; Koninga, C.E.; Heise, A. Optimization of N-carboxyanhydride (NCA) polymerization by variation of reaction temperature and pressure. Polym. Chem. 2011, 2, 1322–1330. [Google Scholar] [CrossRef]
- Aliferis, T.; Iatrou, H.; Hadjichristidis, N. Living Polypeptides. Biomacromolecules 2004, 5, 1653–1656. [Google Scholar] [CrossRef]
- Jenkins, A.D.; Kratochvĺl, P.; Stepto, R.F.T.; Suter, U.W. Glossary of Basic Terms in Polymer Science. IUPAC Stand. Online 2016, 68, 2287–2311. [Google Scholar] [CrossRef]
- Vayaboury, W.; Giani, O.; Cottet, H.; Deratani, A.; Schué, F. Living Polymerization ofα-Amino AcidN-Carboxyanhydrides(NCA) upon Decreasing the Reaction Temperature. Macromol. Rapid Commun. 2004, 25, 1221–1224. [Google Scholar] [CrossRef]
- Dmitrovic, V.; Habraken, G.J.; Hendrix, M.M.; Habraken, W.J.; Heise, A.; De With, G.; Sommerdijk, N.A. Random Poly(Amino Acid)s Synthesized by Ring Opening Polymerization as Additives in the Biomimetic Mineralization of CaCO3. Polymers 2012, 4, 1195–1210. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. J. Organ. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef] [PubMed]
- Friedman, M. Applications of the Ninhydrin Reaction for Analysis of Amino Acids, Peptides, and Proteins to Agricultural and Biomedical. J. Agric. Food Chem. 2004, 52, 385–406. [Google Scholar] [CrossRef]
- Hermanson, G.T. Bioconjugate Techniques, 3rd ed.; Audet, J., Preap, M., Eds.; Elsevier Inc.: London, UK, 2013. [Google Scholar]
- Li, R.; McCoy, B.J. Inter- and Intramolecular Crosslinking Kinetics: Partitioning According to Number of Cross-links. Macromol. Rapid Commun. 2004, 25, 1059–1063. [Google Scholar] [CrossRef]
- Pekcan, O.; Kara, S. Gelation Mechanisms. Mod. Phys. Lett. B 2012, 26, 30019. [Google Scholar] [CrossRef]
- Raghavan, S.R.; Cipriano, B.H. Gel Formation: Phase diagrams using table top rheology and calorimetry. In Molecular Gels: Materials with Self-Assembled Fibrillar Networks; Weiss, R., Terech, P., Eds.; Springer: Dordrecht, The Netherlands, 2006; pp. 233–244. [Google Scholar]
- Loh, X.J.; Goh, S.H.; Li, J. New Biodegradable Thermogelling Copolymers Having Very Low Gelation Concentrations. Biomacromolecules 2007, 8, 585–593. [Google Scholar] [CrossRef]
- Xu, Y.; Shen, Y.; Xiong, Y.; Li, C.; Sun, C.; Ouahab, A.; Tu, J. Synthesis, characterization, biodegradability and biocompatibility of a temperature-sensitive PBLA-PEG-PBLA hydrogel as protein delivery system with low critical gelation concentration. Drug Dev. Ind. Pharm. 2013, 40, 1264–1275. [Google Scholar] [CrossRef] [PubMed]
- Zuidema, J.; Rivet, C.J.; Gilbert, R.J.; Morrison, F.A. A protocol for rheological characterization of hydrogels for tissue engineering strategies. J. Biomed. Mater. Res. Part B Appl. Biomater. 2014, 102, 1063–1073. [Google Scholar] [CrossRef] [PubMed]
- Vedadghavami, A.; Minooei, F.; Mohammadi, M.H.; Khetani, S.; Kolahchi, A.R.; Mashayekhan, S.; Sanati-Nezhad, A. Manufacturing of hydrogel biomaterials with controlled mechanical properties for tissue engineering applications. Acta Biomater. 2017, 62, 42–63. [Google Scholar] [CrossRef]
- Hadjipanayi, E.; Mudera, V.; Brown, R.A. Guiding cell migration in 3D: A collagen matrix with graded directional stiffness. Cell Motil. Cytoskelet. 2009, 66, 121–128. [Google Scholar] [CrossRef]
- Sheikholeslam, M.; Wright, M.E.E.; Jeschke, M.G.; Amini-Nik, S. Biomaterials for Skin Substitutes. Adv. Healthc. Mater. 2018, 7, 1700897. [Google Scholar] [CrossRef]
- Singh, B.R. Basic Aspects of the Technique and Applications of Infrared Spectroscopy of Peptides and Proteins; ACS Symposium; American Chemical Society: Washington, DC, USA, 1999. [Google Scholar]
- Rozenberg, M.; Shoham, G. FTIR spectra of solid poly-l-lysine in the stretching NH mode range. Biophys. Chem. 2007, 125, 166–171. [Google Scholar] [CrossRef]
- Kendall, P.A. Use of the Ninhydrin Reaction for Quantitative Estimation of Amino Groups in Insoluble Specimens. Nat. Cell Biol. 1963, 197, 1305–1306. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Ranjha, N.M. Effect of degree of cross-linking on swelling and on drug release of low viscous chi-tosan/poly(vinyl alcohol) hydrogels. Polym. Bull. 2014, 71, 2133–2158. [Google Scholar] [CrossRef]
- Shet, R.; Ashton, M.; Dodou, K. Effect of Crosslinking Agent Concentration on the Properties of Unmedicated Hydrogels. Pharmaceutics 2015, 7, 305–319. [Google Scholar]
- Annabi, N.; Nichol, J.W.; Zhong, X.; Ji, C.; Koshy, S.T.; Khademhosseini, A.; Dehghani, F. Controlling the Porosity and Microarchitecture of Hydrogels for Tissue Engineering. Tissue Eng. Part B Rev. 2010, 16, 371–383. [Google Scholar] [CrossRef]
- Peng, Z.; Chen, F. Hydroxyethyl Cellulose-Based Hydrogels with Various Pore Sizes Prepared by Freeze-Drying. J. Macromol. Sci. Part B 2010, 50, 340–349. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, A.; Deng, A.; Yang, Y.; Gao, L.; Zhong, Z.; Yang, S. Pore architecture and cell viability on freeze dried 3D recombinant human collagen-peptide (RHC)–chitosan scaffolds. Mater. Sci. Eng. C 2015, 49, 174–182. [Google Scholar] [CrossRef]
- Sun, K.; Li, R.; Jiang, W.; Sun, Y.; Li, H. Comparison of three-dimensional printing and vacuum freeze-dried techniques for fabricating composite scaffolds. Biochem. Biophys. Res. Commun. 2016, 477, 1085–1091. [Google Scholar] [CrossRef]
- Reys, L.; Silva, S.S.; Pirraco, R.P.; Marques, A.; Mano, J.F.; da Silva, T.H.; Reis, R.L. Influence of freezing temperature and deacetylation degree on the performance of freeze-dried chitosan scaffolds towards cartilage tissue engineering. Eur. Polym. J. 2017, 95, 232–240. [Google Scholar] [CrossRef]
- Ho, M.-H.; Kuo, P.-Y.; Hsieh, H.-J.; Hsien, T.-Y.; Hou, L.-T.; Lai, J.-Y.; Wang, D.-M. Preparation of porous scaffolds by using freeze-extraction and freeze-gelation methods. Biomaterials 2004, 25, 129–138. [Google Scholar] [CrossRef]
- Overby, R.J.; Feldman, D.S. Influence of Poly(Ethylene Glycol) End Groups on Poly(Ethylene Glycol)-Albumin System Properties as a Potential Degradable Tissue Scaffold. J. Funct. Biomater. 2018, 10, 1. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Zhou, B.; Xu, D.; Xu, H.; Liang, L.; Feng, X.; Ouyang, P.; Chi, B. Antimicrobial and biocompatible ε-polylysine-γ-poly(glutamic acid)-based hydrogel system for wound healing. J. Bioact. Compat. Polym. 2016, 31, 242–259. [Google Scholar] [CrossRef]
- Wang, Y.X.; Robertson, J.L.; Spillman, W.B., Jr.; Claus, R.O. Effects of the chemical structure and the surface properties of polymeric biomaterials on their bio-compatibility. Pharm. Res. 2004, 21, 1362–1373. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.S.S.; Hsieh, P.-L.; Chen, P.-S.; Chen, Y.-T.; Jan, J.-S. Genipin-cross-linked poly(l-lysine)-based hydrogels: Synthesis, characterization, and drug encapsulation. Colloids Surf. B Biointerfaces 2013, 111, 423–431. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total Polymer Concentration | Molar Ratio | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1:1 | 2:1 | 4:1 | 8:1 | 16:1 | ||||||
| P(KA) | 4-PEG | P(KA) | 4-PEG | P(KA) | 4-PEG | P(KA) | 4-PEG | P(KA) | 4-PEG | |
| 1 wt% | 0.16 | 0.84 | 0.28 | 0.72 | 0.44 | 0.57 | 0.61 | 0.39 | 0.76 | 0.24 |
| 2 wt% | 0.32 | 1.68 | 0.56 | 1.44 | 0.87 | 1.13 | 1.21 | 0.79 | 1.51 | 0.49 |
| 4 wt% | 0.65 | 3.36 | 1.11 | 2.89 | 1.74 | 2.26 | 2.42 | 1.58 | 3.02 | 0.98 |
| 6 wt% | 0.97 | 5.03 | 1.67 | 4.33 | 2.61 | 3.39 | 3.64 | 2.36 | 4.53 | 1.47 |
| 8 wt% | 1.29 | 6.71 | 2.22 | 5.78 | 3.48 | 4.52 | 4.85 | 3.15 | 6.04 | 1.96 |
| Entry | DPexp a | K/A Ratio a | Mn Target (g·mol−1) | Mn NMR (g·mol−1) | Mn SEC (g·mol−1) b | Đ b | Free Amine (μmol·mg−1) |
|---|---|---|---|---|---|---|---|
| #1 | 79 | 0.63:0.37 | 18,342 | 14,862 | 20,928 | 1.33 | 2.55 ± 0.14 |
| #2 | 96 | 0.60:0.40 | 18,342 | 17,608 | 26,933 | 1.18 | 2.02 ± 0.11 |
| Hydrogel | Average Pore Size (μm) a | ||
|---|---|---|---|
| 1:1 | 2:1 | 4:1 | |
| 4 wt% | 91 ± 39 b,c,k,l | 239 ± 68 b,d,n,o | 255 ± 51 c,d,q,r |
| 6 wt% | 132 ± 58 e,f,k,m | 157 ± 48 e,g,n,p | 181 ± 64 f,g,q,s |
| 8 wt% | 128 ± 55 h,i,l,m | 125 ± 45 h,j,o,p | 165 ± 67 i,j,r,s |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giliomee, J.; du Toit, L.C.; Kumar, P.; Klumperman, B.; Choonara, Y.E. Evaluation of Composition Effects on the Physicochemical and Biological Properties of Polypeptide-Based Hydrogels for Potential Application in Wound Healing. Polymers 2021, 13, 1828. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13111828
Giliomee J, du Toit LC, Kumar P, Klumperman B, Choonara YE. Evaluation of Composition Effects on the Physicochemical and Biological Properties of Polypeptide-Based Hydrogels for Potential Application in Wound Healing. Polymers. 2021; 13(11):1828. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13111828
Chicago/Turabian StyleGiliomee, Johnel, Lisa C. du Toit, Pradeep Kumar, Bert Klumperman, and Yahya E. Choonara. 2021. "Evaluation of Composition Effects on the Physicochemical and Biological Properties of Polypeptide-Based Hydrogels for Potential Application in Wound Healing" Polymers 13, no. 11: 1828. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13111828