
Effect of Chemical Agents on the Morphology and Chemical Structures of Microplastics
 , ,
, ,
Abstract
:1. Introduction
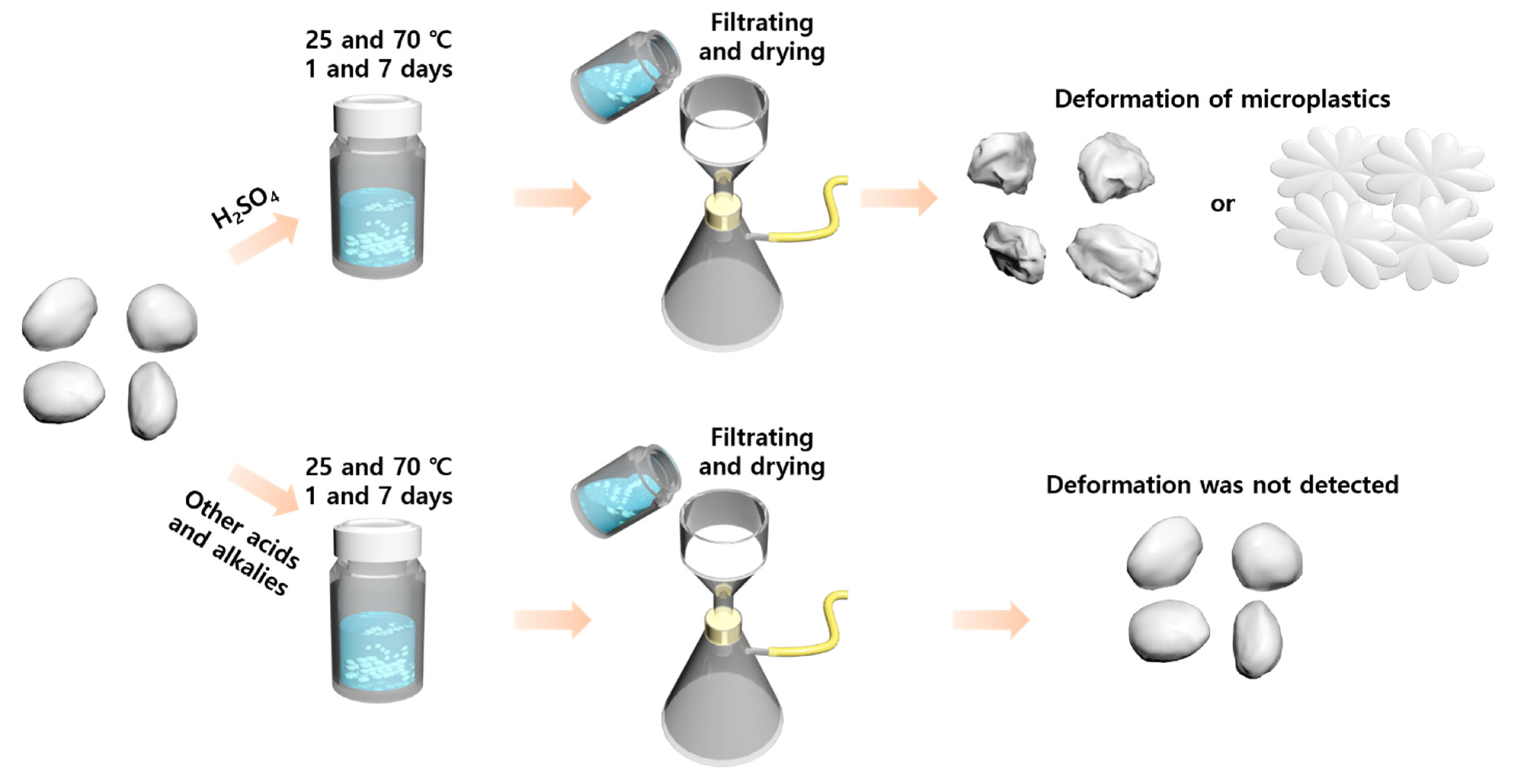
2. Materials and Methods
2.1. Materials
2.2. Experiment Procedure of Chemical Reactions of Microplastics with Acids, Bases, and Oxidizing Agents
2.3. Characteristics of MP Samples before and after Exposure to the Acids, Bases, and Oxidizing Agents
3. Results
3.1. Chemical Stability Tests of Microplastic Samples
3.2. Chemical Stability of HDPE against Acids, Bases, and Oxidizing Agents
3.3. Chemical Stability of LDPE against Acids, Alkalis, and Oxidizing Agents
3.4. Chemical Stability of PS against Acids, Bases, and Oxidizing Agents
3.5. Chemical Stability of PET against Acids, Alkalis, and Oxidizing Agents
3.6. Chemical Stability of PP against Acids, Bases, and Oxidizing Agents
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rillig, M.C. Microplastic in terrestrial ecosystems and the soil? Environ. Sci. Technol. 2012, 46, 6453–6454. [Google Scholar] [CrossRef] [PubMed]
- Barnes, D.K.A.; Galgani, F.; Thompson, R.C.; Barlaz, M. Accumulation and fragmentation of plastic debris in global environments. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 1985–1998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napper, I.E.; Bakir, A.; Rowland, S.J.; Thompson, R.C. Characterisation, quantity and sorptive properties of microplastics extracted from cosmetics. Mar. Pollut. Bull. 2015, 99, 178–185. [Google Scholar] [CrossRef] [Green Version]
- Cole, M.; Lindeque, P.; Halsband, C.; Galloway, T.S. Microplastics as contaminants in the marine environment: A review. Mar. Pollut. Bull. 2011, 62, 2588–2597. [Google Scholar] [CrossRef] [PubMed]
- Duis, K.; Coors, A. Microplastics in the aquatic and terrestrial environment: Sources (with a specific focus on personal care products), fate and effects. Environ. Sci. Eur. 2016, 28, 1–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lusher, A.L.; Welden, N.A.; Sobral, P.; Cole, M. Sampling, isolating and identifying microplastics ingested by fish and invertebrates. Anal. Methods 2017, 9, 1346–1360. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Liu, H.; Paul Chen, J. Microplastics in freshwater systems: A review on occurrence, environmental effects, and methods for microplastics detection. Water Res. 2018, 137, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Eerkes-Medrano, D.; Thompson, R.C.; Aldridge, D.C. Microplastics in freshwater systems: A review of the emerging threats, identification of knowledge gaps and prioritisation of research needs. Water Res. 2015, 75, 63–82. [Google Scholar] [CrossRef] [PubMed]
- Suaria, G.; Avio, C.G.; Mineo, A.; Lattin, G.L.; Magaldi, M.G.; Belmonte, G.; Moore, C.J.; Regoli, F.; Aliani, S. The Mediterranean plastic soup: Synthetic polymers in Mediterranean surface waters. Sci. Rep. 2016, 6, 37551. [Google Scholar] [CrossRef] [Green Version]
- Van Cauwenberghe, L.; Vanreusel, A.; Mees, J.; Janssen, C.R. Microplastic pollution in deep-sea sediments. Environ. Pollut. 2013, 182, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Quinn, B.; Murphy, F.; Ewins, C. Validation of density separation for the rapid recovery of microplastics from sediment. Anal. Methods 2017, 9, 1491–1498. [Google Scholar] [CrossRef] [Green Version]
- Zobkov, M.B.; Esiukova, E.E. Microplastics in a marine environment: Review of methods for sampling, processing, and analyzing microplastics in water, bottom sediments, and coastal deposits. Oceanology 2018, 58, 137–143. [Google Scholar] [CrossRef]
- Prata, J.C.; da Costa, J.P.; Duarte, A.C.; Rocha-Santos, T. Methods for sampling and detection of microplastics in water and sediment: A critical review. TrAC-Trends Anal. Chem. 2019, 110, 150–159. [Google Scholar] [CrossRef]
- Munno, K.; Helm, P.A.; Jackson, D.A.; Rochman, C.; Sims, A. Impacts of temperature and selected chemical digestion methods on microplastic particles. Environ. Toxicol. Chem. 2018, 37, 91–98. [Google Scholar] [CrossRef]
- Corcoran, P.L.; Norris, T.; Ceccanese, T.; Walzak, M.J.; Helm, P.A.; Marvin, C.H. Hidden plastics of Lake Ontario, Canada and their potential preservation in the sediment record. Environ. Pollut. 2015, 204, 17–25. [Google Scholar] [CrossRef]
- Ballent, A.; Corcoran, P.L.; Madden, O.; Helm, P.A.; Longstaffe, F.J. Sources and sinks of microplastics in Canadian Lake Ontario nearshore, tributary and beach sediments. Mar. Pollut. Bull. 2016, 110, 383–395. [Google Scholar] [CrossRef] [Green Version]
- Nuelle, M.T.; Dekiff, J.H.; Remy, D.; Fries, E. A new analytical approach for monitoring microplastics in marine sediments. Environ. Pollut. 2014, 184, 161–169. [Google Scholar] [CrossRef]
- Hidalgo-Ruz, V.; Gutow, L.; Thompson, R.C.; Thiel, M. Microplastics in the marine environment: A review of the methods used for identification and quantification. Environ. Sci. Technol. 2012, 46, 3060–3075. [Google Scholar] [CrossRef]
- Claessens, M.; Van Cauwenberghe, L.; Vandegehuchte, M.B.; Janssen, C.R. New techniques for the detection of microplastics in sediments and field collected organisms. Mar. Pollut. Bull. 2013, 70, 227–233. [Google Scholar] [CrossRef]
- Masura, J.; Baker, J.; Foster, G.; Authur, C. Laboratory Methods for the Analysis of Microplastics in the Marine environment: Recommendations for Quantifying Synthetic Particles in Waters and Sediments. NOAA Tech. Memo. NOS-OR&R 2015. Available online: https://repository.library.noaa.gov/view/noaa/10296 (accessed on 20 August 2022).
- Foekema, E.M.; De Gruijter, C.; Mergia, M.T.; Van Franeker, J.A.; Murk, A.J.; Koelmans, A.A. Plastic in north sea fish. Environ. Sci. Technol. 2013, 47, 8818–8824. [Google Scholar] [CrossRef]
- Cole, M.; Webb, H.; Lindeque, P.K.; Fileman, E.S.; Halsband, C.; Galloway, T.S. Isolation of microplastics in biota-rich seawater samples and marine organisms. Sci. Rep. 2014, 4, 4528. [Google Scholar] [CrossRef] [Green Version]
- Roch, S.; Brinker, A. Rapid and Efficient Method for the Detection of Microplastic in the Gastrointestinal Tract of Fishes. Environ. Sci. Technol. 2017, 51, 4522–4530. [Google Scholar] [CrossRef]
- Avio, C.G.; Gorbi, S.; Regoli, F. Experimental development of a new protocol for extraction and characterization of microplastics in fish tissues: First observations in commercial species from Adriatic Sea. Mar. Environ. Res. 2015, 111, 18–26. [Google Scholar] [CrossRef]
- Imhof, H.K.; Ivleva, N.P.; Schmid, J.; Niessner, R.; Laforsch, C. Contamination of beach sediments of a subalpine lake with microplastic particles. Curr. Biol. 2013, 23, R867–R868. [Google Scholar] [CrossRef] [Green Version]
- Catarino, A.I.; Thompson, R.; Sanderson, W.; Henry, T.B. Development and optimization of a standard method for extraction of microplastics in mussels by enzyme digestion of soft tissues. Environ. Toxicol. Chem. 2017, 36, 947–951. [Google Scholar] [CrossRef]
- Reimonn, G.; Lu, T.; Gandhi, N.; Chen, W.T. Review of microplastic pollution in the environment and emerging recycling solutions. J. Renew. Mater. 2019, 7, 1251–1268. [Google Scholar] [CrossRef] [Green Version]
- Kuwabara, K.; Horii, F. Solid-state 13C NMR analyses of the orthorhombic-to-hexagonal phase transition for constrained ultradrawn polyethylene fibers. Macromolecules 1999, 32, 5600–5605. [Google Scholar] [CrossRef]
- Kitamura, R.; Horii, F.; Murayama, K. Erratum: Phase structure of lamellar crystalline polyethylene by solid-state high-resolution 13C NMR: Detection of the crystalline-amorphous interphase (Macromolecules (1986) 19, 3, (636)). Macromolecules 1986, 19, 1778. [Google Scholar]
- Gulmine, J.V.; Janissek, P.R.; Heise, H.M.; Akcelrud, L. Polyethylene characterization by FTIR. Polym. Test. 2002, 21, 557–563. [Google Scholar] [CrossRef]
- Kaneko, M.; Kumagai, S.; Nakamura, T.; Sato, H. Study of sulfonation mechanism of low-density polyethylene films with fuming sulfuric acid. J. Appl. Polym. Sci. 2004, 91, 2435–2442. [Google Scholar] [CrossRef]
- Grassi, A.; Longo, P.; Guerra, G. Solid-state high-resolution 13C NMR spectra of syndiotactic polystyrene. Makromol. Chem., Rapid Commun. 1989, 10, 687–690. [Google Scholar] [CrossRef]
- Joseph, R.; Ford, W.T.; Zhang, S.; Tsyurupa, M.P.; Pastukhov, A.V.; Davankov, V.A. Solid-state 13C-NMR analysis of hypercrosslinked polystyrene. J. Polym. Sci. A. Polym. Chem. 1996, 35, 695–701. [Google Scholar] [CrossRef]
- Coughlin, J.E.; Reisch, A.; Markarian, M.Z.; Schlenoff, J.B. Sulfonation of polystyrene: Toward the “ideal” polyelectrolyte. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 2416–2424. [Google Scholar] [CrossRef]
- Jalal, N.M.; Jabur, A.R.; Hamza, M.S.; Allami, S. Sulfonated electrospun polystyrene as cation exchange membranes for fuel cells. Energy Rep. 2020, 6, 287–298. [Google Scholar] [CrossRef]
- Tabekh, H.; Al Kurdi, M.H.; Ajji, Z. Sulphonation of expanded polystyrene waste with commercial sulphuric acid for potential use in removal of heavy metals from contaminated waters. Polimeri 2015, 36, 11–14. [Google Scholar]
- Noritake, A.; Hori, M.; Shigematsu, M.; Tanahashi, M. Recycling of polyethylene terephthalate using high-pressure steam treatment. Polym. J. 2008, 40, 498–502. [Google Scholar] [CrossRef] [Green Version]
- Al-Sabagh, A.M.; Yehia, F.Z.; Eshaq, G.; Rabie, A.M.; ElMetwally, A.E. Greener routes for recycling of polyethylene terephthalate. Egypt. J. Pet. 2016, 25, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Liu, R.; Li, C.; Song, Y.; Hu, C. Hydrolysis of waste polyethylene terephthalate catalyzed by easily recyclable terephthalic acid. Waste Manag. 2021, 135, 267–274. [Google Scholar] [CrossRef]
- Chinchillas-Chinchillas, M.J.; Orozco-Carmona, V.M.; Alvarado-Beltrán, C.G.; Almaral-Sánchez, J.L.; Sepulveda-Guzman, S.; Jasso-Ramos, L.E.; Castro-Beltrán, A. Synthesis of recycled poly(ethylene terephthalate)/polyacrylonitrile/styrene composite nanofibers by electrospinning and their mechanical properties evaluation. J. Polym. Environ. 2019, 27, 659–669. [Google Scholar] [CrossRef]
- Valh, J.V.; Vončina, B.; Lobnik, A.; Zemljič, L.F.; Škodič, L.; Vajnhandl, S. Conversion of polyethylene terephthalate to high-quality terephthalic acid by hydrothermal hydrolysis: The study of process parameters. Text. Res. J. 2020, 90, 1446–1461. [Google Scholar] [CrossRef]
- Bunn, A.; Cudby, M.E.A.; Harris, R.K.; Packer, K.J.; Say, B.J. High resolution 13C n.m.r. spectra of solid isotactic polypropylene. Polymer (Guildf) 1982, 23, 694–698. [Google Scholar] [CrossRef]
- Morent, R.; De Geyter, N.; Leys, C.; Gengembre, L.; Payen, E. Comparison between XPS- and FTIR-analysis of plasma-treated polypropylene film surfaces. Surf. Interface Anal. 2008, 40, 597–600. [Google Scholar] [CrossRef]
- Tada, H.; Ito, S. Conformational change restricted selectivity in the surface sulfonation of polypropylene with sulfuric acid. Langmuir 1997, 13, 3982–3989. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H2SO4 | HCl | H2O2 | KOH | NaOH | |
|---|---|---|---|---|---|
| HDPE | Morphologies, functionalities | No effects | No effects | No effects | No effects |
| LDPE | Morphologies, functionalities | No effects | No effects | No effects | No effects |
| PS | Morphologies, functionalities, carbon structures | No effects | No effects | No effects | No effects |
| PET | Morphologies, functionalities, carbon structures | No effects | No effects | No effects | No effects |
| PP | Morphologies, functionalities | No effects | No effects | No effects | No effects |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.B.; Lee, K.S.; Kim, S.J.; Choi, B.I.; Go, B.R.; Rhu, C.J.; Han, T.H. Effect of Chemical Agents on the Morphology and Chemical Structures of Microplastics. Polymers 2022, 14, 4353. https://0-doi-org.brum.beds.ac.uk/10.3390/polym14204353
Lee HB, Lee KS, Kim SJ, Choi BI, Go BR, Rhu CJ, Han TH. Effect of Chemical Agents on the Morphology and Chemical Structures of Microplastics. Polymers. 2022; 14(20):4353. https://0-doi-org.brum.beds.ac.uk/10.3390/polym14204353
Chicago/Turabian StyleLee, Hak Bong, Kyong Sub Lee, Seok Jun Kim, Byung Il Choi, Byung Rye Go, Chan Joo Rhu, and Tae Hee Han. 2022. "Effect of Chemical Agents on the Morphology and Chemical Structures of Microplastics" Polymers 14, no. 20: 4353. https://0-doi-org.brum.beds.ac.uk/10.3390/polym14204353