
Custom-Made Poly(urethane) Coatings Improve the Mechanical Properties of Bioactive Glass Scaffolds Designed for Bone Tissue Engineering

 , , ,
, , ,  and
and
Abstract
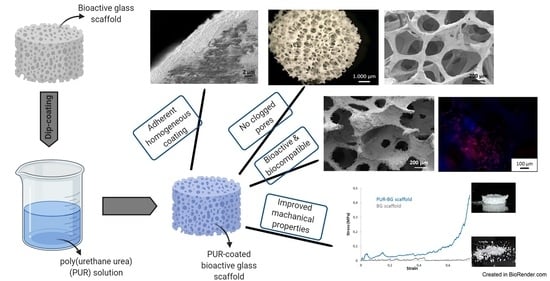
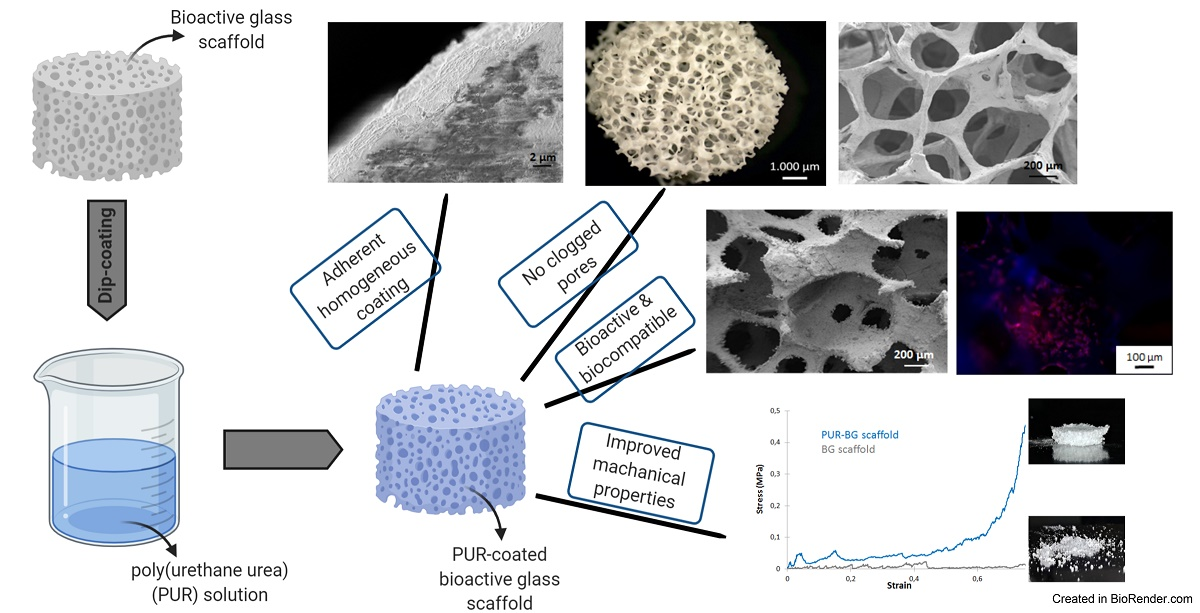
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Poly(urethane) Synthesis
2.3. Poly(urethane) Nomenclature
2.4. Poly(urethane) Characterization
2.4.1. Attenuated Total Reflectance Fourier Transform Infrared (ATR-FTIR) Spectroscopy
2.4.2. Size Exclusion Chromatography (SEC)
2.4.3. Static Contact Angle Measurements
2.4.4. Mechanical Tensile Tests
2.4.5. Degradation/Dissolution Tests
2.5. Scaffold Fabrication
2.6. Scaffold Coating
2.7. Study of Polymer-BG Bond Strength
2.8. Scaffold Characterization
2.8.1. Scaffold Morphological Characterization
2.8.2. Scaffold Bioactivity
2.8.3. Degradation Tests
2.8.4. Mechanical Tests
2.9. Biological Tests
2.9.1. Preparation of Scaffolds
2.9.2. Cell Seeding and Cultivation
2.9.3. Cell Viability and Staining
2.10. Statistical Analysis
3. Results and Discussion
3.1. Poly(urethane) Characterization
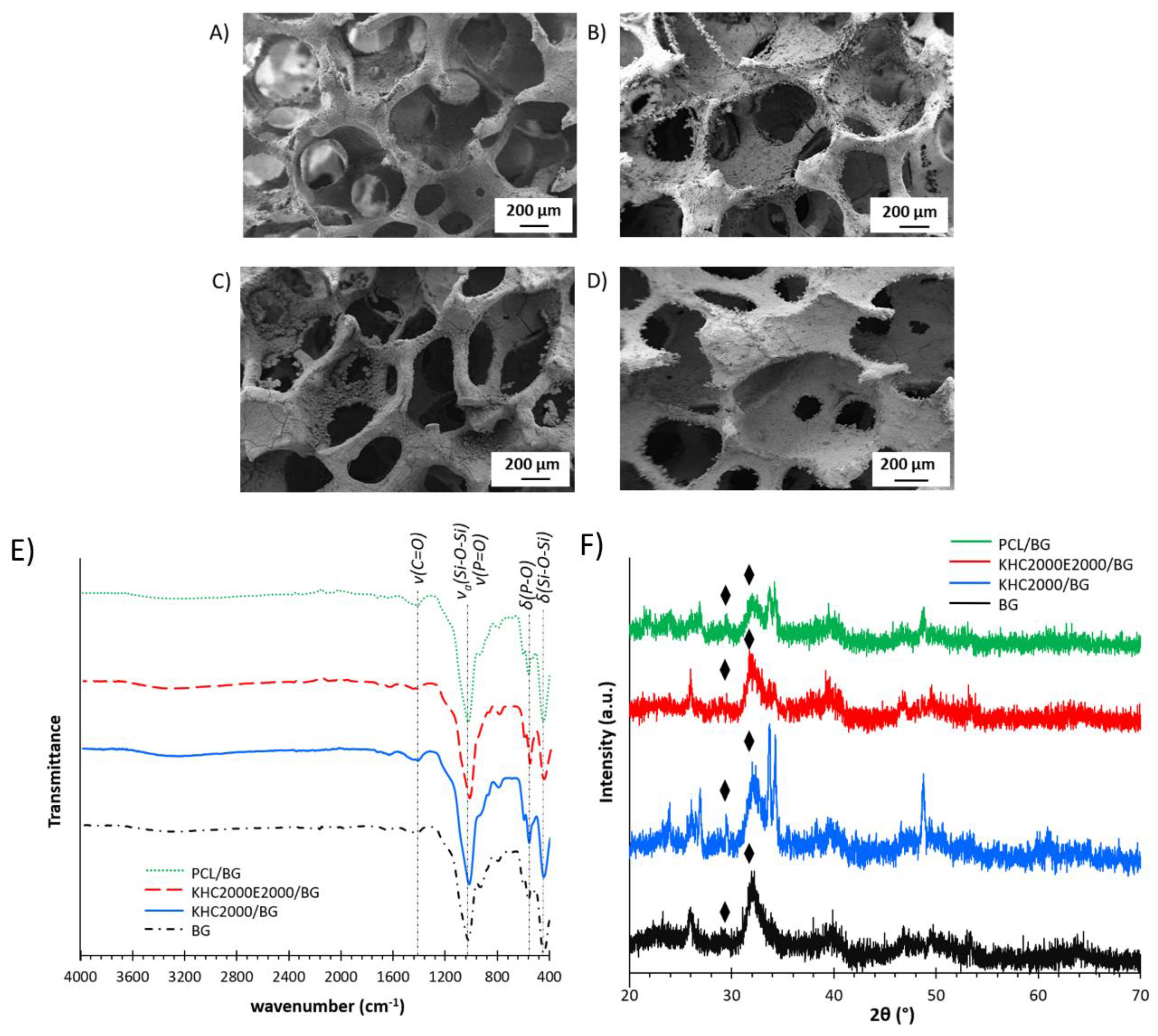
3.1.1. Chemical Characterization
3.1.2. Contact Angle Measurements
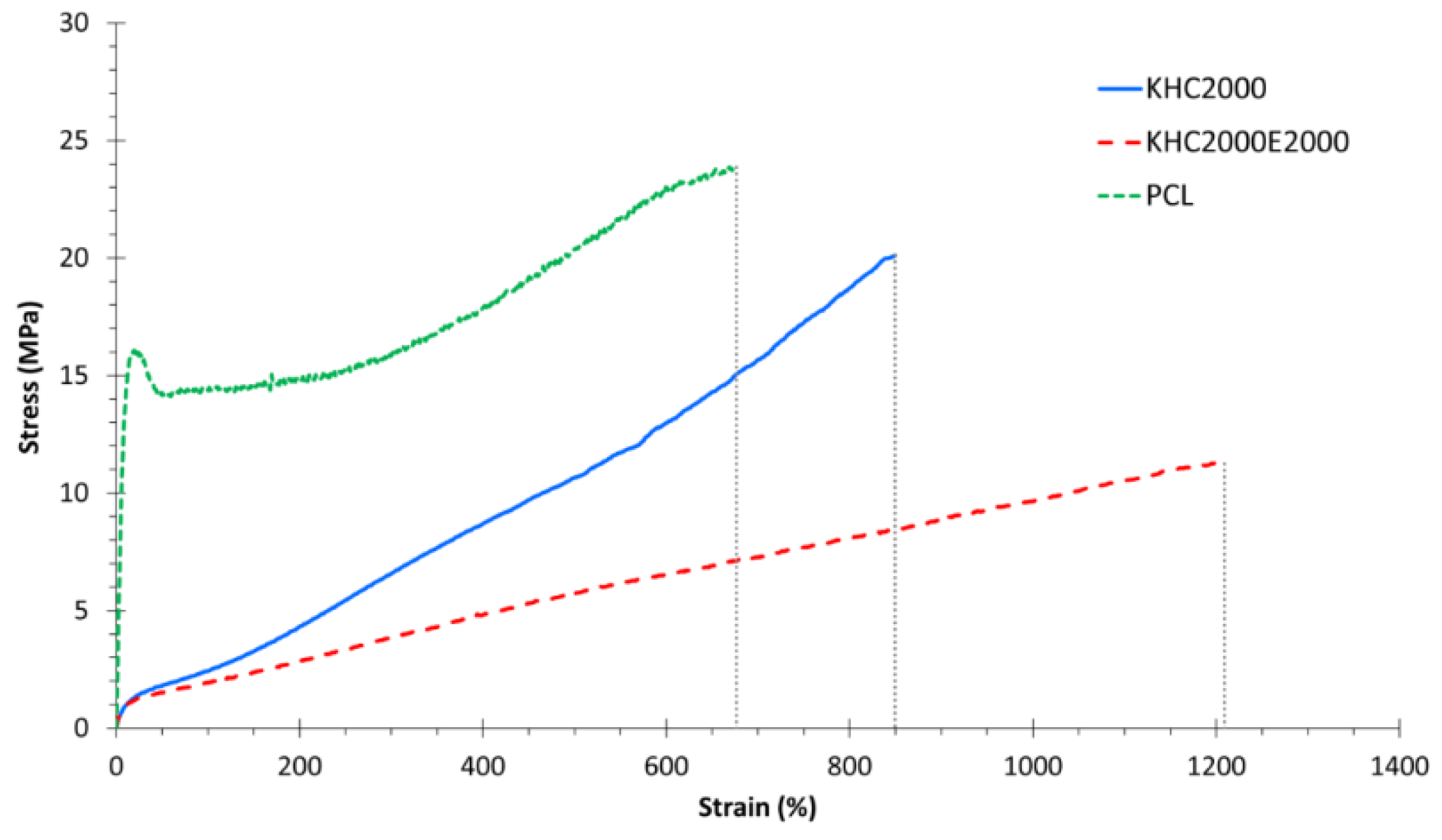
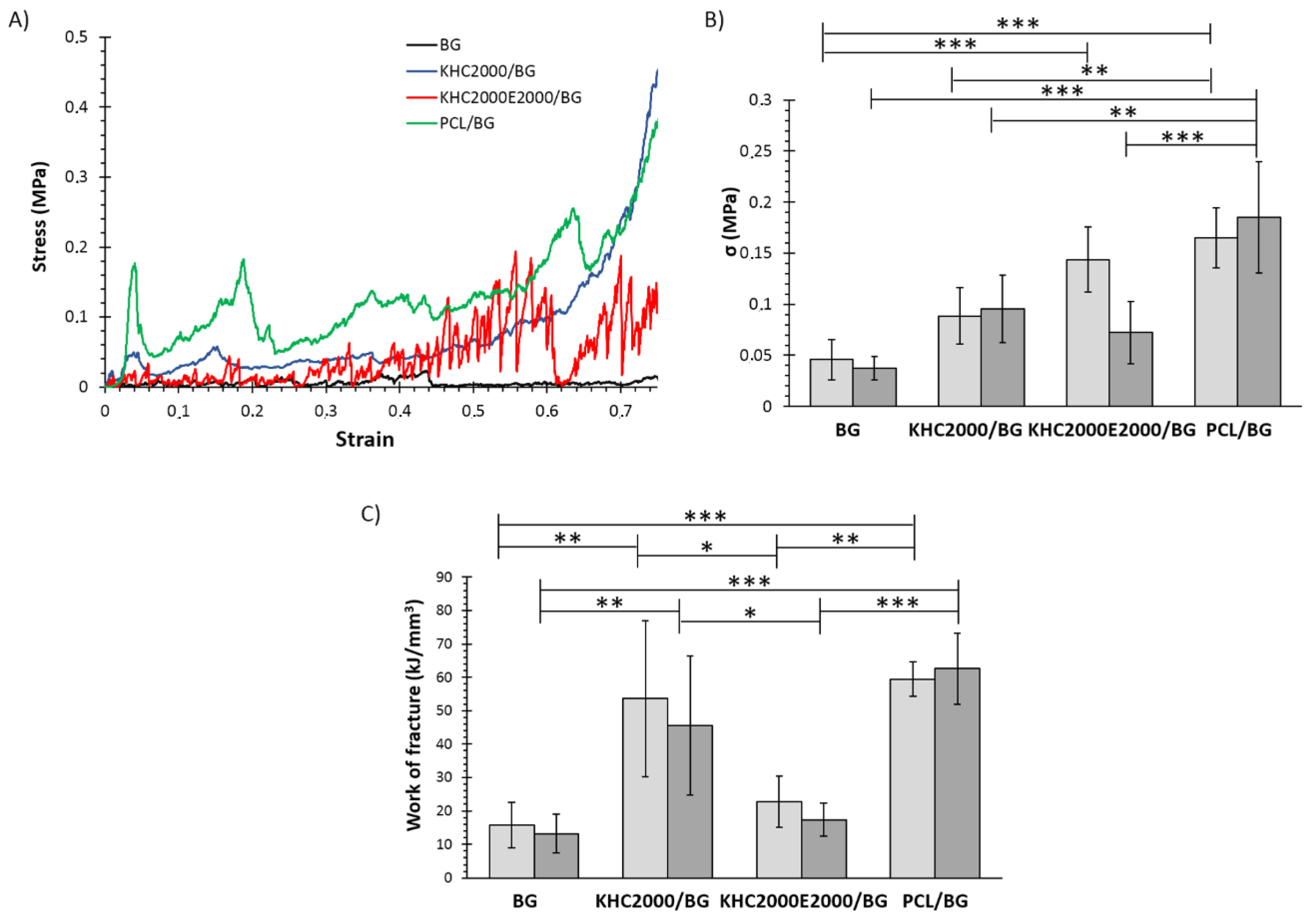
3.1.3. Mechanical Characterization
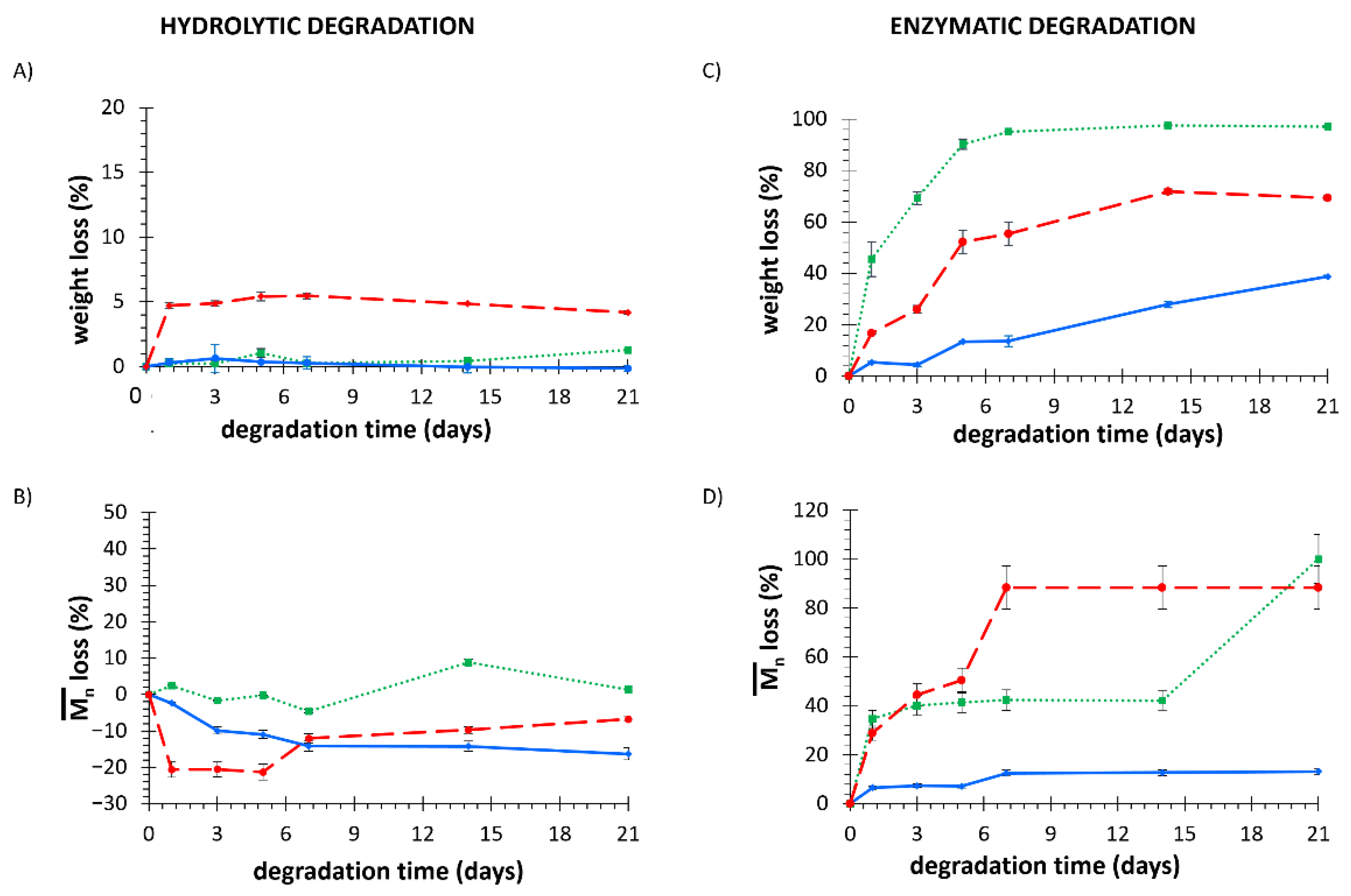
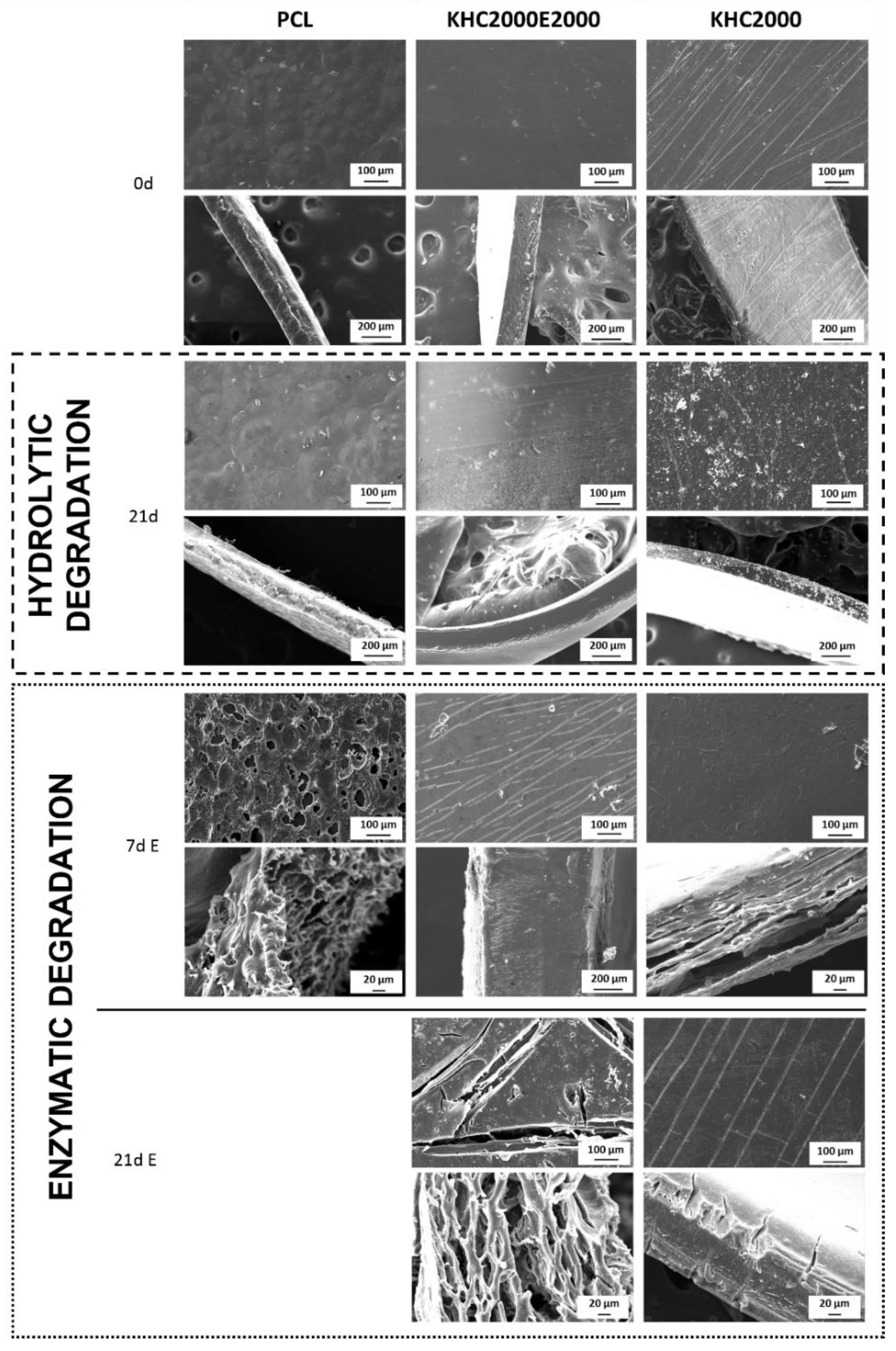
3.1.4. Degradation/Dissolution Tests
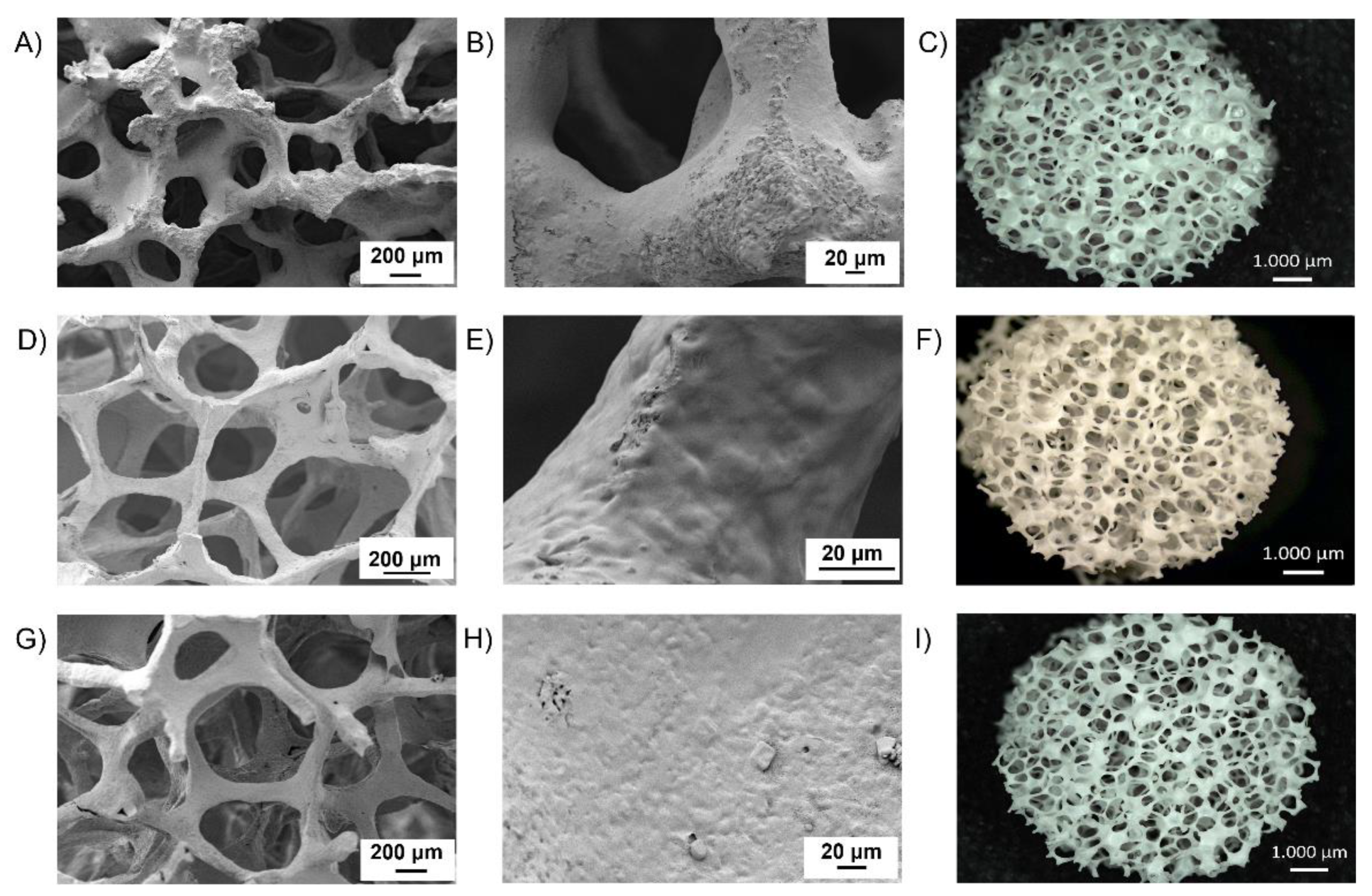
3.2. Pure BG Scaffolds Characterization
3.3. Optimization of BG Scaffold Coating with PURs
3.4. Investigation of Polymer/BG Adhesion
3.5. Characterization of Polymer-Coated BG Scaffolds
3.5.1. Morphological Analysis
3.5.2. Bioactivity Tests
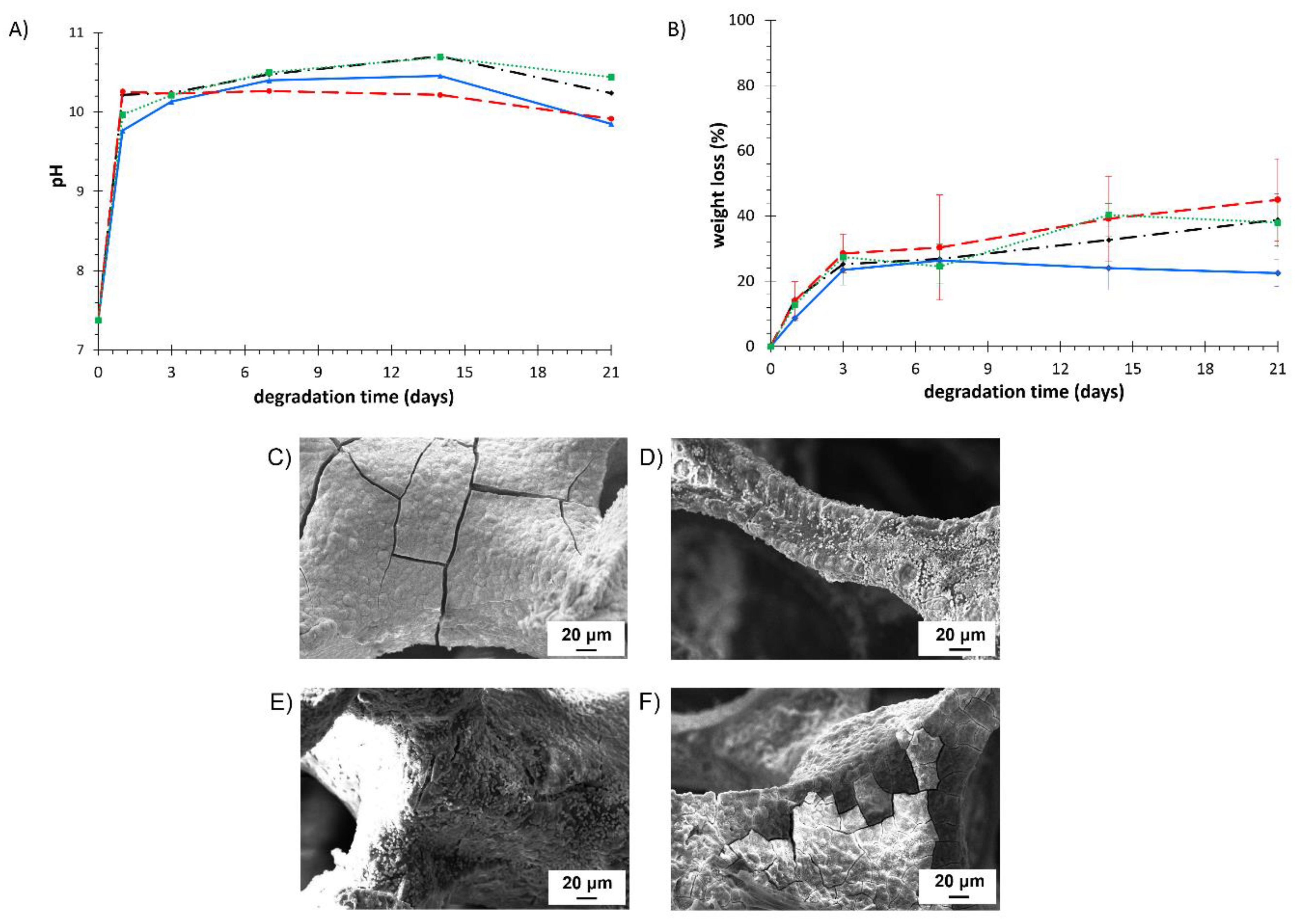
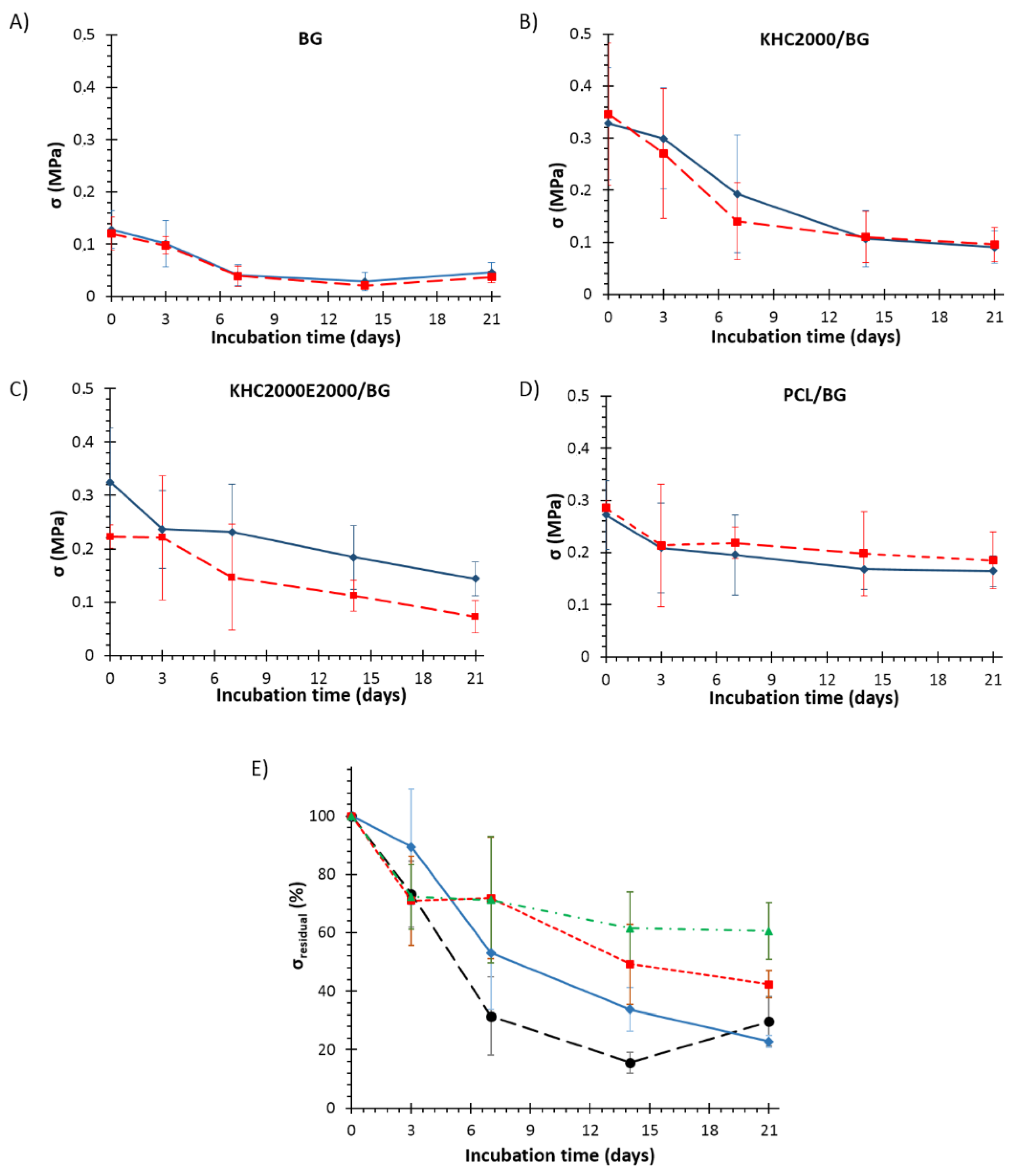
3.5.3. Degradation Tests
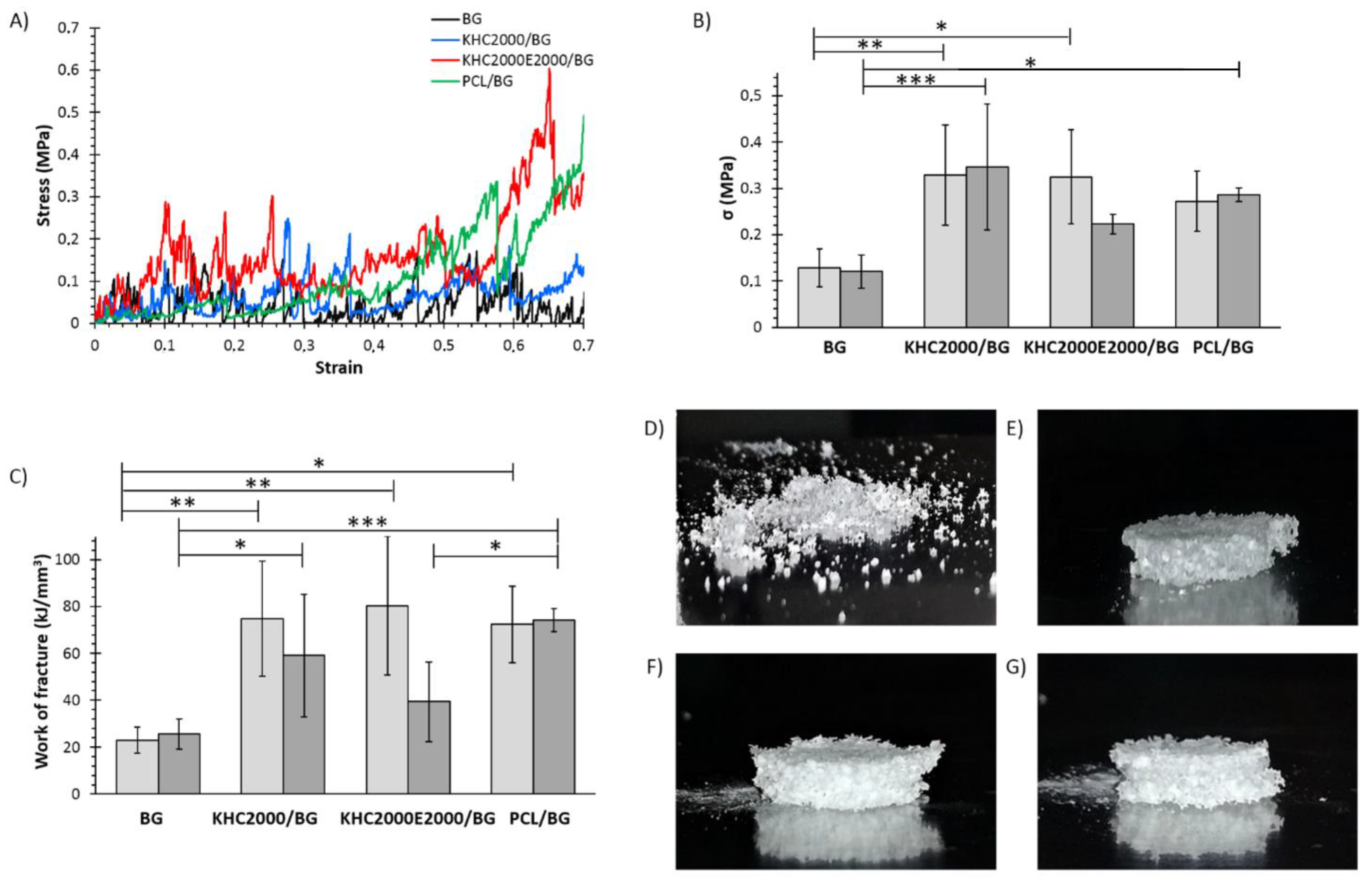
3.5.4. Mechanical Tests
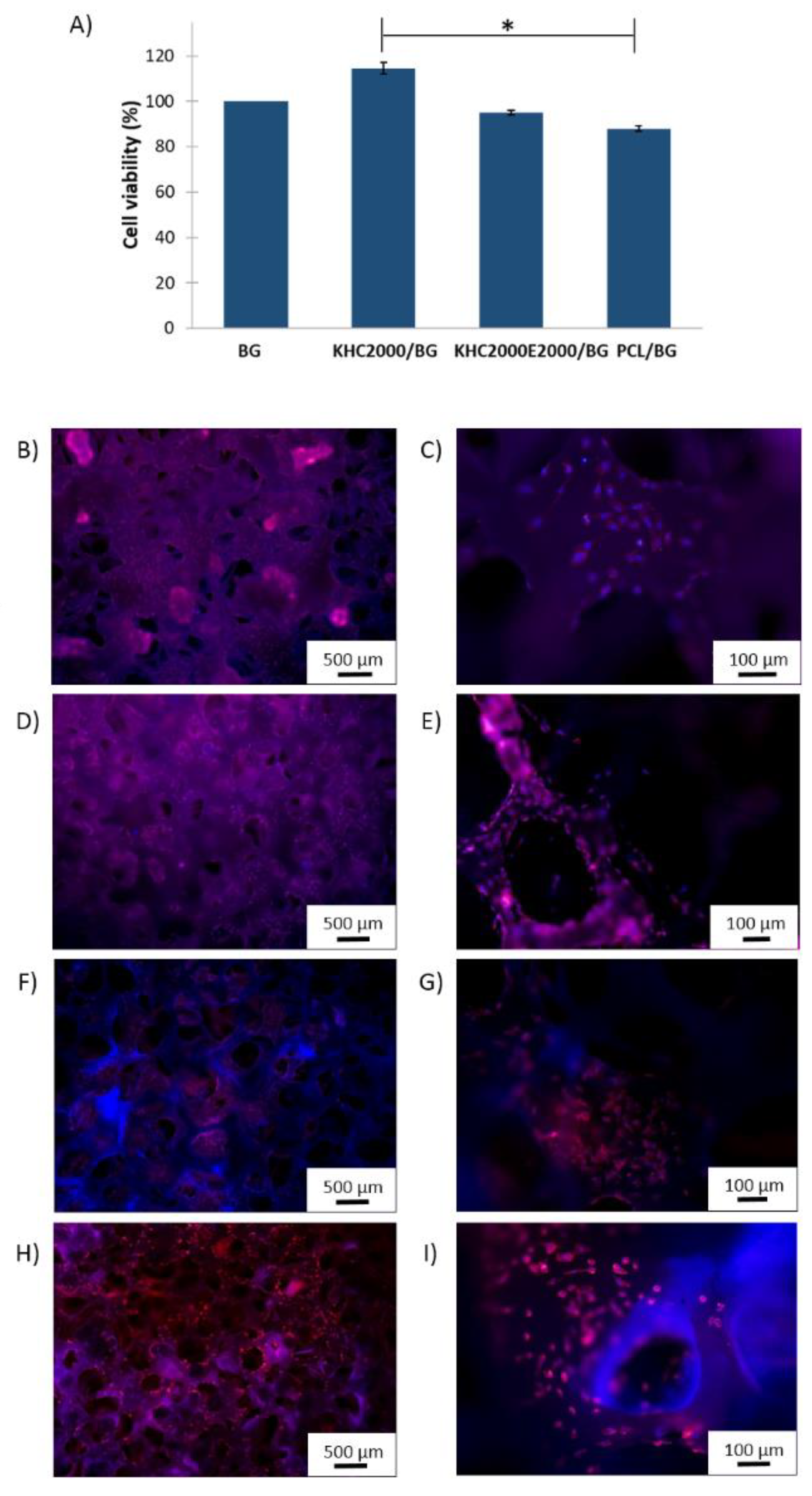
3.5.5. Biological Tests
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- United States Bone and Joint Initiative, U.S. Bone Jt. Initiat. (n.d.). Available online: https://www.usbji.org/ (accessed on 10 September 2021).
- Hsiong, S.X.; Mooney, D.J. Regeneration of vascularized bone. Periodontol. 2000 2006, 41, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Hernlund, E.; Svedbom, A.; Ivergård, M.; Compston, J.; Cooper, C.; Stenmark, J.; McCloskey, E.V.; Jönsson, B.; Kanis, J.A. Osteoporosis in the European Union: Medical management, epidemiology and economic burden. A report prepared in collaboration with the International Osteoporosis Foundation (IOF) and the European Federation of Pharmaceutical Industry Associations (EFPIA). Arch. Osteoporos. 2013, 8, 136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prolo, D.J.; Rodrigo, J.J. Contemporary bone graft physiology and surgery. Clin. Orthop. 1985, 200, 322–342. [Google Scholar] [CrossRef]
- Damien, C.J.; Parsons, J.R. Bone graft and bone graft substitutes: A review of current technology and applications. J. Appl. Biomater. Off. J. Soc. Biomater. 1991, 2, 187–208. [Google Scholar] [CrossRef]
- Baldwin, P.; Li, D.J.; Auston, D.A.; Mir, H.S.; Yoon, R.S.; Koval, K.J. Autograft, allograft, and bone graft substitutes: Clinical evidence and indications for use in the setting of orthopaedic trauma surgery. J. Orthop. Trauma 2019, 33, 203–213. [Google Scholar] [CrossRef]
- Shibuya, N.; Jupiter, D.C. Bone graft substitute: Allograft and xenograft. Clin. Podiatr. Med. Surg. 2015, 32, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Henkel, J.; Woodruff, M.A.; Epari, D.R.; Steck, R.; Glatt, V.; Dickinson, I.C.; Choong, P.F.M.; Schuetz, M.A.; Hutmacher, D.W. Bone Regeneration Based on Tissue Engineering Conceptions—A 21st Century Perspective. Bone Res. 2013, 1, 216–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langer, R.; Vacanti, J. Tissue engineering. Science 1993, 260, 920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hench, L.L. Bioceramics: From concept to clinic. J. Am. Ceram. Soc. 1991, 74, 1487–1510. [Google Scholar] [CrossRef]
- Srinath, P.; Abdul Azeem, P.; Venugopal Reddy, K. Review on calcium silicate-based bioceramics in bone tissue engineering. Int. J. Appl. Ceram. Technol. 2020, 17, 2450–2464. [Google Scholar] [CrossRef]
- Hench, L.L. Bioceramics. J. Am. Ceram. Soc. 2005, 81, 1705–1728. [Google Scholar] [CrossRef]
- Baino, F.; Novajra, G.; Vitale-Brovarone, C. Bioceramics and scaffolds: A winning combination for tissue engineering. Front. Bioeng. Biotechnol. 2015, 3, 202. [Google Scholar] [CrossRef] [Green Version]
- Suchanek, W.; Yoshimura, M. Processing and properties of hydroxyapatite-based biomaterials for use as hard tissue replacement implants. J. Mater. Res. 1998, 13, 94–117. [Google Scholar] [CrossRef]
- Boccaccini, A.; Blaker, J. Bioactive composite materials for bone tissue engineering scaffolds. Expert Rev. Med. Devices 2005, 2, 303–317. [Google Scholar] [CrossRef]
- Kokubo, T. Apatite formation on surfaces of ceramics, metals and polymers in body environment. Acta Mater. 1998, 46, 2519–2527. [Google Scholar] [CrossRef]
- Xynos, I.D.; Edgar, A.J.; Buttery, L.D.K.; Hench, L.L.; Polak, J.M. Gene-expression profiling of human osteoblasts following treatment with the ionic products of Bioglass® 45S5 dissolution. J. Biomed. Mater. Res. 2001, 55, 151–157. [Google Scholar] [CrossRef]
- Hoppe, A.; Güldal, N.S.; Boccaccini, A.R. A review of the biological response to ionic dissolution products from bioactive glasses and glass-ceramics. Biomaterials 2011, 32, 2757–2774. [Google Scholar] [CrossRef]
- Fiume, E.; Ciavattini, S.; Verné, E.; Baino, F. Foam replica method in the manufacturing of bioactive glass scaffolds: Out-of-date technology or still underexploited potential? Materials 2021, 14, 2795. [Google Scholar] [CrossRef]
- Chen, Q.Z.; Thompson, I.D.; Boccaccini, A.R. 45S5 Bioglass®-derived glass–ceramic scaffolds for bone tissue engineering. Biomaterials 2006, 27, 2414–2425. [Google Scholar] [CrossRef]
- Fu, H.; Fu, Q.; Zhou, N.; Huang, W.; Rahaman, M.N.; Wang, D.; Liu, X. In vitro evaluation of borate-based bioactive glass scaffolds prepared by a polymer foam replication method. Mater. Sci. Eng. C 2009, 29, 2275–2281. [Google Scholar] [CrossRef]
- Baino, F.; Ferraris, M.; Bretcanu, O.; Verné, E.; Vitale-Brovarone, C. Optimization of composition, structure and mechanical strength of bioactive 3-D glass-ceramic scaffolds for bone substitution. J. Biomater. Appl. 2013, 27, 872–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clupper, D.C.; Hench, L.L. Crystallization kinetics of tape cast bioactive glass 45S5. J. Non-Cryst. Solids 2003, 318, 43–48. [Google Scholar] [CrossRef]
- Navarro, M.; Aparicio, C.; Charles-Harris, M.; Ginebra, M.P.; Engel, E.; Planell, J.A. Development of a biodegradable composite scaffold for bone tissue engineering: Physicochemical, topographical, mechanical, degradation, and biological properties. In Ordered Polymeric Nanostructures at Surfaces; Vancso, G.J., Ed.; Springer: Berlin, Germany, 2006; pp. 209–231. [Google Scholar]
- Boccaccini, A.R.; Maquet, V. Bioresorbable and bioactive polymer/Bioglass® composites with tailored pore structure for tissue engineering applications. Compos. Sci. Technol. 2003, 63, 2417–2429. [Google Scholar] [CrossRef]
- Komlev, V.; Barinov, S.; Rustichelli, F. Strength enhancement of porous hydroxyapatite ceramics by polymer impregnation. J. Mater. Sci. Lett. 2003, 22, 1215–1217. [Google Scholar] [CrossRef]
- Abdala, A.; Milius, D.; Adamson, D.; Aksay, I.; Prud’homme, R. Inspired by abalone shell: Strengthening of porous ceramics with polymers. Polym. Mater. Sci. Eng. 2004, 90, 384–385. [Google Scholar]
- Miao, X.; Lim, W.K.; Huang, X.; Chen, Y. Preparation and characterization of interpenetrating phased TCP/HA/PLGA composites. Mater. Lett. 2005, 59, 4000–4005. [Google Scholar] [CrossRef] [Green Version]
- Arango-Ospina, M.; Lasch, K.; Weidinger, J.; Boccaccini, A.R. Manuka honey and zein coatings impart bioactive glass bone tissue scaffolds antibacterial properties and superior mechanical properties. Front. Mater. 2021, 7, 449. [Google Scholar] [CrossRef]
- Wang, X.; Bank, R.A.; Tekoppele, J.M.; Agrawal, C.M. The role of collagen in determining bone mechanical properties. J. Orthop. Res. 2001, 19, 1021–1026. [Google Scholar] [CrossRef]
- Bertolla, L.; Chlup, Z.; Stratil, L.; Boccaccini, A.R.; Dlouhý, I. Effect of hybrid polymer coating of Bioglass® foams on mechanical response during tensile loading. Adv. Appl. Ceram. 2015, 114, S63–S69. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Nooeaid, P.; Roether, J.A.; Schubert, D.W.; Boccaccini, A.R. Preparation and characterization of vancomycin releasing PHBV coated 45S5 Bioglass®-based glass–ceramic scaffolds for bone tissue engineering. J. Eur. Ceram. Soc. 2014, 34, 505–514. [Google Scholar] [CrossRef]
- Yao, Q.; Nooeaid, P.; Roether, J.A.; Dong, Y.; Zhang, Q.; Boccaccini, A.R. Bioglass®-based scaffolds incorporating polycaprolactone and chitosan coatings for controlled vancomycin delivery. Ceram. Int. 2013, 39, 7517–7522. [Google Scholar] [CrossRef]
- Fereshteh, Z.; Nooeaid, P.; Fathi, M.; Bagri, A.; Boccaccini, A.R. The effect of coating type on mechanical properties and controlled drug release of PCL/zein coated 45S5 bioactive glass scaffolds for bone tissue engineering. Mater. Sci. Eng. C 2015, 54, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Bretcanu, O.; Misra, S.; Roy, I.; Renghini, C.; Fiori, F.; Boccaccini, A.R.; Salih, V. In vitro biocompatibility of 45S5 Bioglass®—derived glass-ceramic scaffolds coated with poly(3-hydroxybutyrate). J. Tissue Eng. Regen. Med. 2009, 3, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Hum, J.; Luczynski, K.W.; Nooeaid, P.; Newby, P.; Lahayne, O.; Hellmich, C.; Boccaccini, A.R. Stiffness improvement of 45S5 Bioglass®-based scaffolds through natural and synthetic biopolymer coatings: An ultrasonic study. Strain 2013, 49, 431–439. [Google Scholar] [CrossRef]
- Chen, Q.Z.; Boccaccini, A.R. Poly(D,L-lactic acid) coated 45S5 Bioglass®-based scaffolds: Processing and characterization. J. Biomed. Mater. Res. A. 2006, 77A, 445–457. [Google Scholar] [CrossRef]
- Li, W.; Wang, H.; Ding, Y.; Scheithauer, E.C.; Goudouri, O.M.; Grünewald, A.; Detsch, R.; Agarwal, S.; Boccaccini, A.R. Antibacterial 45S5 Bioglass®-based scaffolds reinforced with genipin cross-linked gelatin for bone tissue engineering. J. Mater. Chem. B 2015, 3, 3367–3378. [Google Scholar] [CrossRef] [Green Version]
- Metze, A.L.; Grimm, A.; Nooeaid, P.; Roether, J.A.; Hum, J.; Newby, P.J.; Schubert, D.W.; Boccaccini, A.R. Gelatin coated 45S5 Bioglass®-derived scaffolds for bone tissue engineering. Key Eng. Mater. 2013, 541, 31–39. [Google Scholar] [CrossRef]
- Westhauser, F.; Weis, C.; Prokscha, M.; Bittrich, L.; Li, W.; Xiao, K.; Kneser, U.; Kauczor, H.U.; Schmidmaier, G.; Boccaccini, A.; et al. Three-dimensional polymer coated 45S5-type bioactive glass scaffolds seeded with human mesenchymal stem cells show bone formation in vivo. J. Mater. Sci. Mater. Med. 2016, 27, 119. [Google Scholar] [CrossRef] [PubMed]
- Sartori, S.; Boffito, M.; Serafini, P.; Caporale, A.; Silvestri, A.; Bernardi, E.; Sassi, M.P.; Boccafoschi, F.; Ciardelli, G. Synthesis and structure–property relationship of polyester-urethanes and their evaluation for the regeneration of contractile tissues. React. Funct. Polym. 2013, 73, 1366–1376. [Google Scholar] [CrossRef]
- Silvestri, A.; Sartori, S.; Boffito, M.; Mattu, C.; Di Rienzo, A.M.; Boccafoschi, F.; Ciardelli, G. Biomimetic myocardial patches fabricated with poly(ɛ-caprolactone) and polyethylene glycol-based polyurethanes. J. Biomed. Mater. Res. B Appl. Biomater. 2014, 102, 1002–1013. [Google Scholar] [CrossRef]
- Sartori, S.; Rechichi, A.; Vozzi, G.; D’Acunto, M.; Heine, E.; Giusti, P.; Ciardelli, G. Surface modification of a synthetic polyurethane by plasma glow discharge: Preparation and characterization of bioactive monolayers. React. Funct. Polym. 2008, 68, 809–821. [Google Scholar] [CrossRef]
- Boffito, M.; Di Meglio, F.; Mozetic, P.; Giannitelli, S.M.; Carmagnola, I.; Castaldo, C.; Nurzynska, D.; Sacco, A.M.; Miraglia, R.; Montagnani, S.; et al. Surface functionalization of polyurethane scaffolds mimicking the myocardial microenvironment to support cardiac primitive cells. PLoS ONE 2018, 13, e0199896. [Google Scholar] [CrossRef] [PubMed]
- Chiono, V.; Sartori, S.; Calzone, S.; Boffito, M.; Tonda-Turo, C.; Mattu, C.; Gentile, P.; Ciardelli, G. 6—Synthetic biodegradable medical polyurethanes. In Science and Principles of Biodegradable and Bioresorbable Medical Polymers; Zhang, X., Ed.; Woodhead Publishing: Shaxton, UK, 2017; pp. 189–216. [Google Scholar]
- Kokubo, T.; Takadama, H. How useful is SBF in predicting in vivo bone bioactivity? Biomaterials 2006, 27, 2907–2915. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Garmendia, N.; Pérez de Larraya, U.; Ding, Y.; Detsch, R.; Grünewald, A.; Roether, J.A.; Schubert, D.W.; Boccaccini, A.R. 45S5 bioactive glass-based scaffolds coated with cellulose nanowhiskers for bone tissue engineering. RSC Adv. 2014, 4, 56156–56164. [Google Scholar] [CrossRef] [Green Version]
- Boffito, M.; Gioffredi, E.; Chiono, V.; Calzone, S.; Ranzato, E.; Martinotti, S.; Ciardelli, G. Novel polyurethane-based thermosensitive hydrogels as drug release and tissue engineering platforms: Design and in vitro characterization. Polym. Int. 2016, 65, 756–769. [Google Scholar] [CrossRef]
- Guan, J.; Sacks, M.S.; Beckman, E.J.; Wagner, W.R. Synthesis, characterization, and cytocompatibility of elastomeric, biodegradable poly(ester-urethane)ureas based on poly(caprolactone) and putrescine. J. Biomed. Mater. Res. 2002, 61, 493–503. [Google Scholar] [CrossRef]
- Zhang, C.; Ren, Z.; Yin, Z.; Jiang, L.; Fang, S. Experimental FTIR and simulation studies on H-bonds of model polyurethane in solutions. I: In dimethylformamide (DMF). Spectrochim. Acta A Mol. Biomol. Spectrosc. 2011, 81, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Russo, V.; Tammaro, L.; Di Marcantonio, L.; Sorrentino, A.; Ancora, M.; Valbonetti, L.; Turriani, M.; Martelli, A.; Camma, C.; Barboni, B. Amniotic epithelial stem cell biocompatibility for electrospun poly(lactide-co-glycolide), poly(ε-caprolactone), poly(lactic acid) scaffolds. Mater. Sci. Eng. C 2016, 69, 321–329. [Google Scholar] [CrossRef]
- Jung, S.C. Biological effect of gas plasma treatment on CO2 gas foaming/salt leaching fabricated porous polycaprolactone scaffolds in bone tissue engineering. J. Nanomater. 2014, 2014, 657542. [Google Scholar]
- Allegrezzajr, A.; Seymour, R.W.; Ng, H.N.; Cooper, S.L. Segmental orientation studies of block copolymers: 2. Non-hydrogen bonded polyurethanes. Polymer 1974, 15, 433–440. [Google Scholar] [CrossRef]
- Gan, Z.; Liang, Q.; Zhang, J.; Jing, X. Enzymatic degradation of poly(ε-caprolactone) film in phosphate buffer solution containing lipases. Polym. Degrad. Stab. 1997, 56, 209–213. [Google Scholar] [CrossRef]
- Ponjavic, M.; Nikolic, M.; Nikodinovic-Runic, J.; Jeremic, S.; Stevanovic, S.; Đonlagić, J. Degradation behavior of PCL/PEO/PCL and PCL/PEO block copolymers under controlled hydrolytic, enzymatic and composting conditions. Polym. Test. 2016, 57, 67–77. [Google Scholar] [CrossRef]
- Trathnigg, B. Size-exclusion chromatography of polymers. In Encyclopedia of Analytical Chemistry: Applications, Theory and Instrumentation; Meyers, R.A., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2006; p. a2032. [Google Scholar]
- Li, S.; Garreau, H.; Pauvert, B.; McGrath, J.; Toniolo, A.; Vert, M. Enzymatic degradation of block copolymers prepared from ε-caprolactone and poly(ethylene glycol). Biomacromolecules 2002, 3, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Hong, Y.; Nelson, D.M.; Pichamuthu, J.E.; Leeson, C.E.; Wagner, W.R. Biodegradable polyurethane ureas with variable polyester or polycarbonate soft segments: Effects of crystallinity, molecular weight, and composition on mechanical properties. Biomacromolecules 2011, 12, 3265–3274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felfel, R.M.; Hossain, K.M.Z.; Parsons, A.J.; Rudd, C.D.; Ahmed, I. Accelerated in vitro degradation properties of polylactic acid/phosphate glass fibre composites. J. Mater. Sci. 2015, 50, 3942–3955. [Google Scholar] [CrossRef]
- Heimowska, A.; Morawska, M.; Bocho-Janiszewska, A. Biodegradation of poly(ε-caprolactone) in natural water environments. Pol. J. Chem. Technol. 2017, 19, 120–126. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.M. Control of pore geometry on influencing the mechanical property of porous hydroxyapatite bioceramic. J. Mater. Sci. Lett. 1996, 15, 419–421. [Google Scholar]
- Chen, Q.; Mohn, D.; Stark, W.J. Optimization of Bioglass® scaffold fabrication process. J. Am. Ceram. Soc. 2011, 94, 4184–4190. [Google Scholar] [CrossRef]
- Bretcanu, O.; Chatzistavrou, X.; Paraskevopoulos, K.; Conradt, R.; Thompson, I.; Boccaccini, A.R. Sintering and crystallization of 45S5 Bioglass® powder. J. Eur. Ceram. Soc. 2009, 29, 3299–3306. [Google Scholar] [CrossRef]
- Lin, K.S.K.; Tseng, Y.H.; Mou, Y.; Hsu, Y.C.; Yang, C.M.; Chan, J.C.C. Mechanistic study of apatite formation on bioactive glass surface using 31P solid-state NMR spectroscopy. Chem. Mater. 2005, 17, 4493–4501. [Google Scholar] [CrossRef]
- Farag, M.M.; Rüssel, C. Glass-ceramic scaffolds derived from Bioglass® and glass with low crystallization affinity for bone regeneration. Mater. Lett. 2012, 73, 161–165. [Google Scholar] [CrossRef]
- Fu, Q.; Rahaman, M.N.; Day, D.E. Accelerated conversion of silicate bioactive glass (13–93) to hydroxyapatite in aqueous phosphate solution containing polyanions. J. Am. Cer. Soc. 2009, 92, 2870–2876. [Google Scholar] [CrossRef]
- Peroglio, M.; Gremillard, L.; Chevalier, J.; Chazeau, L.; Gauthier, C.; Hamaide, T. Toughening of bioceramics scaffolds by polymer coating. J. Eur. Ceram. Soc. 2007, 27, 2679–2685. [Google Scholar] [CrossRef]
- Pezzotti, G.; Asmus, S.M.F. Fracture behaviour of hydroxyapatite/polymer inter-penetrating network composites prepared by in situ polymerization process. Mater. Sci. Eng. A 2001, 316, 231–237. [Google Scholar] [CrossRef]
- Morgan, E.F.; Unnikrisnan, G.U.; Hussein, A.I. Bone mechanical properties in healthy and diseased states. Ann. Rev. Biomed. Eng. 2018, 20, 119–143. [Google Scholar] [CrossRef]
- Gatti, A.M.; Valdre, G.; Andersson, O.H. Analysis of the in vivo reactions of a bioactive glass in soft and hard tissue. Biomaterials 1994, 15, 208–212. [Google Scholar] [CrossRef]
- Matsuda, T.; Davies, J.E. The in vitro response of osteoblast to bioactive glass. Biomaterials 1987, 8, 275–284. [Google Scholar] [CrossRef]
- Lobel, K.D.; Hench, L.L. In vitro adsorption and activity of enzymes on reaction layers of bioactive glass substrates. J. Biomed. Mater. Res. 1998, 39, 575–579. [Google Scholar] [CrossRef]
- Ohgushi, H.; Dohi, Y.; Yoshikawa, T.; Tamai, S.; Tabata, S.; Okunaga, K.; Shibuya, T. Osteogenic differentiation of cultured marrow stromal stem cells on the surface of bioactive glass ceramics. J. Biomed. Mater. Res. 1996, 32, 341–348. [Google Scholar] [CrossRef]
- Chen, Q.; Efthymiou, A.; Salih, V.; Boccaccini, A. Bioglass®-derived glass–ceramic scaffolds: Study of cell proliferation and scaffold degradation in vitro. J. Biomed. Mater. Res. A 2008, 84, 1049–1060. [Google Scholar] [CrossRef] [PubMed]
- Tonda-Turo, C.; Boffito, M.; Cassino, C.; Gentile, P.; Ciardelli, G. Biomimetic polyurethane-based fibrous scaffolds. Mater. Lett. 2016, 167, 9–12. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | |
|---|---|
| PCL | 1.145 [34] |
| KHC2000 | 1.1 ± 0.04 |
| KHC2000E2000 | 1.07 ± 0.03 |
| Young’s Modulus (MPa) | Stress at Break (MPa) | Strain at Break (%) | |
|---|---|---|---|
| PCL | 210.3 ± 28.8 | 26.8 ± 4.0 | 683.3 ± 9.7 |
| KHC2000 | 13.8 ± 1.0 | 20.3 ± 3.1 | 822.5 ± 79.7 |
| KHC2000E2000 | 11.8 ± 2.6 | 11.2 ± 0.7 | 1145.0 ± 117.4 |
| Parameter | |
|---|---|
| Weight | 0.052 ± 0.01 g |
| Diameter | 7.8 ± 0.2 mm |
| Height | 5.1 ± 0.1 mm |
| Porosity | 92.1 ± 1.6% |
| Pore size | 200 ÷ 600 μm |
| Amount of Polymer in the Coating (mg) | Porosity (%) | |
|---|---|---|
| PCL/BG | 1.5 ± 0.3 | 90.7 ± 1.6 |
| KHC2000/BG | 0.9 ± 0.2 | 91.1 ± 1.7 |
| KHC2000E2000/BG | 1.5 ± 0.5 | 91.7 ± 1.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boffito, M.; Servello, L.; Arango-Ospina, M.; Miglietta, S.; Tortorici, M.; Sartori, S.; Ciardelli, G.; Boccaccini, A.R. Custom-Made Poly(urethane) Coatings Improve the Mechanical Properties of Bioactive Glass Scaffolds Designed for Bone Tissue Engineering. Polymers 2022, 14, 151. https://0-doi-org.brum.beds.ac.uk/10.3390/polym14010151
Boffito M, Servello L, Arango-Ospina M, Miglietta S, Tortorici M, Sartori S, Ciardelli G, Boccaccini AR. Custom-Made Poly(urethane) Coatings Improve the Mechanical Properties of Bioactive Glass Scaffolds Designed for Bone Tissue Engineering. Polymers. 2022; 14(1):151. https://0-doi-org.brum.beds.ac.uk/10.3390/polym14010151
Chicago/Turabian StyleBoffito, Monica, Lucia Servello, Marcela Arango-Ospina, Serena Miglietta, Martina Tortorici, Susanna Sartori, Gianluca Ciardelli, and Aldo R. Boccaccini. 2022. "Custom-Made Poly(urethane) Coatings Improve the Mechanical Properties of Bioactive Glass Scaffolds Designed for Bone Tissue Engineering" Polymers 14, no. 1: 151. https://0-doi-org.brum.beds.ac.uk/10.3390/polym14010151