
Thermoplastic Starch with Poly(butylene adipate-co-terephthalate) Blends Foamed by Supercritical Carbon Dioxide

 , , , ,
, , , ,
Abstract
:
1. Introduction
2. Experimental Preparations
2.1. Materials and Procedure
2.2. Functional Group Chemical Modification TPS Experiment
2.3. The (SA/TPS)/PBAT Biodegradable Polyester Composite Mixing
2.4. The (SA/TPS)/PBAT Composite Foam Test Piece and Supercritical Foaming Experiment
2.5. The (SA/TPS)/PBAT Composite Foam Appearance and Internal Structure SEM Analysis
2.6. Chemical Modification and Analysis of TPS Functional Group
2.7. Supercritical Batch Foaming Process
3. Results and Discussions
3.1. FT-IR Analysis of TPS and SA/TPS
H NMR Analysis of TPS and SA/TPS
3.2. The Composite Foam Density of TPS/PBAT
3.2.1. The Density of Foamed Material of PBAT
3.2.2. The Density of TPS/PBAT Composite Foam
3.2.3. The Density of (SA/TPS)/PBAT Composite Foam
3.2.4. The Influence of Foaming Temperature on the Density of Composite Foams with PBAT in Different Ratios (SA/TPS)
3.3. Appearance and Internal Structure of TPS/PBAT Composites Foam
3.3.1. Appearance and Internal Microstructure of PBAT Foam
3.3.2. The Appearance and Internal Structure of TPS/PBAT Composites Foam
3.4. Appearance and Internal Structure of (SA/TPS)/PBAT Composites Foam
3.4.1. The (SA/TPS)/PBAT Composite Foam Appearance and Internal Structure
3.4.2. The Appearance and Internal Structure of (SA/TPS)/PBAT Foams with Different (SA/TPS) Ratios
3.5. Mechanical Properties of the TPS/PBAT and the (SA/TPS)/PBAT Composites Foam
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cho, C.-J.; Lu, S.-T.; Kuo, C.-C.; Liang, F.-C.; Chen, B.-Y.; Chu, C.-C. Pyrene or rhodamine derivative–modified surfaces of electrospun nanofibrous chemosensors for colorimetric and fluorescent determination of Cu2+, Hg2+, and pH. React. Funct. Polym. 2016, 108, 137–147. [Google Scholar] [CrossRef]
- Venkatesan, M.; Veeramuthu, L.; Liang, F.-C.; Chen, W.-C.; Cho, C.-J.; Chen, C.-W.; Chen, J.-Y.; Yan, Y.; Chang, S.-H.; Kuo, C.-C. Evolution of electrospun nanofibers fluorescent and colorimetric sensors for environmental toxicants, pH, temperature, and cancer cells—A review with insights on applications. Chem. Eng. J. 2020, 397, 125431. [Google Scholar] [CrossRef]
- Cho, C.-J.; Chen, S.-Y.; Kuo, C.-C.; Veeramuthu, L.; Au-Duong, A.-N.; Chiu, Y.-C.; Chang, S.-H. Morphology and optoelectronic characteristics of organic field-effect transistors based on blends of polylactic acid and poly(3-hexylthiophene). Polym. J. 2018, 50, 975–987. [Google Scholar] [CrossRef]
- He, C.; Liang, F.; Veeramuthu, L.; Cho, C.; Benas, J.; Tzeng, Y.; Tseng, Y.; Chen, W.; Rwei, A.; Kuo, C. Super Tough and Spontaneous Water-Assisted Autonomous Self-Healing Elastomer for Underwater Wearable Electronics. Adv. Sci. 2021, 8, 2102275. [Google Scholar] [CrossRef]
- Lin, C.C.; Jiang, D.-H.; Kuo, C.-C.; Cho, C.-J.; Tsai, Y.-H.; Satoh, T.; Su, C. Water-Resistant Efficient Stretchable Perovskite-Embedded Fiber Membranes for Light-Emitting Diodes. ACS Appl. Mater. Interfaces 2018, 10, 2210–2215. [Google Scholar] [CrossRef]
- Jao, C.; Chang, J.; Ya, C.; Chen, W.; Cho, C.; Lin, J.; Chiu, Y.; Zhou, Y.; Kuo, C. Novel stretchable light-emitting diodes based on conjugated-rod block elastic-coil copolymers. Polym. Int. 2020, 70, 426–431. [Google Scholar] [CrossRef]
- Veeramuthu, L.; Venkatesan, M.; Benas, J.-S.; Cho, C.-J.; Lee, C.-C.; Lieu, F.-K.; Lin, J.-H.; Lee, R.-H.; Kuo, C.-C. Recent Progress in Conducting Polymer Composite/Nanofiber-Based Strain and Pressure Sensors. Polymers 2021, 13, 4281. [Google Scholar] [CrossRef]
- Venkatesan, M.; Chen, W.-C.; Cho, C.-J.; Veeramuthu, L.; Chen, L.-G.; Li, K.-Y.; Tsai, M.-L.; Lai, Y.-C.; Lee, W.-Y.; Chen, W.-C.; et al. Enhanced piezoelectric and photocatalytic performance of flexible energy harvester based on CsZn0.75Pb0.25I3/CNC–PVDF composite nanofibers. Chem. Eng. J. 2022, 433, 133620. [Google Scholar] [CrossRef]
- Mao, H.-I.; Wang, L.-Y.; Chen, C.-W.; Hsu, K.-H.; Tsai, C.-H.; Cho, C.-J.; Yu, Y.-Y.; Rwei, S.-P.; Kuo, C.-C. Enhanced crystallization rate of bio-based poly(butylene succinate-co-propylene succinate) copolymers motivated by glycerol. J. Polym. Res. 2021, 28, 92. [Google Scholar] [CrossRef]
- Chen, C.-W.; Liu, P.-H.; Lin, F.-J.; Cho, C.-J.; Wang, L.-Y.; Mao, H.-I.; Chiu, Y.-C.; Chang, S.-H.; Rwei, S.-P.; Kuo, C.-C. Influence of Different Molecular Weights and Concentrations of Poly(glycidyl methacrylate) on Recycled Poly(ethylene terephthalate): A Thermal, Mechanical, and Rheological Study. J. Polym. Environ. 2020, 28, 2880–2892. [Google Scholar] [CrossRef]
- Chan, H.; Cho, C.; Hsu, K.; He, C.; Kuo, C.; Chu, C.; Chen, Y.; Chen, C.; Rwei, S. Smart Wearable Textiles with Breathable Properties and Repeatable Shaping in In Vitro Orthopedic Support from a Novel Biomass Thermoplastic Copolyester. Macromol. Mater. Eng. 2019, 304, 1900103. [Google Scholar] [CrossRef]
- Cho, C.-J.; Chang, Y.-S.; Lin, Y.-Z.; Jiang, D.-H.; Chen, W.-H.; Lin, W.-Y.; Chen, C.-W.; Rwei, S.-P.; Kuo, C.-C. Green electrospun nanofiber membranes filter prepared from novel biomass thermoplastic copolyester: Morphologies and filtration properties. J. Taiwan Inst. Chem. Eng. 2020, 106, 206–214. [Google Scholar] [CrossRef]
- Hsu, K.-H.; Chen, C.-W.; Wang, L.-Y.; Chan, H.-W.; He, C.-L.; Cho, C.-J.; Rwei, S.-P.; Kuo, C.-C. Bio-based thermoplastic poly(butylene succinate-co-propylene succinate) copolyesters: Effect of glycerol on thermal and mechanical properties. Soft Matter 2019, 15, 9710–9720. [Google Scholar] [CrossRef]
- Lu, W.-C.; Chen, C.-Y.; Cho, C.-J.; Venkatesan, M.; Chiang, W.-H.; Yu, Y.-Y.; Lee, C.-H.; Lee, R.-H.; Rwei, S.-P.; Kuo, C.-C. Antibacterial Activity and Protection Efficiency of Polyvinyl Butyral Nanofibrous Membrane Containing Thymol Prepared through Vertical Electrospinning. Polymers 2021, 13, 1122. [Google Scholar] [CrossRef]
- Lu, W.-C.; Chuang, F.-S.; Venkatesan, M.; Cho, C.-J.; Chen, P.-Y.; Tzeng, Y.-R.; Yu, Y.-Y.; Rwei, S.-P.; Kuo, C.-C. Synthesis of Water Resistance and Moisture-Permeable Nanofiber Using Sodium Alginate–Functionalized Waterborne Polyurethane. Polymers 2020, 12, 2882. [Google Scholar] [CrossRef]
- Benas, J.-S.; Veeramuthu, L.; Chuang, Y.-Y.; Chuang, S.-Y.; Liang, F.-C.; Cho, C.-J.; Lee, W.-Y.; Yan, Y.; Zhou, Y.; Kuo, C.-C. Eco-friendly collagen-based bio-organic field effect transistor with improved memory characteristics. Org. Electron. 2020, 86, 105925. [Google Scholar] [CrossRef]
- Chen, P.-Y.; Hsu, C.; Venkatesan, M.; Tseng, Y.-L.; Cho, C.-J.; Han, S.-T.; Zhou, Y.; Chiang, W.-H.; Kuo, C.-C. Enhanced electrical and thermal properties of semi-conductive PANI-CNCs with surface modified CNCs. RSC Adv. 2021, 11, 11444–11456. [Google Scholar] [CrossRef]
- Motloung, M.P.; Ojijo, V.; Bandyopadhyay, J.; Ray, S.S. Cellulose Nanostructure-Based Biodegradable Nanocomposite Foams: A Brief Overview on the Recent Advancements and Perspectives. Polymers 2019, 11, 1270. [Google Scholar] [CrossRef] [Green Version]
- Nanda, S.; Patra, B.R.; Patel, R.; Bakos, J.; Dalai, A.K. Innovations in applications and prospects of bioplastics and biopolymers: A review. Environ. Chem. Lett. 2021, 20, 379–395. [Google Scholar] [CrossRef]
- Rai, P.; Mehrotra, S.; Priya, S.; Gnansounou, E.; Sharma, S.K. Recent advances in the sustainable design and applications of biodegradable polymers. Bioresour. Technol. 2021, 325, 124739. [Google Scholar] [CrossRef]
- Abe, M.; Branciforti, M.; Brienzo, M. Biodegradation of Hemicellulose-Cellulose-Starch-Based Bioplastics and Microbial Polyesters. Recycling 2021, 6, 22. [Google Scholar] [CrossRef]
- Arvanitoyannis, I.; Biliaderis, C.; Ogawa, H.; Kawasaki, N. Biodegradable films made from low-density polyethylene (LDPE), rice starch and potato starch for food packaging applications: Part Carbohydr. Polym. 1998, 36, 89–104. [Google Scholar] [CrossRef]
- Arvanitoyannis, I.; Nakayama, A.; Aiba, S.-I. Edible films made from hydroxypropyl starch and gelatin and plasticized by polyols and water. Carbohydr. Polym. 1998, 36, 105–119. [Google Scholar] [CrossRef]
- Bilck, A.P.; Grossmann, M.V.; Yamashita, F. Biodegradable mulch films for strawberry production. Polym. Test. 2010, 29, 471–476. [Google Scholar] [CrossRef]
- Huneault, M.A.; Li, H. Morphology and properties of compatibilized polylactide/thermoplastic starch blends. Polymer 2007, 48, 270–280. [Google Scholar] [CrossRef]
- Lourdin, D.; Della Valle, G.; Colonna, P. Influence of amylose content on starch films and foams. Carbohydr. Polym. 1995, 27, 261–270. [Google Scholar] [CrossRef]
- Phothisarattana, D.; Wongphan, P.; Promhuad, K.; Promsorn, J.; Harnkarnsujarit, N. Biodegradable Poly(Butylene Adipate-Co-Terephthalate) and Thermoplastic Starch-Blended TiO2 Nanocomposite Blown Films as Functional Active Packaging of Fresh Fruit. Polymers 2021, 13, 4192. [Google Scholar] [CrossRef]
- Raquez, J.-M.; Nabar, Y.; Narayan, R.; Dubois, P. In situ compatibilization of maleated thermoplastic starch/polyester melt-blends by reactive extrusion. Polym. Eng. Sci. 2008, 48, 1747–1754. [Google Scholar] [CrossRef]
- Ekielski, A.; Żelaziński, T.; Mishra, P.K.; Skudlarski, J. Properties of Biocomposites Produced with Thermoplastic Starch and Digestate: Physicochemical and Mechanical Characteristics. Materials 2021, 14, 6092. [Google Scholar] [CrossRef]
- Gutiérrez, T.J.; Álvarez, V. Bionanocomposite films developed from corn starch and natural and modified nano-clays with or without added blueberry extract. Food Hydrocoll. 2018, 77, 407–420. [Google Scholar] [CrossRef] [Green Version]
- Haurat, M.; Dumon, M. Amorphous Polymers’ Foaming and Blends with Organic Foaming-Aid Structured Additives in Supercritical CO2, a Way to Fabricate Porous Polymers from Macro to Nano Porosities in Batch or Continuous Processes. Molecules 2020, 25, 5320. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Mi, J.; Zhou, H.; Wang, X.; Zhang, Y. Chain extension of poly (butylene adipate-co-terephthalate) and its microcellular foaming behaviors. Polym. Degrad. Stab. 2018, 157, 143–152. [Google Scholar] [CrossRef]
- Bhatnagar, S.; Hanna, M.A. Starch-based plastic foams from various starch sources. Cereal Chemistry 1996, 73, 601–604. [Google Scholar]
- Chen, L.; Gordon, S.H.; Imam, S.H. Starch Graft Poly(methyl acrylate) Loose-Fill Foam: Preparation, Properties and Degradation. Biomacromolecules 2004, 5, 238–244. [Google Scholar] [CrossRef]
- Diyana, Z.; Jumaidin, R.; Selamat, M.; Ghazali, I.; Julmohammad, N.; Huda, N.; Ilyas, R. Physical Properties of Thermoplastic Starch Derived from Natural Resources and Its Blends: A Review. Polymers 2021, 13, 1396. [Google Scholar] [CrossRef]
- Nabar, Y.; Raquez, J.M.; Dubois, A.P.; Narayan, R. Production of Starch Foams by Twin-Screw Extrusion: Effect of Maleated Poly(butylene adipate-co-terephthalate) as a Compatibilizer. Biomacromolecules 2005, 6, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Thanpitcha, T.; Kritchayanon, N.; Pentrakoon, D.; Pimpan, V. An Initial Study of Starch-g-polystyrene Foam Prepared by a Steaming Process. J. Met. Mater. Miner. 2003, 12, 1–6. [Google Scholar]
- Willett, J.; Shogren, R. Processing and properties of extruded starch/polymer foams. Polymer 2002, 43, 5935–5947. [Google Scholar] [CrossRef]
- Tatarka, P.D.; Cunningham, R.L. Properties of protective loose-fill foams. J. Appl. Polym. Sci. 1998, 67, 1157–1176. [Google Scholar] [CrossRef]
- Fazeli, M.; Florez, J.P.; Simão, R.A. Improvement in adhesion of cellulose fibers to the thermoplastic starch matrix by plasma treatment modification. Compos. Part B Eng. 2019, 163, 207–216. [Google Scholar] [CrossRef]
- Nazrin, A.; Sapuan, S.M.; Zuhri, M.Y.M.; Ilyas, R.; Syafiq, R.; Sherwani, S.F.K. Nanocellulose Reinforced Thermoplastic Starch (TPS), Polylactic Acid (PLA), and Polybutylene Succinate (PBS) for Food Packaging Applications. Front. Chem. 2020, 8, 213. [Google Scholar] [CrossRef] [PubMed]
- Boonprasertpoh, A.; Pentrakoon, D.; Junkasem, J. Effect of PBAT on physical, morphological, and mechanical properties of PBS/PBAT foam. Cell. Polym. 2019, 39, 31–41. [Google Scholar] [CrossRef]
- Fourati, Y.; Tarrés, Q.; Mutjé, P.; Boufi, S. PBAT/thermoplastic starch blends: Effect of compatibilizers on the rheological, mechanical and morphological properties. Carbohydr. Polym. 2018, 199, 51–57. [Google Scholar] [CrossRef]
- Shi, X.; Qin, J.; Wang, L.; Ren, L.; Rong, F.; Li, D.; Wang, R.; Zhang, G. Introduction of stereocomplex crystallites of PLA for the solid and microcellular poly(lactide)/poly(butylene adipate-co-terephthalate) blends. RSC Adv. 2018, 8, 11850–11861. [Google Scholar] [CrossRef] [Green Version]
- Tian, H.-L.; Wang, Z.-P.; Jia, S.-L.; Pan, H.-W.; Han, L.-J.; Bian, J.-J.; Li, Y.; Yang, H.-L.; Zhang, H.-L. Biodegradable Foaming Material of Poly(butylene adipate-co-terephthalate) (PBAT)/Poly(propylene carbonate) (PPC). Chin. J. Polym. Sci. 2021, 40, 208–219. [Google Scholar] [CrossRef]
- Lackner, M.; Ivanič, F.; Kováčová, M.; Chodák, I. Mechanical properties and structure of mixtures of poly(butylene-adipate-co-terephthalate) (PBAT) with thermoplastic starch (TPS). Int. J. Biobased Plast. 2021, 3, 126–138. [Google Scholar] [CrossRef]
- Masina, N.; Choonara, Y.E.; Kumar, P.; du Toit, L.C.; Govender, M.; Indermun, S.; Pillay, V. A review of the chemical modification techniques of starch. Carbohydr. Polym. 2017, 157, 1226–1236. [Google Scholar] [CrossRef]
- Dehouche, N.; Idres, C.; Kaci, M.; Zembouai, I.; Bruzaud, S. Effects of various surface treatments on Aloe Vera fibers used as reinforcement in poly(3-hydroxybutyrate-co-3-hydroxyhexanoate) (PHBHHx) biocomposites. Polym. Degrad. Stab. 2020, 175, 109131. [Google Scholar] [CrossRef]
- Petzold, K.; Einfeldt, L.; Günther, W.; Stein, A.; Klemm, D. Regioselective Functionalization of Starch: Synthesis and 1H NMR Characterization of 6-O-Silyl Ethers. Biomacromolecules 2001, 2, 965–969. [Google Scholar] [CrossRef]
- Pothan, L.A.; Simon, F.; Spange, A.S.; Thomas, S. XPS Studies of Chemically Modified Banana Fibers. Biomacromolecules 2006, 7, 892–898. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
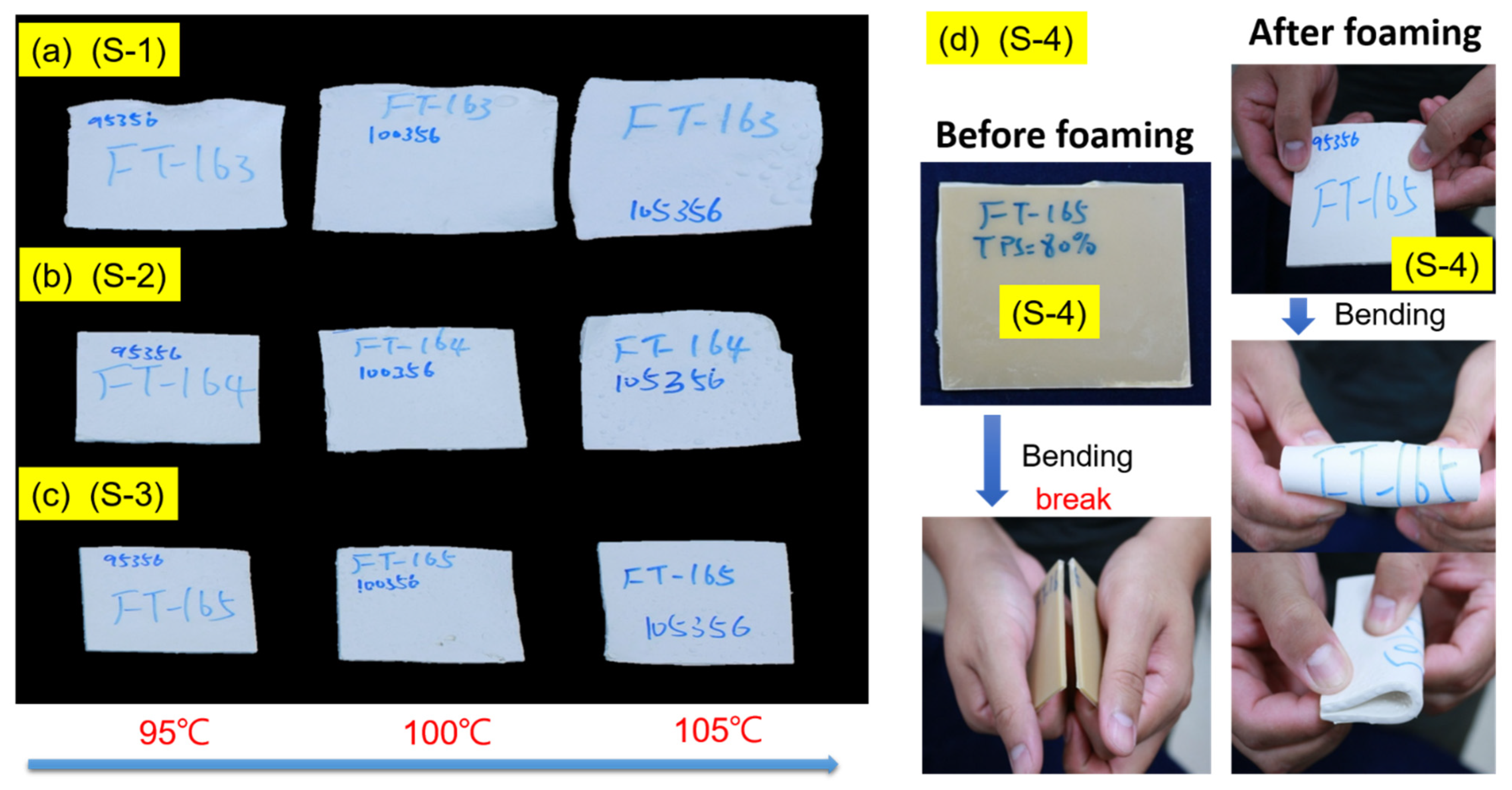{kind=link}
{kind=link}
| Symbols | Blends (by Weight) |
|---|---|
| [N-1] | 50% TPS/50% PBAT |
| [N-2] | 60% TPS/40% PBAT |
| [N-3] | 70% TPS/30% PBAT |
| [S-0.5] | 50% (TPS with 5PHR SA)/50% PBAT |
| [S-1] | 50% (TPS with 10PHR SA)/50% PBAT |
| [S-2] | 60% (TPS with 10PHR SA)/40% PBAT |
| [S-3] | 70% (TPS with 10PHR SA)/30% PBAT |
| [S-4] | 80% (TPS with 10PHR SA)/20% PBAT |
| [N-1] (F) | [N-2] (F) | [N-3] (F) | [S-1] (F) | [S-2] (F) | [S-3] (F) | [S-4] (F) | |
|---|---|---|---|---|---|---|---|
| Tensile strength (kPa) | |||||||
| Elongation (%) | 22 ± 2 | ||||||
| Foam density (kg/m3) |
| [N-1] | [N-2] | [N-3] | [S-1] | [S-2] | [S-3] | [S-4] | |
|---|---|---|---|---|---|---|---|
| Tensile strength (Breaking point) (kPa) | |||||||
| Elongation (%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, C.-J.; Venkatesan, M.; Cho, C.-J.; Chung, P.-Y.; Chandrasekar, J.; Lee, C.-H.; Wang, H.-T.; Wong, C.-M.; Kuo, C.-C. Thermoplastic Starch with Poly(butylene adipate-co-terephthalate) Blends Foamed by Supercritical Carbon Dioxide. Polymers 2022, 14, 1952. https://0-doi-org.brum.beds.ac.uk/10.3390/polym14101952
Chang C-J, Venkatesan M, Cho C-J, Chung P-Y, Chandrasekar J, Lee C-H, Wang H-T, Wong C-M, Kuo C-C. Thermoplastic Starch with Poly(butylene adipate-co-terephthalate) Blends Foamed by Supercritical Carbon Dioxide. Polymers. 2022; 14(10):1952. https://0-doi-org.brum.beds.ac.uk/10.3390/polym14101952
Chicago/Turabian StyleChang, Chih-Jen, Manikandan Venkatesan, Chia-Jung Cho, Ping-Yu Chung, Jayashree Chandrasekar, Chen-Hung Lee, Hsin-Ta Wang, Chang-Ming Wong, and Chi-Ching Kuo. 2022. "Thermoplastic Starch with Poly(butylene adipate-co-terephthalate) Blends Foamed by Supercritical Carbon Dioxide" Polymers 14, no. 10: 1952. https://0-doi-org.brum.beds.ac.uk/10.3390/polym14101952