Thermomechanical Analysis of Isora Nanofibril Incorporated Polyethylene Nanocomposites


 ,
, 
 , , ,
, , , 
Abstract
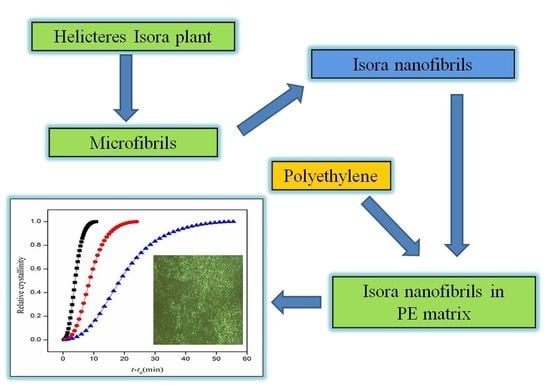
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of the Isora Nanofibril (INF)-Reinforced PE Composite
2.3. Differential Scanning Calorimetry (DSC)
2.4. Scanning Electron Microscopy (SEM)
2.5. Atomic Force Microscopy (AFM)
2.6. Optical Microscopy (OM)
2.7. Mechanical Characterization
2.8. Dynamic Mechanical Analysis
2.9. Rheological Characterization
2.10. Contact Angle Measurement
2.11. Fourier Transform Infrared Spectroscopy (FTIR)
3. Results and Discussion
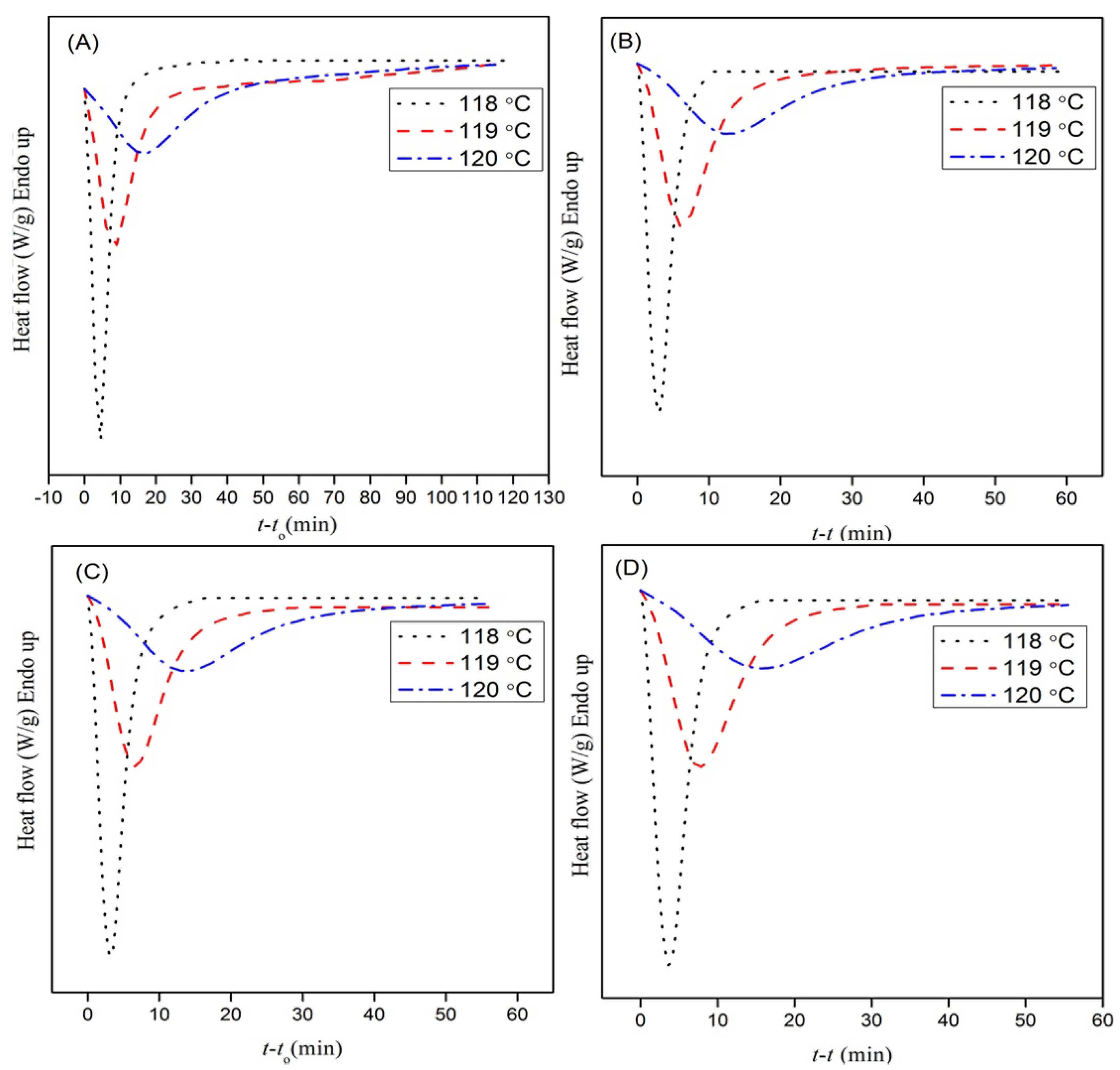
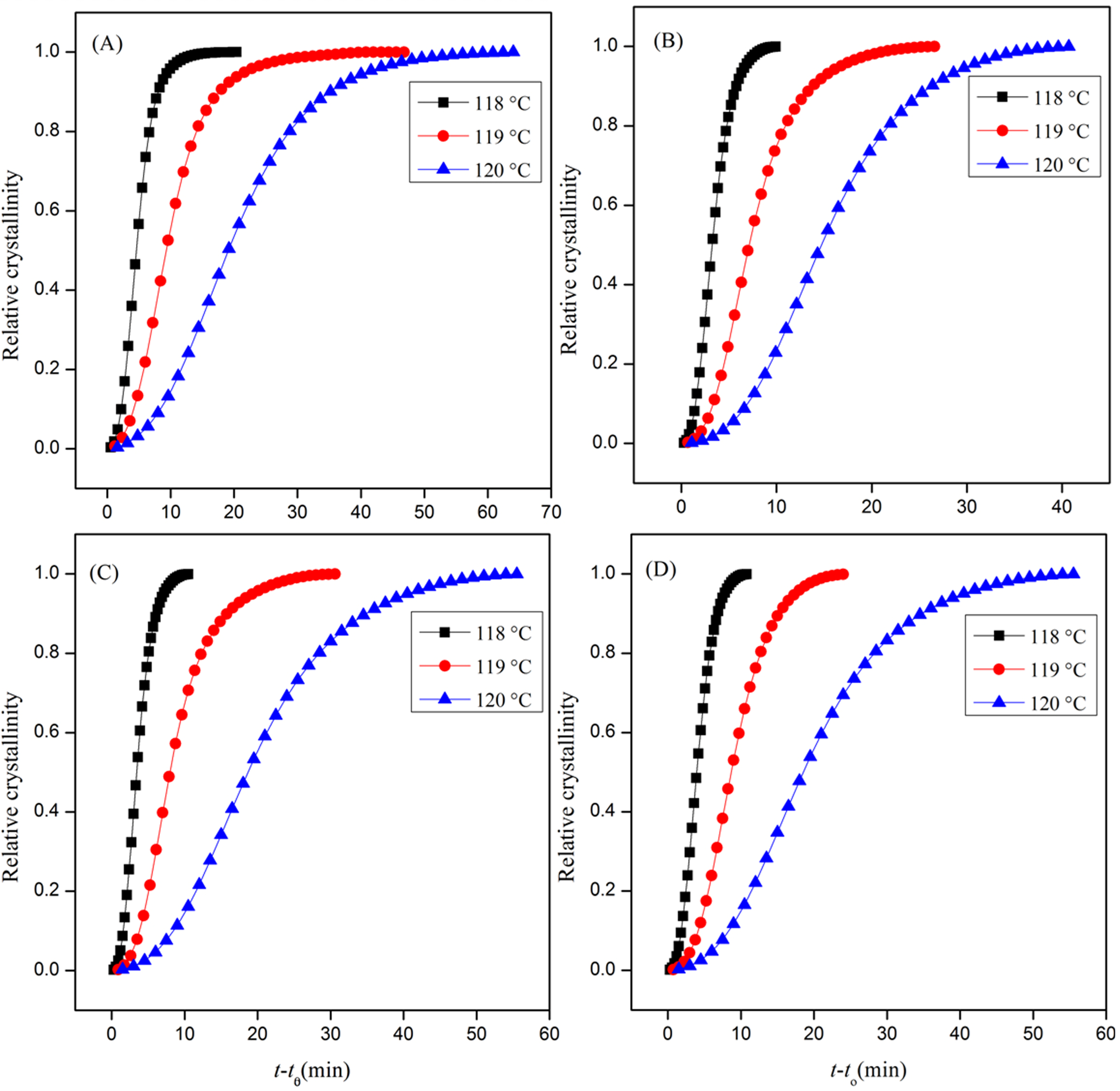
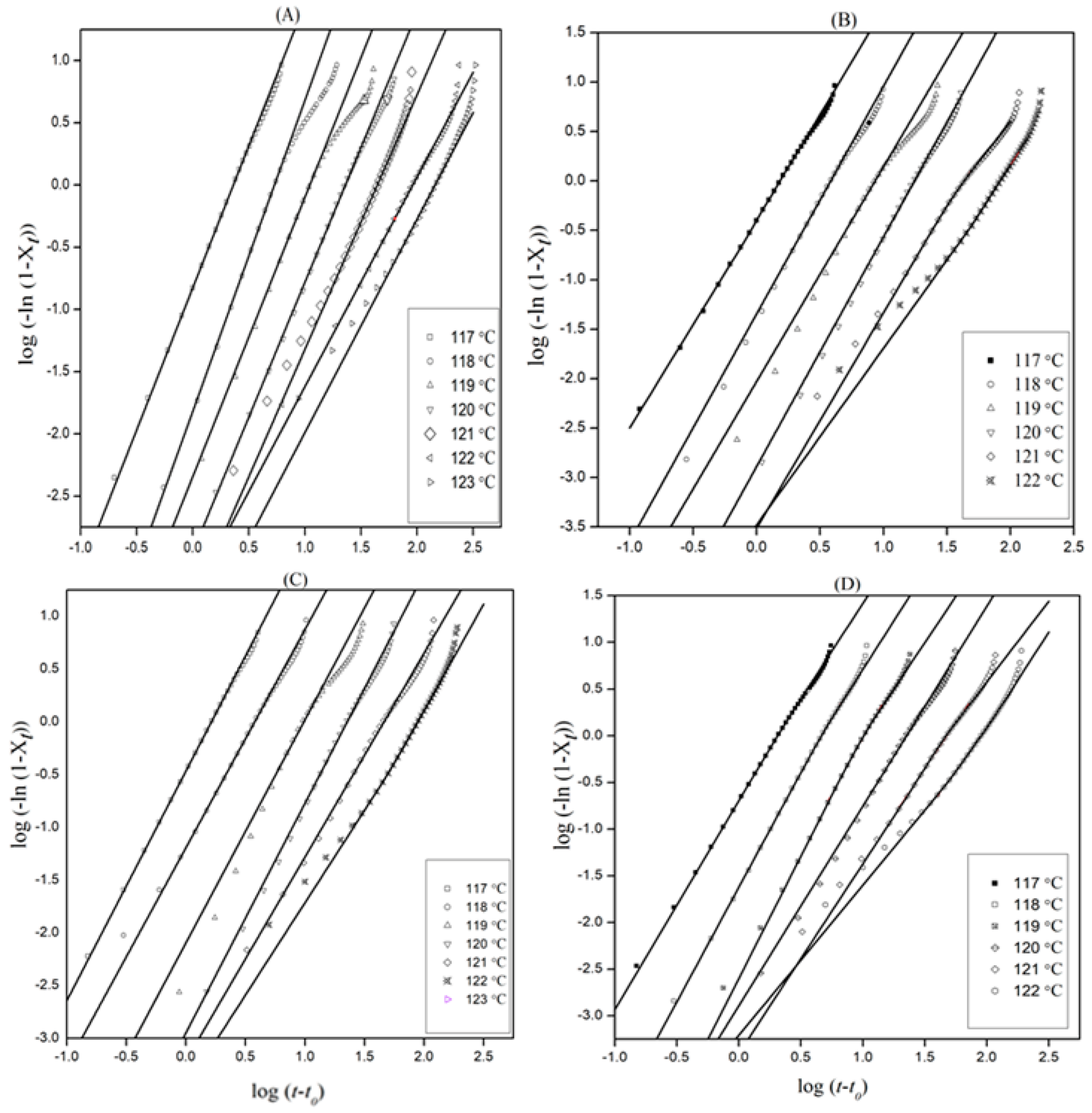
3.1. Isothermal Crystallization Behavior
3.1.1. Evaluation of Relative Crystallinity
3.1.2. The Avrami Model
3.1.3. Activation Energy
3.2. Morphological Analysis of the Nanocomposites
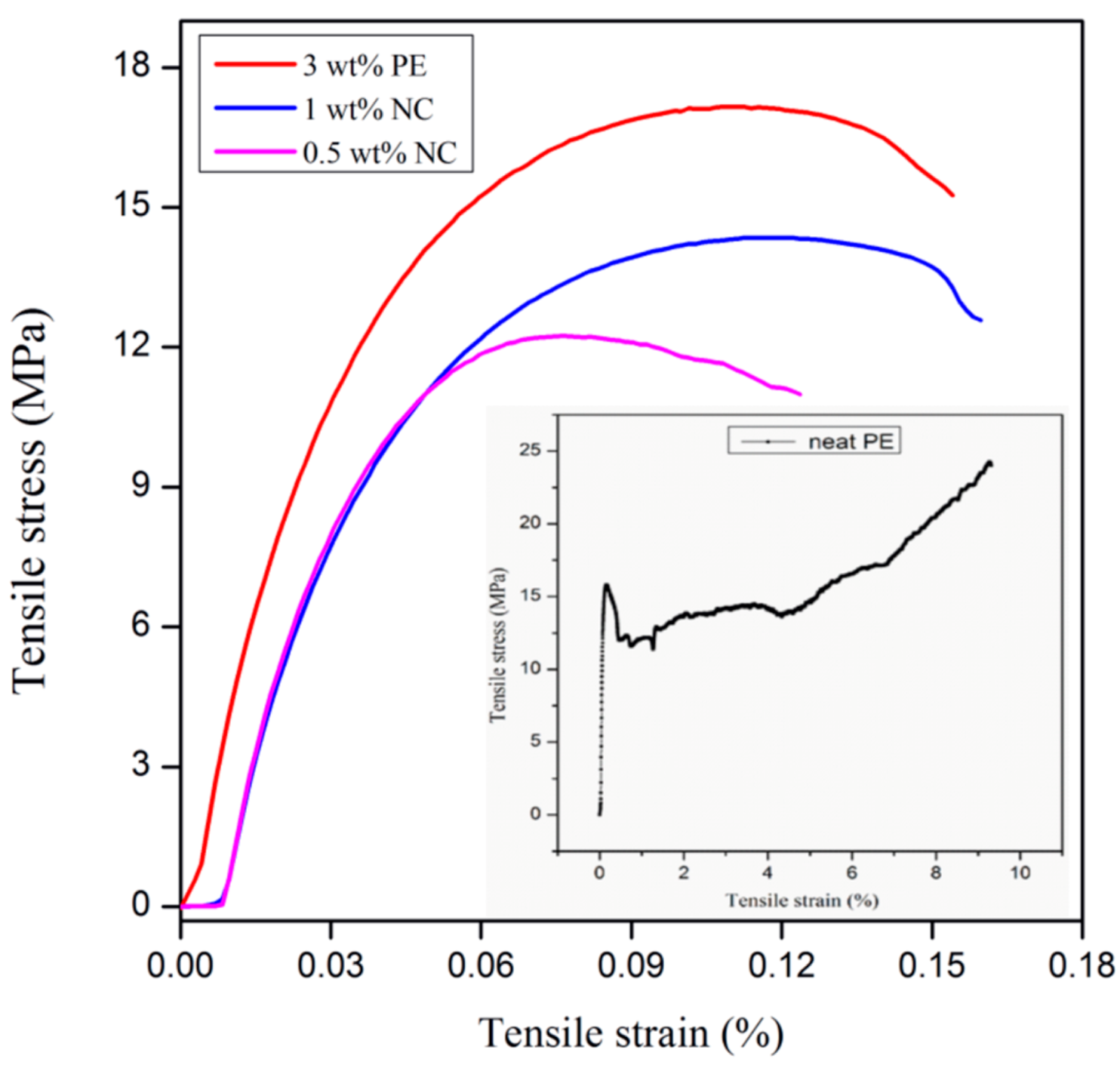
3.3. Tensile Characterization
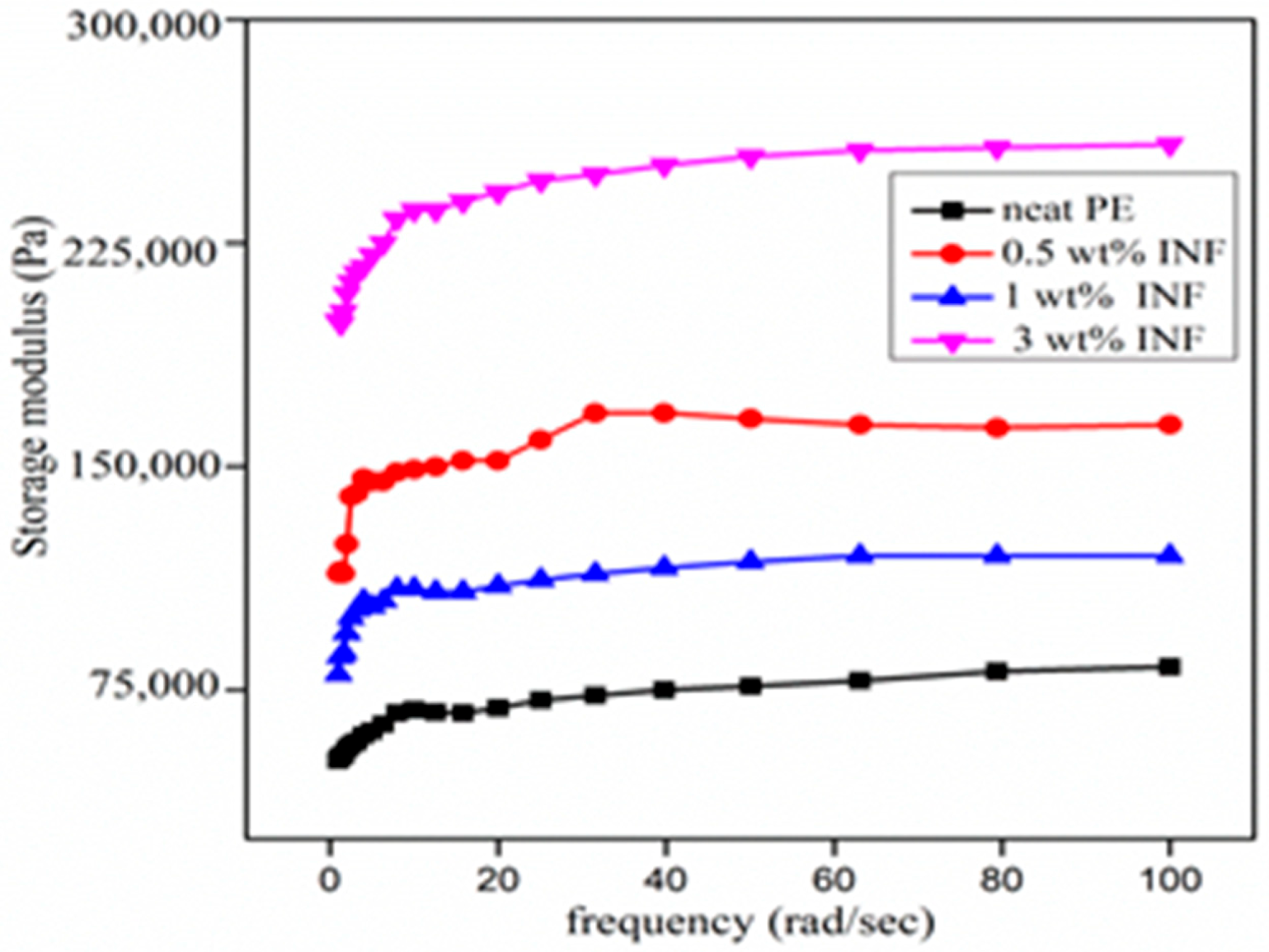
3.4. Rheological Characterization
3.4.1. Viscoelastic Properties versus Strain
3.4.2. Viscoelastic Properties versus Frequency
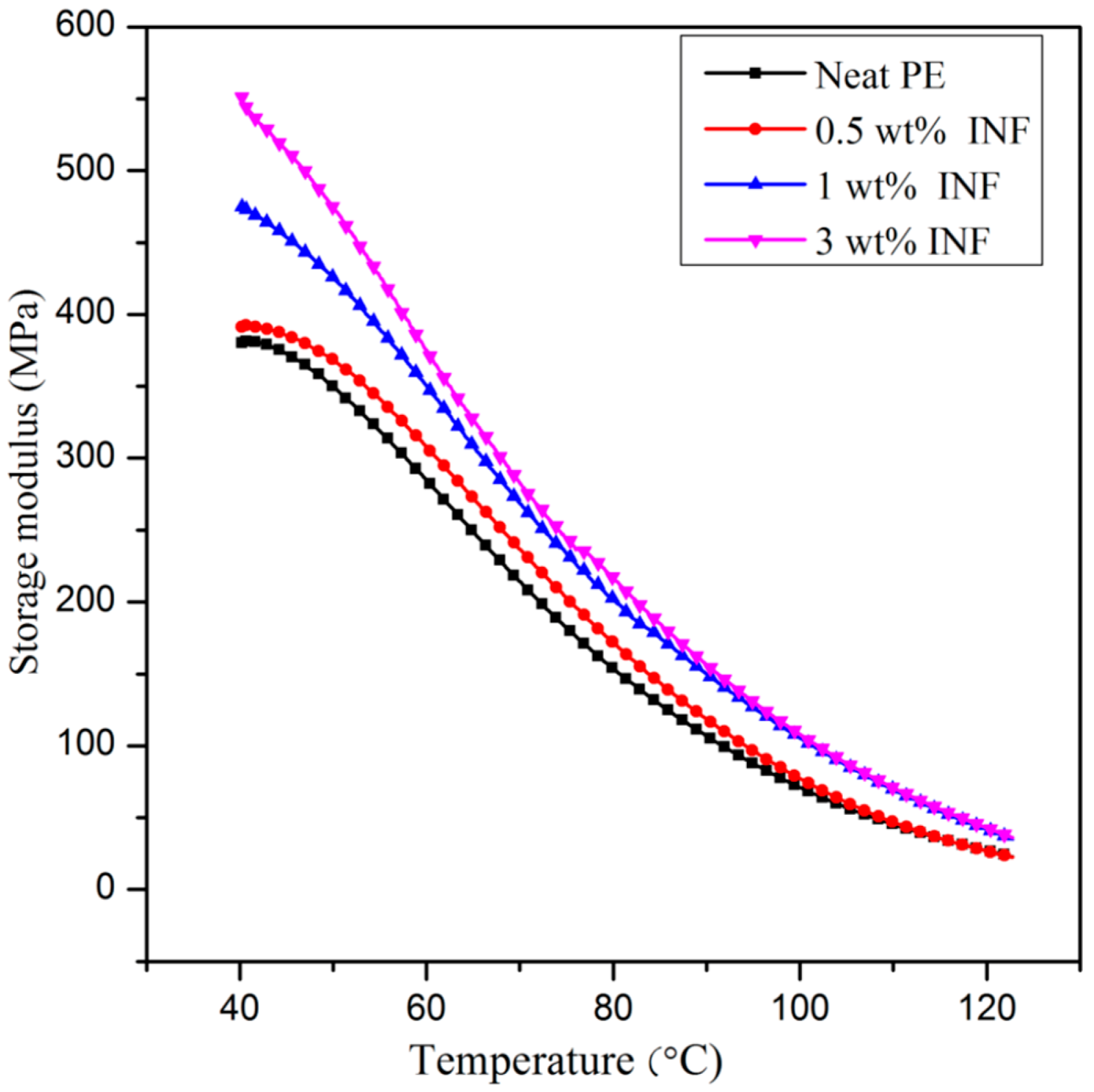
3.5. Dynamic Mechanical Analysis
3.6. Contact Angle Measurements
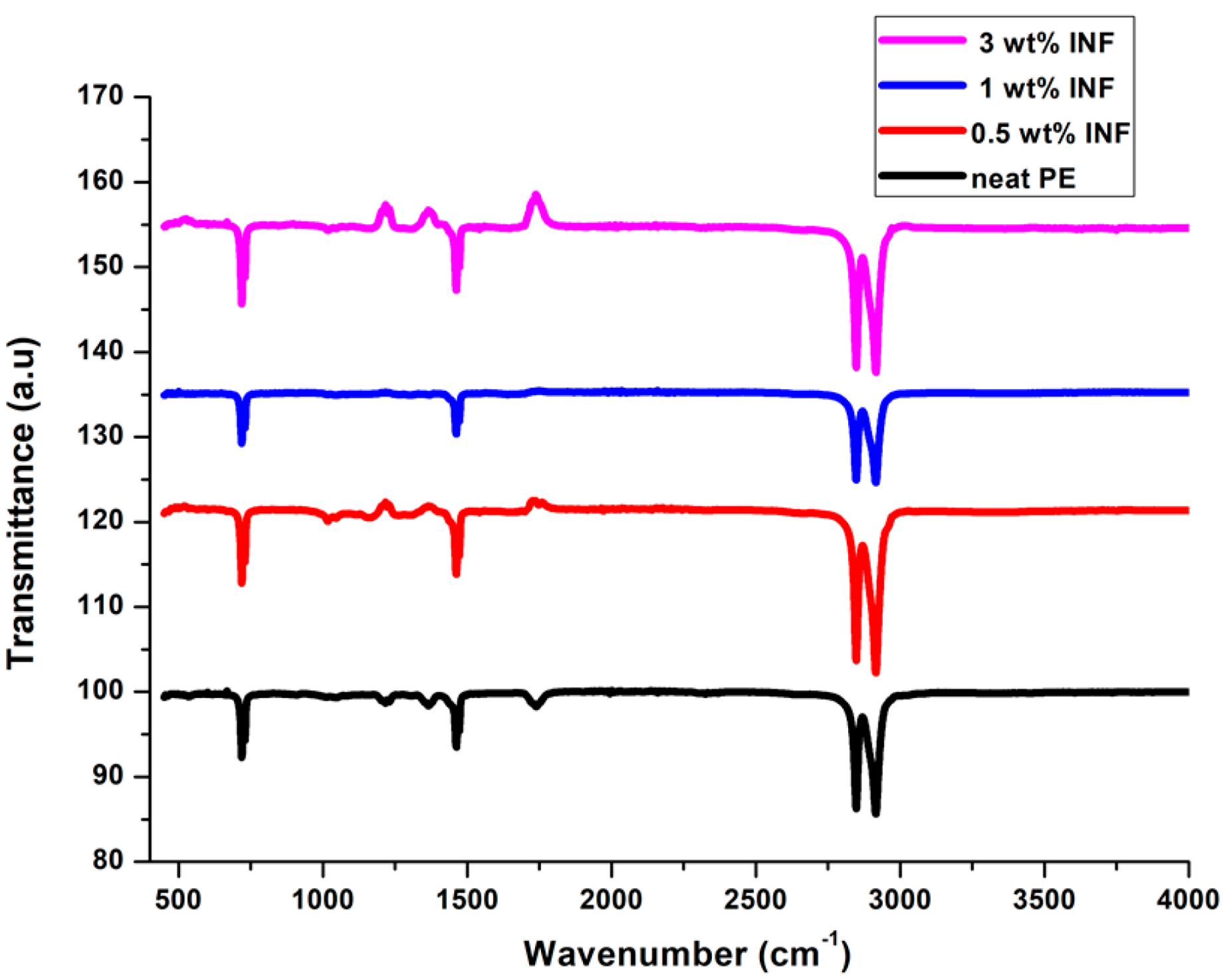
3.7. FTIR Spectra
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Joseph, B.; K, S.V.; Sabu, C.; Kalarikkal, N.; Thomas, S. Cellulose nanocomposites: Fabrication and biomedical applications. J. Bioresour. Bioprod. 2020, 5, 223–237. [Google Scholar] [CrossRef]
- Kathirselvam, M.; Kumaravel, A.; Arthanarieswaran, V.P.; Saravanakumar, S.S. Isolation and characterization of cellulose fibers from Thespesia populnea barks: A study on physicochemical and structural properties. Int. J. Biol. Macromol. 2019, 129, 396–406. [Google Scholar] [CrossRef]
- Grégoire, M.; Barthod-Malat, B.; Labonne, L.; Evon, P.; De Luycker, E.; Ouagne, P. Investigation of the potential of hemp fibre straws harvested using a combine machine for the production of technical load-bearing textiles. Ind. Crops Prod. 2020, 145, 111988. [Google Scholar] [CrossRef]
- Mokhena, T.C.; John, M.J. Cellulose nanomaterials: New generation materials for solving global issues. Cellulose 2020, 27, 1149–1194. [Google Scholar] [CrossRef]
- Amalraj, A.; Gopi, S.; Thomas, S.; Haponiuk, J.T. Cellulose Nanomaterials in Biomedical, Food, and Nutraceutical Applications: A Review. Macromol. Symp. 2018, 380, 1800115. [Google Scholar] [CrossRef]
- Kargarzadeh, H.; Mariano, M.; Gopakumar, D.; Ahmad, I.; Thomas, S.; Dufresne, A.; Huang, J.; Lin, N. Advances in cellulose nanomaterials. Cellulose 2018, 25, 2151–2189. [Google Scholar] [CrossRef]
- Kargarzadeh, H.; Ahmad, I.; Thomas, S.; Dufresne, A. (Eds.) Handbook of Nanocellulose and Cellulose Nanocomposites; Wiley-VCH Verlag GmbH & Company: Weinheim, Germany, 2017. [Google Scholar]
- Gurunathan, T.; Mohanty, S.; Nayak, S.K. A review of the recent developments in biocomposites based on natural fibres and their application perspectives. Compos. Part A Appl. Sci. Manuf. 2015, 77, 1–25. [Google Scholar] [CrossRef]
- Dos Santos Rosa, D.; Maria, D. Biocomposites: Influence of Matrix Nature and Additives on the Properties and Biodegradation Behaviour. In Biodegradation—Engineering and Technology; InTech: Rijeka, Croatia, 2013; pp. 433–475. [Google Scholar]
- Hina, K.; Zou, H.; Qian, W.; Zuo, D.; Yi, C. Preparation and performance comparison of cellulose-based activated carbon fibres. Cellulose 2018, 25, 607–617. [Google Scholar] [CrossRef]
- Rong, M.Z.; Zhang, M.Q.; Ruan, W.H. Surface modification of nanoscale fillers for improving properties of polymer nanocomposites: A review. Mater. Sci. Technol. 2006, 22, 787–796. [Google Scholar] [CrossRef]
- Šupová, M.; Martynková, G.S.; Barabaszová, K. Effect of nanofillers dispersion in polymer matrices: A review. Sci. Adv. Mater. 2011, 3, 1–25. [Google Scholar] [CrossRef]
- Jose, J.P.; Chazeau, L.; Cavaillé, J.Y.; Varughese, K.T.; Thomas, S. Nucleation and nonisothermal crystallization kinetics in cross-linked polyethylene/zinc oxide nanocomposites. RSC Adv. 2014, 4, 31643–31651. [Google Scholar] [CrossRef]
- Tharayil, A.; Banerjee, S.; Kar, K.K. Dynamic mechanical properties of zinc oxide reinforced linear low density polyethylene composites. Mater. Res. Express 2019, 6, 055301. [Google Scholar] [CrossRef]
- Pasquini, D.; Teixeira, E.d.M.; Curvelo, A.A.d.S.; Belgacem, M.N.; Dufresne, A. Surface esterification of cellulose fibres: Processing and characterisation of low-density polyethylene/cellulose fibres composites. Compos. Sci. Technol. 2008, 68, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, F.V.; Trindade, G.N.; Lona, L.M.F.; Bernardes, J.S.; Gouveia, R.F. LDPE-based composites reinforced with surface modified cellulose fibres: 3D morphological and morphometrical analyses to understand the improved mechanical performance. Eur. Polym. J. 2019, 117, 105–113. [Google Scholar] [CrossRef]
- Chartrand, A.; Lavoie, J.M.; Huneault, M.A. Surface modification of microcrystalline cellulose (MCC) and its application in LDPE-based composites. J. Appl. Polym. Sci. 2017, 134. [Google Scholar] [CrossRef] [Green Version]
- Roy, D.; Kotula, A.P.; Natarajan, B.; Gilman, J.W.; Fox, D.M.; Migler, K.B. Effect of cellulose nanocrystals on crystallization kinetics of polycaprolactone as probed by Rheo-Raman. Polymer (Guildf.) 2018, 153, 70–77. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, H.R.; Huang, C.; Xiong, L.; Luo, J.; Chen, X. De Study on non-isothermal crystallization behavior of isotactic polypropylene/bacterial cellulose composites. RSC Adv. 2017, 7, 42113–42122. [Google Scholar] [CrossRef] [Green Version]
- Chirayil, C.J.; Joy, J.; Mathew, L.; Mozetic, M.; Koetz, J.; Thomas, S. Isolation and characterization of cellulose nanofibrils from Helicteres isora plant. Ind. Crops Prod. 2014, 59, 27–34. [Google Scholar] [CrossRef]
- Zhang, M.C.; Guo, B.H.; Xu, J. A review on polymer crystallization theories. Crystals 2017, 7, 4. [Google Scholar] [CrossRef]
- Tang, X.; Chen, W.; Li, L. The Tough Journey of Polymer Crystallization: Battling with Chain Flexibility and Connectivity. Macromolecules 2019, 52, 3575–3591. [Google Scholar] [CrossRef]
- Hao, W.; Yang, W.; Cai, H.; Huang, Y. Non-isothermal crystallization kinetics of polypropylene/silicon nitride nanocomposites. Polym. Test. 2010, 29, 527–533. [Google Scholar] [CrossRef]
- Jeon, K.; Lumata, L.; Tokumoto, T.; Steven, E.; Brooks, J.; Alamo, R.G. Low electrical conductivity threshold and crystalline morphology of single-walled carbon nanotubes—High density polyethylene nanocomposites characterized by SEM, Raman spectroscopy and AFM. Polymer 2007, 48, 4751–4764. [Google Scholar] [CrossRef]
- Chen, Y.; Yao, X.; Gu, Q.; Pan, Z. Non-isothermal crystallization kinetics of poly (lactic acid)/graphene nanocomposites. J. Polym. Eng. 2013, 33, 163–171. [Google Scholar] [CrossRef]
- Jha, S.K.; Karthika, S.; Radhakrishnan, T.K. Modelling and control of crystallization process. Resour. Technol. 2017, 3, 94–100. [Google Scholar] [CrossRef]
- Keridou, I.; del Valle, L.J.; Funk, L.; Turon, P.; Yousef, I.; Franco, L.; Puiggalí, J. Isothermal crystallization kinetics of poly(4-hydroxybutyrate) biopolymer. Materials 2019, 12, 2488. [Google Scholar] [CrossRef] [Green Version]
- Bai, J.; Fang, H.; Zhang, Y.; Wang, Z. Studies on crystallization kinetics of bimodal long chain branched polylactides. CrystEngComm 2014, 16, 2452–2461. [Google Scholar] [CrossRef]
- Avrami, M. Kinetics of phase change. I: General theory. J. Chem. Phys. 1939, 7, 1103–1112. [Google Scholar] [CrossRef]
- Avrami, M. Kinetics of phase change. II Transformation-time relations for random distribution of nuclei. J. Chem. Phys. 1940, 8, 212–224. [Google Scholar] [CrossRef]
- Avrami, M. Granulation, phase change, and microstructure kinetics of phase change. III. J. Chem. Phys. 1941, 9, 177–184. [Google Scholar] [CrossRef]
- Papageorgiou, G.; Bikiaris, D.N.; Chrissafis, K. A different approach for the study of the crystallization kinetics in polymers. Key study: Poly(ethylene terephthalate)/ SiO2 nanocomposites. Polym. Int. 2010, 59, 1630–1638. [Google Scholar] [CrossRef]
- Hoffman, J.D.; Lauritzen, J.I. Crystallization of bulk polymers with chain folding: Theory of growth of lamellar spherulites. J. Res. Natl. Bur. Stand. Sect. A Phys. Chem. 1961, 65, 297. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.R.; Lee, H.Y.; Chang, F.C. Crystallization kinetics and crystallization behavior of syndiotactic polystyrene/clay nanocomposites. J. Polym. Sci. Part B Polym. Phys. 2001, 39, 2097–2107. [Google Scholar] [CrossRef]
- Chirayil, C.J. Development of High Performance Polymer Nanocomposites Using Cellulose Nanofibrils from Isora Fibers: A Wood Substitute; Mahatma Gandhi University: Kottayam, India, 2014. [Google Scholar]
- Harnisch, K.; Muschik, H. Determination of the Avrami exponent of partially crystallized polymers by DSC- (DTA-) analyses. Colloid Polym. Sci. 1983, 261, 908–913. [Google Scholar] [CrossRef]
- Kamal, M.R.; Chu, E. Isothermal and nonisothermal crystallization of polyethylene. Polym. Eng. Sci. 1983, 23, 27–31. [Google Scholar] [CrossRef]
- Tarani, E.; Papageorgiou, G.Z.; Bikiaris, D.N.; Chrissafis, K. Kinetics of crystallization and thermal degradation of an isotactic polypropylene matrix reinforced with graphene/glass-fiber filler. Molecules 2019, 24, 1984. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Niu, Y.; Yang, L.; Xie, W.; Li, H.; Gan, Z.; Wang, Z. Morphology, rheology and crystallization behavior of polylactide composites prepared through addition of five-armed star polylactide grafted multiwalled carbon nanotubes. Polymer 2010, 51, 730–737. [Google Scholar] [CrossRef]
- Seven, K.M.; Cogen, J.M.; Gilchrist, J.F. Nucleating agents for high-density polyethylene—A review. Polym. Eng. Sci. 2016, 56, 541–554. [Google Scholar] [CrossRef] [Green Version]
- Mistretta, M.C.; Fontana, P.; Ceraulo, M.; Morreale, M.; La Mantia, F.P. Effect of compatibilization on the photo-oxidation behaviour of polyethylene/polyamide 6 blends and their nanocomposites. Polym. Degrad. Stab. 2015, 112, 192–197. [Google Scholar] [CrossRef]
- Pothan, L.A.; Thomas, S. Effect of hybridization and chemical modification on the water-absorption behavior of banana fiber-reinforced polyester composites. J. Appl. Polym. Sci. 2004, 91, 3865. [Google Scholar] [CrossRef]
- Menard, K.P.; Noah, R. Dynamic mechanical analysis in the analysis of polymers and rubbers. In Encyclopedia of Polymer Science and Technology; John Wiley & Sons: Hoboken, NJ, USA, 2015; pp. 1–33. ISBN 9780471440260. [Google Scholar]
- Anderson, B.J.; Zukoski, C.F. Rheology and microstructure of polymer nanocomposite melts: Variation of polymer segment-surface interaction. Langmuir 2010, 26, 8709–8720. [Google Scholar] [CrossRef]
- Sarvestani, A.S. Modeling the solid-like behavior of entangled polymer nanocomposites at low frequency regimes. Eur. Polym. J. 2008, 44, 263–269. [Google Scholar] [CrossRef]
- Kopal, I.; Harničárová, M.; Valíček, J.; Kušnerová, M. Modeling the temperature dependence of dynamic mechanical properties and visco-elastic behavior of thermoplastic polyurethane using artificial neural network. Polymers 2017, 9, 519. [Google Scholar] [CrossRef] [PubMed] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Tc (°C) | t1/2 (min) Exptl | t1/2 (min) Calculated | Slope (n) | Δn | KA1/n (min) | ΔKA 1/n (min) | r2 |
|---|---|---|---|---|---|---|---|---|
| Neat | 117 | 2.0 | 2.0 | 2.28 | 0.03 | 0.433 | −0.0006 | 0.9997 |
| 118 | 4..6 | 4.6 | 2.38 | 0.09 | 0.185 | −0.0003 | 0.9983 | |
| 119 | 9.3 | 9.5 | 2.16 | 0.10 | 0.089 | −0.0003 | 0.9968 | |
| 120 | 19.1 | 19.2 | 2.15 | 0.05 | 0.044 | −0.0001 | 0.9990 | |
| 121 | 37.2 | 37.2 | 2.02 | 0.03 | 0.022 | −0.0001 | 0.9997 | |
| 122 | 74 | 72.9 | 1.75 | 0.04 | 0.011 | −0.0001 | 0.9991 | |
| 123 | 120 | 119.6 | 1.63 | 0.07 | 0.007 | −0.0001 | 0.9974 | |
| 0.5 wt% INF/PE | 117 | 1.3 | 1.3 | 2.13 | 0.03 | 0.652 | 0.0004 | 0.9998 |
| 118 | 3.2 | 3.3 | 2.25 | 0.07 | 0.261 | −0.0002 | 0.9987 | |
| 119 | 7.1 | 7.3 | 2.17 | 0.11 | 0.116 | −0.0005 | 0.9959 | |
| 120 | 14.7 | 14.8 | 2.23 | 0.07 | 0.057 | −0.0048 | 0.9986 | |
| 121 | 35.2 | 35.7 | 1.98 | 0.05 | 0.023 | −0.0002 | 0.9979 | |
| 122 | 69.1 | 69.1 | 1.96 | 0.04 | 0.007 | −0.0001 | 0.9995 | |
| 1 wt% INF/PE | 117 | 1.4 | 1.4 | 2.17 | 0.02 | 0.611 | 0.0001 | 0.9999 |
| 118 | 3.4 | 3.2 | 2.09 | 0.04 | 0.266 | −0.0004 | 0.9995 | |
| 119 | 8.1 | 8.2 | 2.17 | 0.15 | 0.103 | −0.0005 | 0.9932 | |
| 120 | 16.7 | 18.9 | 2.24 | 0.07 | 0.045 | −0.0002 | 0.9986 | |
| 121 | 36.7 | 37.2 | 1.98 | 0.08 | 0.022 | −0.0002 | 0.9977 | |
| 122 | 73.4 | 73.3 | 1.96 | 0.02 | 0.011 | −0.0001 | 0.9999 | |
| 123 | 111.8 | 112.2 | 1.26 | 0.05 | 0.007 | −0.0002 | 0.9963 | |
| 3 wt% INF/PE | 117 | 1.7 | 1.7 | 2.19 | 0.02 | 0.484 | −0.0002 | 0.9999 |
| 118 | 4.0 | 4.0 | 2.34 | 0.03 | 0.214 | −0.0002 | 0.9996 | |
| 119 | 8.6 | 8.8 | 2.35 | 0.07 | 0.097 | −0.0002 | 0.9984 | |
| 120 | 18.7 | 18.8 | 2.17 | 0.07 | 0.045 | −0.0002 | 0.9978 | |
| 121 | 39.0 | 39.4 | 2.01 | 0.04 | 0.021 | −0.0001 | 0.9992 | |
| 122 | 73.9 | 73.8 | 1.82 | 0.03 | 0.011 | −0.0011 | 0.9994 | |
| 123 | 108 | 108.1 | 1.48 | 0.07 | 0.007 | −0.0001 | 0.9954 |
| Sample | Activation Energy (kJ mol−1) | r2 |
|---|---|---|
| Neat PE | −7.71 | 0.9899 |
| 0.5 wt% PE | −17.34 | 0.9968 |
| 1 wt% PE | −21.29 | 0.9873 |
| 3 wt% PE | −14.42 | 0.9848 |
| Sample | Contact Angle (Degrees) | Work of Adhesion (WA, mJ/m2) | Interfacial Energy γsl = γs + γl − WA (mJ/m2) * | Spreading Coefficient Sc = γs − γsl − γl (mJ/m2) * | Interaction Parameter Φ = [(1 + cosθ)γl]/2(γs γl)1/2 * |
|---|---|---|---|---|---|
| Neat PE | 87.5 | 75.92 | 21.76 | −69.68 | 1.63 |
| 0.5 wt% | 81 | 83.07 | 19.78 | −62.53 | 1.45 |
| 1 wt% | 75 | 91.75 | 16.66 | −53.85 | 1.29 |
| 3 wt% | 68 | 100.04 | 15.22 | −45.56 | 1.14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jose, C.; Chan, C.H.; Winie, T.; Joseph, B.; Tharayil, A.; Maria, H.J.; Volova, T.; La Mantia, F.P.; Rouxel, D.; Morreale, M.; et al. Thermomechanical Analysis of Isora Nanofibril Incorporated Polyethylene Nanocomposites. Polymers 2021, 13, 299. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13020299
Jose C, Chan CH, Winie T, Joseph B, Tharayil A, Maria HJ, Volova T, La Mantia FP, Rouxel D, Morreale M, et al. Thermomechanical Analysis of Isora Nanofibril Incorporated Polyethylene Nanocomposites. Polymers. 2021; 13(2):299. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13020299
Chicago/Turabian StyleJose, Cintil, Chin Han Chan, Tan Winie, Blessy Joseph, Abhimanyu Tharayil, Hanna J Maria, Tatiana Volova, Francesco Paolo La Mantia, Didier Rouxel, Marco Morreale, and et al. 2021. "Thermomechanical Analysis of Isora Nanofibril Incorporated Polyethylene Nanocomposites" Polymers 13, no. 2: 299. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13020299