
Recent Advances in Fluorescence Recovery after Photobleaching for Decoupling Transport and Kinetics of Biomacromolecules in Cellular Physiology


 ,
,
Abstract
:1. Introduction
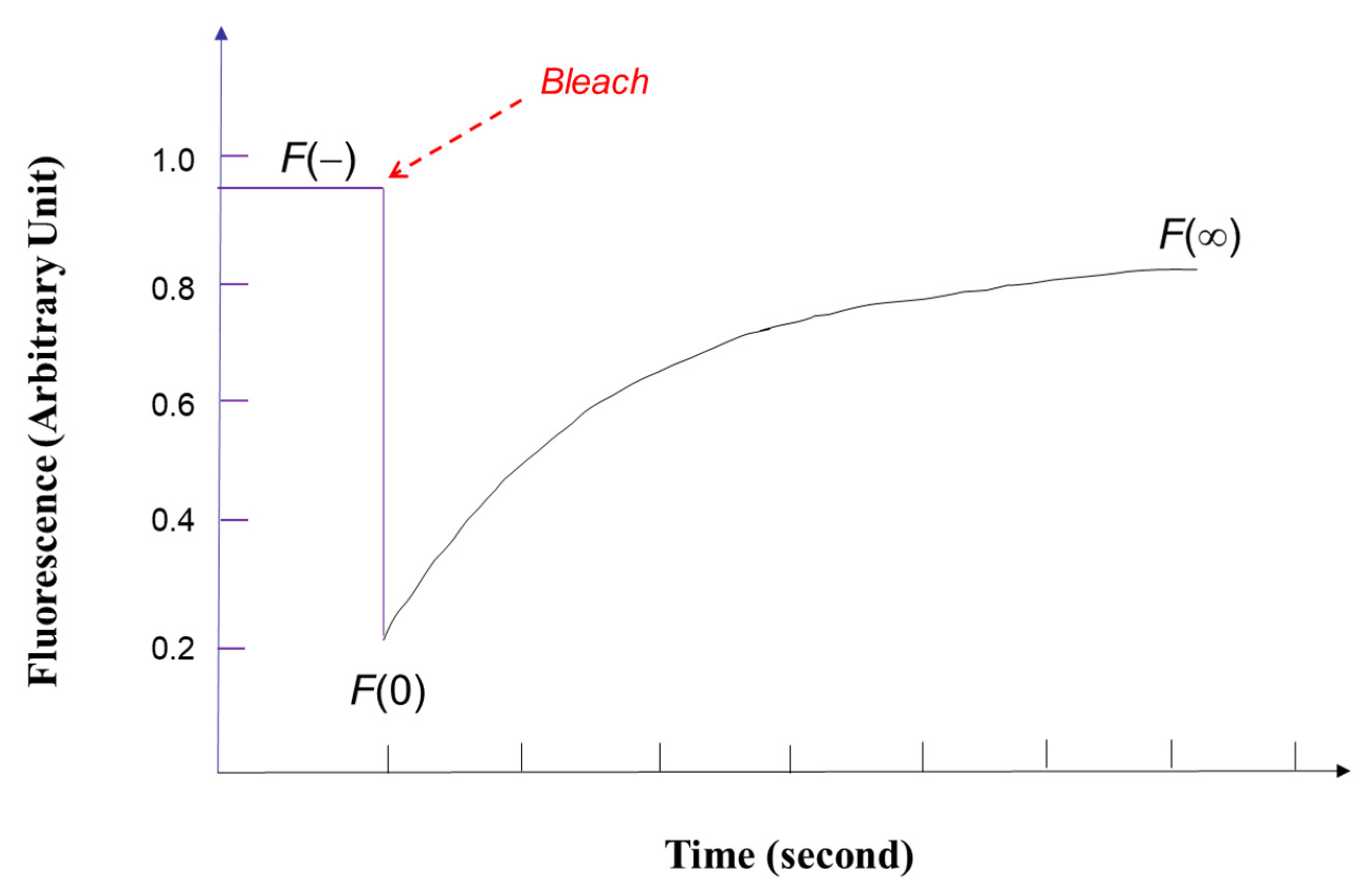
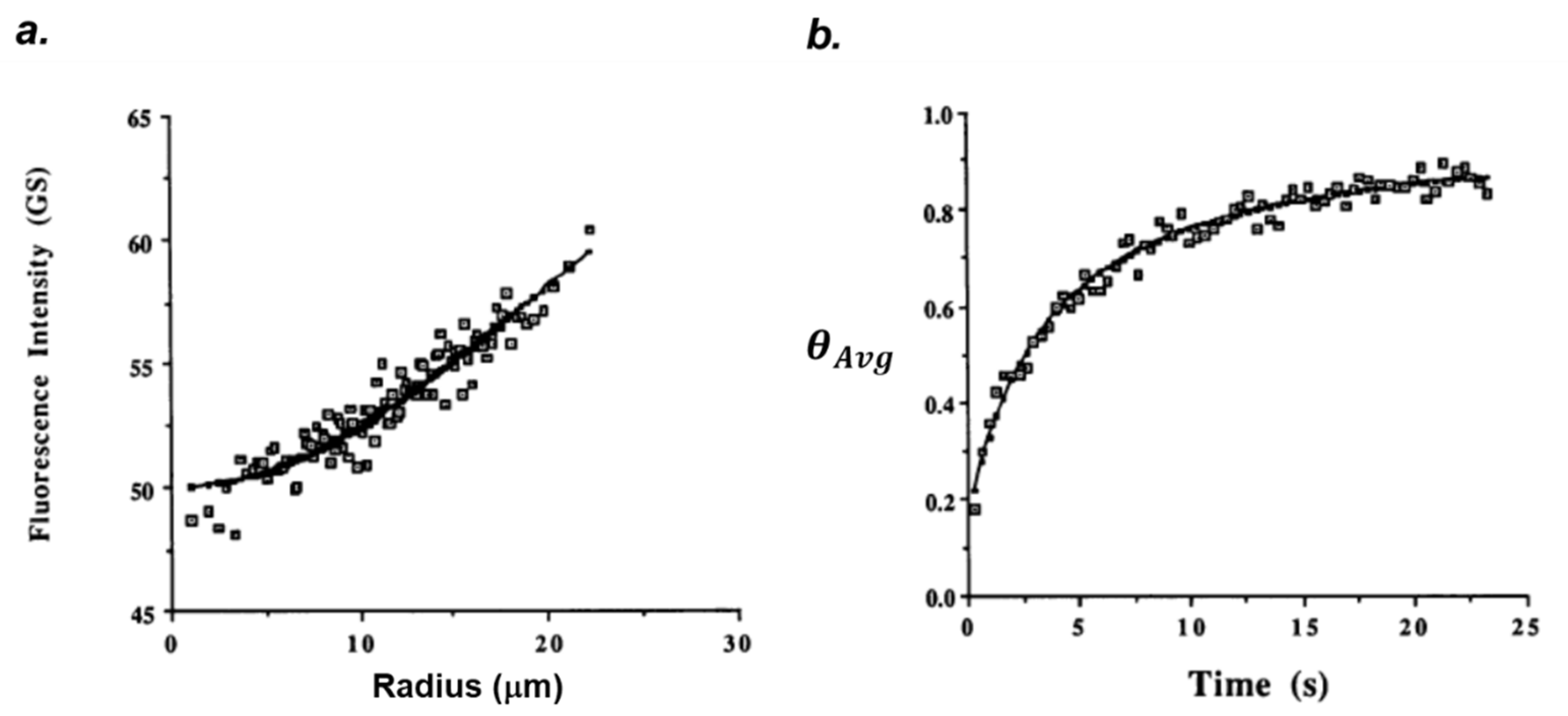
2. FRAP Theory and Data Analysis
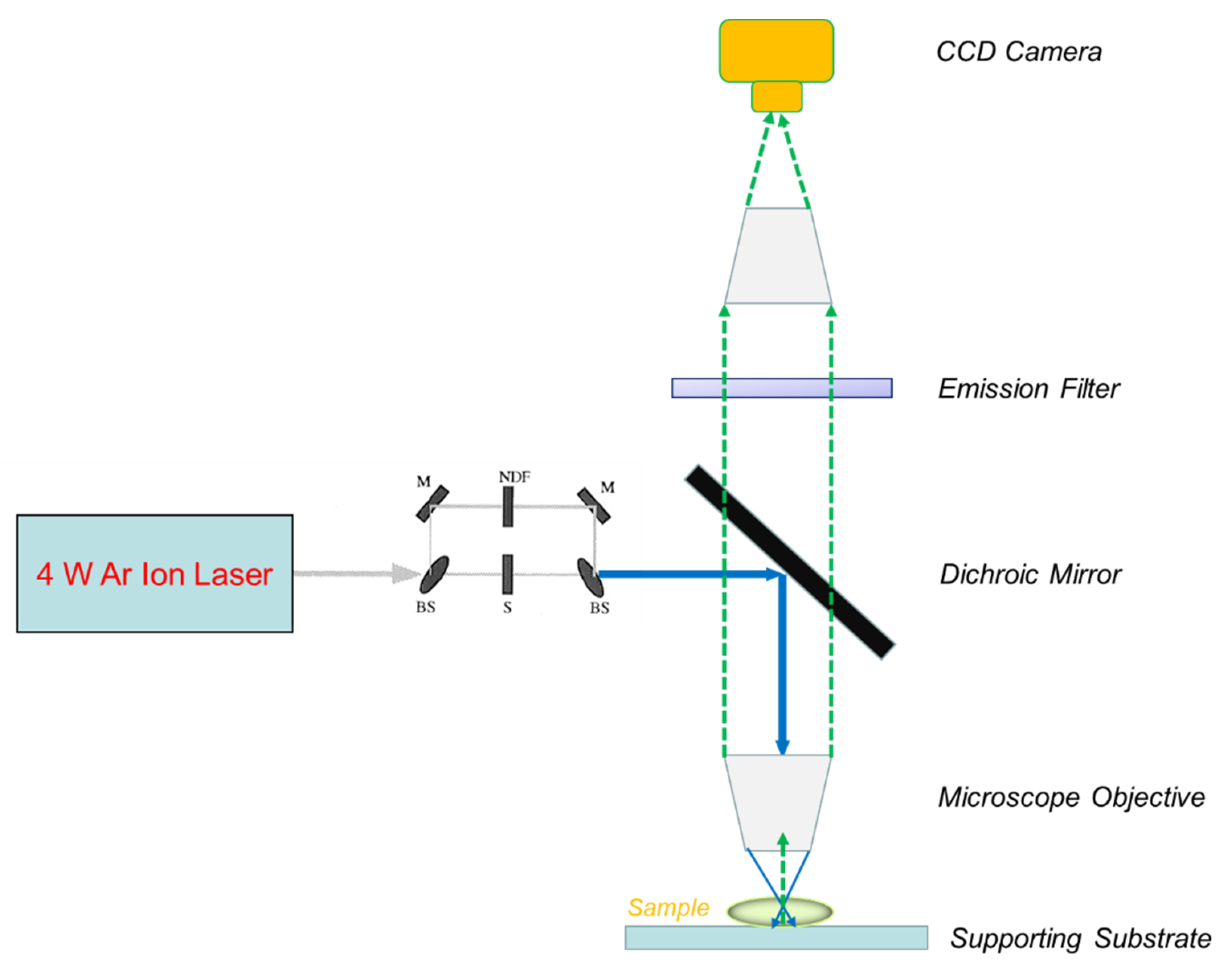
2.1. Epifluorescence Spot Photobleaching
2.2. Total Internal Reflection/Fluorescence Recovery after Photobleaching
2.3. Confocal FRAP
3. Emerging FRAP Applications
3.1. Physiological Transport
3.2. Interfacial Biophysics
3.3. Cellular Dynamics
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Manzoni, C.; Kia, D.A.; Vandrovcova, J.; Hardy, J.; Wood, N.W.; Lewis, P.A.; Ferrari, R. Genome, transcriptome and proteome: The rise of omics data and their integration in biomedical sciences. Brief. Bioinform. 2018, 19, 286–302. [Google Scholar] [CrossRef] [PubMed]
- Hartl, D.; de Luca, V.; Kostikova, A.; Laramie, J.; Kennedy, S.; Ferrero, E.; Siegel, R.; Fink, M.; Ahmed, S.; Millholland, J.; et al. Translational precision medicine: An industry perspective. J. Transl. Med. 2021, 19, 245. [Google Scholar] [CrossRef] [PubMed]
- Ganapathiraju, M.K.; Orii, N. Research prioritization through prediction of future impact on biomedical science: A position paper on inference-analytics. GigaScience 2013, 2, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, A.; Chauhan, P.; Saha, B.; Kubatzky, K.F. Conceptual evolution of cell signaling. Int. J. Mol. Sci. 2019, 20, 3292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarbell, J.M.; Simon, S.I.; Curry, F.-R.E. Mechanosensing at the vascular interface. Annu. Rev. Biomed. Eng. 2014, 16, 505–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allam, M.; Cai, S.; Coskun, A.F. Multiplex bioimaging of single-cell spatial profiles for precision cancer diagnostics and therapeutics. NPJ Precis. Oncol. 2020, 4, 11. [Google Scholar] [CrossRef]
- Miller, H.; Zhou, Z.; Shepherd, J.; Wollma, A.J.M.; Leake, M.C. Single-molecule techniques in biophysics: A review of the progress in methods and applications. Rep. Prog. Phys. 2018, 81, 024601. [Google Scholar] [CrossRef] [Green Version]
- Baghban, R.; Roshangar, L.; Rana Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Pijuan-Galito, S.; Rho, H.S.; Vasilevich, A.S.; Eren, A.D.; Ge, L.; Habibović, P.; Alexander, M.R.; de Boer, J.; Carlier, A.; et al. High-throughput methods in the discovery and study of biomaterials and materiobiology. Chem. Rev. 2021, 121, 4561–4677. [Google Scholar] [CrossRef]
- Sykes, E.A.; Dai, Q.; Sarsons, C.D.; Chen, J.; Rocheleau, J.V.; Hwang, D.M.; Zheng, G.; Cramb, D.T.; Rinker, K.S.; Chan, W.C.W. Tailoring nanoparticle designs to target cancer based on tumor pathophysiology. Proc. Natl. Acad. Sci. USA 2016, 113, E1142–E1151. [Google Scholar] [CrossRef] [Green Version]
- Senapati, S.; Mahanta, A.K.; Kumar, S.; Maiti, P. Controlled drug delivery vehicles for cancer treatment and their performance. Signal Transduct. Target. Ther. 2018, 3, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nugent, L.J.; Jain, R.K. Extravascular diffusion in normal and neoplastic tissues. Cancer Res. 1984, 44, 238–244. [Google Scholar]
- Chary, S.R.; Jain, R.K. Direct measurement of interstitial convection and diffusion of albumin in normal and neoplastic tissues by fluorescence photobleaching. Proc. Natl. Acad. Sci. USA 1989, 86, 5385–5389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clauss, M.A.; Jain, R.K. Interstitial transport of rabbit and sheep antibodies in normal and neoplastic tissues. Cancer Res. 1990, 50, 3487–3492. [Google Scholar] [PubMed]
- Dewhirst, M.W.; Secomb, T.W. Transport of drugs from blood vessels to tumour tissue. Nat. Rev. Cancer 2017, 17, 738–750. [Google Scholar] [CrossRef]
- Padera, T.P.; Meijer, E.F.J.; Munn, L.L. The Lymphatic System in Disease Processes and Cancer Progression. Annu. Rev. Biomed. Eng. 2016, 18, 125–158. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, E.N.; Jain, R.K. Effect of bivalent interaction upon apparent antibody affinity: Experimental confirmation of theory using fluorescence photobleaching and implications for antibody binding assays. Cancer Res. 1992, 52, 4157–4167. [Google Scholar]
- Kaufman, E.N.; Jain, R.K. In vitro measurement and screening of monoclonal antibody affinity using fluorescence photobleaching. J. Immunol. Methods 1992, 155, 1–17. [Google Scholar] [CrossRef]
- Lu, R.-M.; Hwang, Y.-C.; Liu, I.-J.; Lee, C.C.; Tsai, H.Z.; Li, H.-J.; Wu, H.-C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef]
- Kaufman, E.N.; Jain, R.K. Quantification of transport and binding parameters using fluorescence recovery after photobleaching. Potential for in vivo applications. Biophys J. 1990, 58, 873–885. [Google Scholar] [CrossRef] [Green Version]
- Maeda, H.; Khatami, M. Analyses of repeated failures in cancer therapy for solid tumors: Poor tumor-selective drug delivery, low therapeutic efficacy and unsustainable costs. Clin. Transl. Med. 2018, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Rose, K.A.; Molaei, M.; Boyle, M.J.; Lee, D.; Crocker, J.C.; Composto, R.J. Particle tracking of nanoparticles in soft matter. J. Appl. Phys. 2020, 127, 191101. [Google Scholar] [CrossRef]
- Nauman, J.V.; Campbell, P.G.; Lanni, F.; Anderson, J.L. Diffusion of Insulin-Like Growth Factor-I and Ribonuclease through Fibrin Gels. Biophys. J. 2007, 92, 4444–4450. [Google Scholar] [CrossRef] [Green Version]
- Collett, J.A.; Corridon, P.R.; Mehrotra, P.; Kolb, A.L.; Rhodes, G.J.; Miller, C.A.; Molitoris, B.A.; Pennington, J.G.; Sandoval, R.M.; Atkinson, S.J.; et al. Hydrodynamic isotonic fluid delivery ameliorates moderate-to-severe ischemia-reperfusion injury in rat kidneys. J. Am. Soc. Nephrol. 2017, 28, 2081–2092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corridon, P.R.; Karam, S.H.; Khraibi, A.A.; Khan, A.A.; Alhashmi, M.A. Intravital imaging of real-time endogenous actin dysregulation in proximal and distal tubules at the onset of severe ischemia-reperfusion injury. Sci. Rep. 2021, 11, 8280. [Google Scholar] [CrossRef] [PubMed]
- Corridon, P.R.; Rhodes, G.J.; Leonard, E.C.; Basile, D.P.; Gattone, V.H.; Bacallao, R.L.; Atkinson, S.J. A method to facilitate and monitor expression of exogenous genes in the rat kidney using plasmid and viral vectors. Am. J. Physiol. Renal. Physiol. 2013, 304, F1217–F1229. [Google Scholar] [CrossRef] [Green Version]
- Hall, A.M.; Rhodes, G.J.; Sandoval, R.M.; Corridon, P.R.; Molitoris, B.A. In vivo multiphoton imaging of mitochondrial structure and function during acute kidney injury. Kidney Int. 2013, 83, 72–83. [Google Scholar] [CrossRef] [Green Version]
- Kolb, A.L.; Corridon, P.R.; Zhang, S.; Xu, W.; Witzmann, F.A.; Collett, J.A.; Rhodes, G.J.; Winfree, S.; Bready, D.; Pfeffenberger, Z.J.; et al. Exogenous gene transmission of isocitrate dehydrogenase 2 mimics ischemic preconditioning protection. J. Am. Soc. Nephrol. 2018, 29, 1154–1164. [Google Scholar] [CrossRef] [Green Version]
- Jain, R.K.; Munn, L.L.; Fukumura, D. Intravital Microscopy of Normal and Diseased Tissues in the Mouse. In Live Cell Imaging: A Laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory: New York, NY, USA, 2010. [Google Scholar]
- Pluen, A.; Boucher, Y.; Ramanujan, S.; McKee, T.D.; Gohongi, T.; di Tomaso, E.; Brown, E.B.; Izumi, Y.; Campbell, R.B.; Berk, D.A.; et al. Role of tumor–host interactions in interstitial diffusion of macromolecules: Cranial vs. subcutaneous tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 4628–4633. [Google Scholar] [CrossRef] [Green Version]
- Calvert, P.D.; Peet, J.A.; Bragin, A.; Schiesser, W.E.; Pugh, E.N., Jr. Fluorescence relaxation in 3D from diffraction-limited sources of PAGFP or sinks of EGFP created by multiphoton photoconversion. J. Microsc. 2007, 225, 49–71. [Google Scholar] [CrossRef]
- Owens, G.J.; Singh, R.K.; Foroutan, F.; Alqaysi, M.; Han, C.-M.; Mahapatra, C.; Kim, H.-W.; Knowles, J.C. Sol–gel based materials for biomedical applications. Prog. Mater. Sci. 2016, 77, 1–79. [Google Scholar] [CrossRef] [Green Version]
- Jain, R.K.; Stock, R.J.; Chary, S.R.; Rueter, M. Convection and diffusion measurements using fluorescence recovery after photobleaching and video image analysis: In vitro calibration and assessment. Microvasc. Res. 1990, 39, 77–93. [Google Scholar] [CrossRef]
- Fu, B.M.; Adamson, R.H.; Curry, F.-R.E. Determination of microvessel permeability and tissue diffusion coefficient of solutes by laser scanning confocal microscopy. J. Biomech. Eng. 2005, 127, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Shaya, J.; Corridon, P.R.; Al-Omari, B.; Aoudi, A.; Shunnar, A.; Mohideen, M.I.H.; Qurashi, A.; Michel, B.Y.; Burger, A. Design, photophysical properties, and applications of fluorene-based fluorophores in two-photon fluorescence bioimaging: A review. J. Photochem. Photobiol. C: Photochem. Rev. 2022, 52, 100529. [Google Scholar] [CrossRef]
- Ando, D.; Korabel, N.; Huang, K.C.; Gopinathan, A. Cytoskeletal Network Morphology Regulates Intracellular Transport Dynamics. Biophys. J. 2015, 109, 1574–1582. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Ju, L.; Rushdi, M.; Ge, C.; Zhu, C. Receptor-mediated cell mechanosensing. Mol. Biol. Cell 2017, 28, 3133–3470. [Google Scholar] [CrossRef]
- Stewart, P.S.; Zhang, T.; Xu, R.; Pitts, B.; Walters, M.C.; Roe, F.; Kikhney, J.; Moter, A. Reaction–diffusion theory explains hypoxia and heterogeneous growth within microbial biofilms associated with chronic infections. NPJ Biofilms Microbiomes 2016, 2, 16012. [Google Scholar] [CrossRef]
- Lauffenburger, D.A.; Linderman, J.J. Receptors: Models for Binding, Trafficking, and Signaling; Oxford University Press: Oxford, UK, 1993. [Google Scholar]
- Grabon, A.; Bankaitis, V.A.; McDermott, M.I. The interface between phosphatidylinositol transfer protein function and phosphoinositide signaling in higher eukaryotes. J. Lipid Res. 2019, 60, 242–268. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Weinreb, V.; Ridilla, M.; Betts, L.; Patel, P.; de Silva, A.M.; Thompson, N.L.; Jacobson, K. Rapid, directed transport of DC-SIGN clusters in the plasma membrane. Sci. Adv. 2017, 3, eaao1616. [Google Scholar] [CrossRef] [Green Version]
- Burghardt, T.P.; Axelrod, D. Total internal reflection/fluorescence photobleaching recovery study of serum albumin adsorption dynamics. Biophys. J. 1981, 33, 455–467. [Google Scholar] [CrossRef] [Green Version]
- Tallawi, M.; Rosellini, E.; Barbani, N.; Cascone, M.G.; Rai, R.; Saint-Pierre, G.; Boccaccini, A.R. Strategies for the chemical and biological functionalization of scaffolds for cardiac tissue engineering: A review. J. R. Soc. Interface 2015, 12, 20150254. [Google Scholar] [CrossRef] [PubMed]
- Purohit, B.; Vernekar, P.R.; Shetti, N.P.; Chandra, P. Biosensor nanoengineering: Design, operation, and implementation for biomolecular analysis. Sens. Int. 2020, 1, 100040. [Google Scholar] [CrossRef]
- Tilton, R.D.; Gast, A.P.; Robertson, C.R. Surface diffusion of interacting proteins. Effect of concentration on the lateral mobility of adsorbed bovine serum albumin. Biophys. J. 1990, 58, 1321–1326. [Google Scholar] [CrossRef] [Green Version]
- Migliorini, E.; Weidenhaupt, M.; Picart, C. Practical guide to characterize biomolecule adsorption on solid surfaces (Review). Biointerphases 2018, 13, 06D303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rachit Bansal, R.; Elgundi, Z.; Care, A.; Goodchild, S.C.; Lord, M.S.; Rodger, A.; Sunna, A. Elucidating the Binding Mechanism of a Novel Silica-Binding Peptide. Biomolecules 2020, 10, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russev, S.C.; Arguirov, T.V.; Gurkov, T.D. b-Casein adsorption kinetics on air–water and oil–water interfaces studied by ellipsometry. Colloids Surf. B 2000, 19, 89–100. [Google Scholar] [CrossRef]
- Yan, E.C.Y.; Fu, L.; Wang, Z.; Liu, W. Biological macromolecules at interfaces probed by chiral sum frequency generation spectroscopy. Chem. Rev. 2014, 114, 8471. [Google Scholar] [CrossRef] [Green Version]
- Kriechbaumer, V.; Nabok, A.; Widdowson, R.; Smith, D.P.; Abell, B.M. Quantification of ligand binding to G-protein coupled receptors on cell membranes by ellipsometry. PLoS ONE 2012, 7, e46221. [Google Scholar] [CrossRef] [Green Version]
- Triacca, V.; Güç, E.; Kilarski, W.W.; Pisano, M.; Swartz, M.A. Transcellular pathways in lymphatic endothelial cells regulate changes in solute transport by fluid stress. Circ. Res. 2017, 120, 1440–1452. [Google Scholar] [CrossRef]
- Nguyen, H.H.; Park, J.; Kang, S.; Kim, M. Surface plasmon resonance: A versatile technique for biosensor applications. Sensors 2015, 15, 10481–10510. [Google Scholar] [CrossRef] [Green Version]
- Paradiso, F.; Serpelloni, S.; Francis, L.W.; Taraballi, F. Mechanical studies of the third dimension in cancer: From 2D to 3D model. Int. J. Mol. Sci. 2021, 22, 10098. [Google Scholar] [CrossRef] [PubMed]
- Dogra, P.; Butner, J.D.; Nizzero, S.; Ramírez, J.R.; Noureddine, A.; Peláez, M.J.; Elganainy, D.; Yang, Z.; Le, A.-D.; Goel, S.; et al. Image-guided mathematical modeling for pharmacological evaluation of nanomaterials and monoclonal antibodies. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2020, 12, e1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGlynn, J.A.; Wu, N.; Schultz, K.M. Multiple particle tracking microrheological characterization: Fundamentals, emerging techniques and applications. J. Appl. Phys. 2020, 127, 201101. [Google Scholar] [CrossRef]
- Elsner, M.; Hashimoto, H.; Simpson, J.C.; Cassel, D.; Nilsson, T.; Weiss, M. Spatiotemporal dynamics of the COPI vesicle machinery. EMBO Rep. 2003, 4, 1000–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Varela, A.I.; Gaspar, R.; Miranda, A.; Assis, J.L.; Valverde, R.H.F.; Einicker-Lamas, M.; Silva, B.F.B.; De Beule, P.A.A.J. Fluorescence cross-correlation spectroscopy as a valuable tool to characterize cationic liposome-DNA nanoparticle assembly. Biophotonics 2021, 14, e202000200. [Google Scholar] [CrossRef]
- Bacia, K.; Schwille, P. Practical guidelines for dual-color fluorescence cross-correlation spectroscopy. Nat. Protoc. 2007, 2, 2842–2856. [Google Scholar] [CrossRef]
- Krüger, D.; Ebenhan, J.; Werner, S.; Bacia, K. Measuring protein binding to lipid vesicles by fluorescence cross-correlation spectroscopy. Biophys. J. 2017, 113, 1311–1320. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, H.V.; Thompson, N.L. Theory for measuring bivalent surface binding kinetics using total internal reflection with fluorescence photobleaching recovery. Biophys. J. 1994, 66, 898–911. [Google Scholar] [CrossRef] [Green Version]
- Thompson, N.L.; Elsardt, T.P.; Axelrod, D. Measuring surface dynamics of biomolecules by total internal reflection fluorescence with photobleaching recovery or correlation spectroscopy. Biophys. J. 1981, 33, 435–454. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, J.; Sasaki, A. Fluorescence correlation spectroscopy measurement based on fiber optics for biological materials. Appl. Sci. 2021, 11, 6744. [Google Scholar] [CrossRef]
- Ruthardt, N.; Lamb, D.C.; Bräuchle, C. Single-particle tracking as a quantitative microscopy-based approach to unravel cell entry mechanisms of viruses and pharmaceutical nanoparticles. Mol. Ther. 2011, 19, 1199–1211. [Google Scholar] [CrossRef] [PubMed]
- Piliarik, M.; Homola, J. Surface plasmon resonance (SPR) sensors: Approaching their limits? Opt. Express 2009, 17, 16505–16517. [Google Scholar] [CrossRef]
- Ahmed, F.E.; Wiley, J.E.; Weidner, D.A.; Bonnerup, C.; Mota, H. Surface plasmon resonance (SPR) spectrometry as a tool to analyze nucleic acid–protein interactions in crude cellular extracts. Cancer Genom. Proteom. 2010, 7, 303–309. [Google Scholar]
- Sehayek, S.; Yi, X.; Weiss, S.; Wiseman, P.W. Rapid ensemble measurement of protein diffusion and probe blinking dynamics in cells. Biophys. Rep. 2021, 1, 100015. [Google Scholar] [CrossRef]
- Gebhardt, J.C.M.; Suter, D.M.; Roy, R.; Zhao, Z.W.; Chapman, A.R.; Basu, S.; Maniatis, T.; Xie, X.S. Single-molecule imaging of transcription factor binding to DNA in live mammalian cells. Nat. Methods 2013, 10, 421–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achimovich, A.M.; Ai, H.; Gahlmann, A. Enabling technologies in super-resolution fluorescence microscopy: Reporters, labeling, and methods of measurement. Curr. Opin. Struct. Biol. 2019, 58, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Bacia, K.; Kim, S.A.; Schwille, P. Fluorescence cross-correlation spectroscopy in living cells. Nat. Methods 2006, 3, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Briddon, S.J.; Hill, S.J. Pharmacology under the microscope: The use of fluorescence correlation spectroscopy to determine the properties of ligand–receptor complexes. Trends Pharmacol. Sci. 2007, 28, 637–645. [Google Scholar] [CrossRef] [Green Version]
- Axelrod, D.; Koppel, D.E.; Schlessinger, J.; Elson, E.; Webb, W.W. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys. J. 1976, 16, 1055–1069. [Google Scholar] [CrossRef] [Green Version]
- Koppel, E.; Axelrod, D.; Schlessinger, J.; Elson, E.L.; Webb, W.W. Dynamics of fluorescence marker concentration as a probe of mobility. Biophys. J. 1976, 16, 1315–1329. [Google Scholar] [CrossRef] [Green Version]
- Tsay, T.-T.; Jacobson, K.A. Spatial fourier analysis of video photobleaching measurements. Principles and optimization. Biophys. J. 1991, 60, 360–368. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.C.; Wong, D.T.; Discher, D.E. Direct measures of large, anisotropic strains in deformation of the erythrocyte cytoskeleton. Biophys. J. 1999, 77, 853–864. [Google Scholar] [CrossRef] [Green Version]
- Jain, R.K. Delivery of molecular and cellular medicine to solid tumors. Adv. Drug Deliv. Rev. 2012, 64, 353–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramanujan, S.; Pluen, A.; McKee, T.D.; Brown, E.B.; Boucher, Y.; Jain, R.K. Diffusion and convection in collagen gels: Implications for transport in the tumor interstitium. Biophys. J. 2002, 83, 1650–1660. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, V.P.; Lanning, R.M.; Diop-Frimpong, B.; Mok, W.; Brown, E.B.; Padera, T.P.; Boucher, Y.; Jain, R.K. Multiscale measurements distinguish cellular and interstitial hindrances to diffusion in vivo. Biophys. J. 2009, 97, 330–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jönsson, P.; Jonsson, M.P.; Tegenfeldt, J.O.; Höök, F. A Method improving the accuracy of fluorescence recovery after photobleaching analysis. Biophys. J. 2008, 95, 5334–5348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattheyses, A.L.; Simon, S.M.; Rappoport, J.Z. Imaging with total internal reflection fluorescence microscopy for the cell biologist. J. Cell Sci. 2010, 123, 3621–3628. [Google Scholar] [CrossRef] [Green Version]
- Wazawa, T.; Ueda, M. Total internal reflection fluorescence microscopy in single molecule nanobioscience. Adv. Biochem. Eng. Biotechnol. 2005, 95, 77–106. [Google Scholar] [CrossRef]
- Pisarchick, M.L.; Thompson, N.L. Binding of a monoclonal antibody and its Fab fragment to supported phospholipid monolayers measured by total internal reflection fluorescence microscopy. Biophys. J. 1990, 58, 1235–1249. [Google Scholar] [CrossRef] [Green Version]
- Thompson, N.L.; Pearce, K.H.; Hsieh, H.V. Total internal reflection fluorescence microscopy: Application to substrate-supported planar membranes. Eur. Biophys. J. 1993, 22, 367–378. [Google Scholar] [CrossRef]
- Axelrod, D. Total internal reflection fluorescence microscopy in cell biology. Traffic 2001, 2, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Wachsmuth, M. Molecular diffusion and binding analyzed with FRAP. Protoplasma 2014, 251, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Chan, V.; Graves, D.J.; Fortina, P.; McKenzie, S.E. Adsorption and surface diffusion of DNA oligonucleotides at liquid/solid interfaces. Langmuir 1997, 13, 320–329. [Google Scholar] [CrossRef]
- Zadeh, K.S.; Montas, H.J.; Shirmohammadi, A. Identification of biomolecule mass transport and binding rate parameters in living cells by inverse modelling. Theor. Biol. Med. Model. 2006, 3, 36. [Google Scholar] [CrossRef] [Green Version]
- Lippincott-Schwartz, J.; Snapp, E.L.; Phair, R.D. The Development and Enhancement of FRAP as a Key Tool for Investigating Protein Dynamics. Biophys. J. 2018, 115, 1146–1155. [Google Scholar] [CrossRef] [Green Version]
- Mai, J.; Trump, S.; Lehmann, I.; Attinger, S. Parameter importance in FRAP acquisition and analysis: A simulation approach. Biophys. J. 2013, 104, 2089–2097. [Google Scholar] [CrossRef] [Green Version]
- Williamson, D.E.; Sahai, E.; Jenkins, R.P.; O’Dea, R.D.; King, J.R. Parameter estimation in fluorescence recovery after photobleaching: Quantitative analysis of protein binding reactions and diffusion. J. Math. Biol. 2021, 83, 1. [Google Scholar] [CrossRef]
- Berk, D.A.; Yuan, F.; Leunig, M.; Jain, R.K. Fluorescence photobleaching with spatial fourier analysis: Measurement of diffusion in light scattering medium. Biophys. J. 1993, 65, 2428–2436. [Google Scholar] [CrossRef] [Green Version]
- Geiger, A.C.; Smith, C.J.; Takanti, N.; Harmon, D.M.; Carlsen, M.S.; Simpson, G.J. Anomalous diffusion characterization by Fourier transform-FRAP with patterned illumination. Biophys. J. 2020, 119, 737–748. [Google Scholar] [CrossRef]
- Lorén, N.; Hagman, J.; Jonasson, J.K.; Deschout, H.; Bernin, D.; Cella-Zanacchi, F.; Diaspro, A.; McNally, J.G.; Ameloot, M.; Smisdom, N.; et al. Fluorescence recovery after photobleaching in material and life sciences: Putting theory into practice. Q. Rev. Biophys. 2015, 48, 323–387. [Google Scholar] [CrossRef]
- Mueller, F.; Mazza, D.; Stasevich, T.J.; McNally, J.G. FRAP and kinetic modeling in the analysis of nuclear protein dynamics: What do we really know? Curr. Opin. Cell Biol. 2010, 22, 403–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, F.; Morisaki, T.; Mazza, D.; McNally, J.G. Minimizing the impact of photoswitching of fluorescent proteins on FRAP Analysis. Biophys. J. 2012, 102, 1656–1665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufman, E.N.; Jain, R.K. Measurement of mass transport and reaction parameters in bulk solution using photobleaching: Reaction limited binding regime. Biophys. J. 1991, 60, 596–610. [Google Scholar] [CrossRef] [Green Version]
- Chan, V. Reduction of Dimensionality Processes of DNA Hybridization with Immobilized Oligonucleotide Probes. Ph.D. Thesis, University of Pennsylvania, Philadelphia, PA, USA, 1997. [Google Scholar]
- Spada, A.; Emami, J.; Tuszynski, J.A.; Lavasanifar, A. The uniqueness of albumin as a carrier in nanodrug delivery. Mol. Pharm. 2021, 18, 1862–1894. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, Y.; Sedat, J.W.; Agard, D.A. Determination of three dimensional imaging properties of a light microscope system. Biophys. J. 1990, 57, 325–333. [Google Scholar] [CrossRef] [Green Version]
- Sniekers, Y.H.; van Donkelaar, C.C. Determining diffusion coefficients in inhomogeneous tissues Using fluorescence recovery after photobleaching. Biophys. J. 2005, 89, 1302–1307. [Google Scholar] [CrossRef] [Green Version]
- Flamion, B.; Bungay, P.M.; Gibson, C.C.; Spring, K.R. Flow rate measurements in isolated perfused kidney tubules by fluorescence photobleaching recovery. Biophys. J. 1991, 60, 1229–1242. [Google Scholar] [CrossRef] [Green Version]
- Bush, P.G.; Wokosin, D.L.; Hall, A.C. Two-versus one photon excitation laser scanning microscopy: Critical importance of excitation wavelength. Front. Biosci. 2007, 12, 2646–2657. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, K.D.; Sipprell, W.H.; Brown, E.B., Jr.; Brown, E.B. Improved model of fluorescence recovery expands the application of multiphoton fluorescence recovery after photobleaching in vivo. Biophys. J. 2009, 96, 5082–5094. [Google Scholar] [CrossRef] [Green Version]
- De Los Santos, C.; Chang, C.-W.; Mycek, M.-A.; Cardullo, R.A. FRAP, FLIM, and FRET: Detection and analysis of cellular dynamics on a molecular scale using fluorescence microscopy. Mol. Reprod. Dev. 2015, 82, 587–604. [Google Scholar] [CrossRef] [Green Version]
- Black, L.D.; Meyers, J.D.; Weinbaum, J.S.; Shvelidze, Y.A.; Tranquillo, R.T. Cell-induced alignment augments twitch force in fibrin gel–based engineered myocardium via gap junction modification. Tissue Eng. A 2009, 15, 3099–3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corridon, P.R. In vitro investigation of the impact of pulsatile blood flow on the vascular architecture of decellularized porcine kidneys. Sci. Rep. 2021, 11, 16965. [Google Scholar] [CrossRef] [PubMed]
- Corridon, P.R.; Ko, I.K.; Yoo, J.J.; Atala, A. Bioartificial kidneys. Curr. Stem Cell. Rep. 2017, 3, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Travascio, F.; Gu, W.Y. Anisotropic diffusive transport in annulus fibrosus: Experimental determination of the diffusion tensor by FRAP technique. Ann. Biomed. Eng. 2007, 35, 1739–1748. [Google Scholar] [CrossRef] [Green Version]
- Syková, E.; Nicholson, C. Diffusion in brain extracellular space. Physiol. Rev. 2008, 88, 1277–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.; Chen, X.; Hepfer, R.G.; Damon, B.J.; Shi, C.; Yao, J.J.; Coombs, M.C.; Kern, M.J.; Ye, T.; Yao, H. A noninvasive fluorescence imaging-based platform measures 3D anisotropic extracellular diffusion. Nat. Commun. 2021, 12, 1913. [Google Scholar] [CrossRef]
- Yang, N.J.; Hinner, M.J. Getting across the cell membrane: An overview for small molecules, peptides, and proteins. Methods Mol. Biol. 2015, 1266, 29–53. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.L.; Rauh, F.; Morales, A. Particle diffusion as a function of concentration and ionic strength. J. Phys. Chem. 1978, 82, 608–616. [Google Scholar] [CrossRef]
- Saitakis, M.; Tsortos, A.; Gizeli, E. Probing the interaction of a membrane receptor with a surface-attached ligand using whole cells on acoustic biosensors. Biosens. Bioelectron. 2010, 25, 1688–1693. [Google Scholar] [CrossRef]
- Thompson, C.J.; Su, Z.; Vu, V.H.; Wu, Y.; Leckband, D.E.; Schwartz, D.K. Cadherin clusters stabilized by a combination of specific and nonspecific cis-interactions. eLife 2020, 9, e59035. [Google Scholar] [CrossRef]
- Brinkmann, J.; Cavatorta, E.; Sankaran, S.; Schmidt, B.; van Weerd, J.; Jonkheijm, P. About supramolecular systems for dynamically probing cells. Chem. Soc. Rev. 2014, 43, 4449–4469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saftics, A.; Kurunczi, S.; Peter, B.; Szekacs, I.; Ramsden, J.J.; Horvath, R. Data evaluation for surface-sensitive label-free methods to obtain real-time kinetic and structural information of thin films: A practical review with related software packages. Adv. Colloid Interface Sci. 2021, 294, 102431. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.R.; Knecht, M.R. Biointerface structural effects on the properties and applications of bioinspired peptide-based nanomaterials. Chem. Rev. 2017, 117, 12641–12704. [Google Scholar] [CrossRef] [PubMed]
- Tilton, R.D.; Robertson, C.R.; Gast, A.P. Lateral diffusion of bovine serum albumin adsorbed at the solid-liquid interface. J. Colloid Interface Sci. 1990, 137, 192–203. [Google Scholar] [CrossRef]
- Sun, M.; Deng, J.; Tang, Z.; Wu, J.; Li, D.; Chen, H.; Gao, C. A correlation study of protein adsorption and cell behaviors on substrates with different densities of PEG chains. Colloids Surf. B Biointerfaces 2014, 122, 134–142. [Google Scholar] [CrossRef]
- Saxton, M.J. Lateral diffusion in an archipelago: Effects of impermeable patches on diffusion in a cell membrane. Biophys. J. 1982, 39, 165–173. [Google Scholar] [CrossRef] [Green Version]
- Koop, J.; Merz, J.; Schembecker, G. Hydrophobicity, amphilicity, and flexibility: Relation between molecular protein properties and the macroscopic effects of surface activity. J. Biotechnol. 2021, 334, 11–25. [Google Scholar] [CrossRef]
- Sonesson, A.W.; Callisen, T.H.; Brismar, H.; Elofsson, U.M. Lipase surface diffusion studied by fluorescence recovery after photobleaching. Langmuir 2005, 21, 11949–11956. [Google Scholar] [CrossRef]
- Zimmerberg, J. Membrane biophysics. Curr. Biol. 2006, 16, R272–R276. [Google Scholar] [CrossRef] [Green Version]
- Reddy, A.S.; Warshaviak, D.T.; Chachisvilis, M. Effect of membrane tension on the physical properties of DOPC lipid bilayer membrane. Biochim. Biophys. Acta 2012, 1818, 2271–2281. [Google Scholar] [CrossRef] [Green Version]
- Ladha, S.; Mackie, A.R.; Harvey, L.J.; Clark, D.C.; Lea, E.J.A.; Brullemans, M.; Duclohier, H. Lateral diffusion in planar lipid bilayers: A fluorescence recovery after photobleaching investigation of its modulation by lipid composition, cholesterol, or alamethicin content and divalent cations. Biophys. J. 1996, 71, 1364–1373. [Google Scholar] [CrossRef] [Green Version]
- Esposito, C.; Tian, A.; Melamed, S.; Johnson, C.; Tee, S.-Y.; Baumgart, T. Flicker spectroscopy of thermal lipid bilayer domain boundary fluctuations. Biophys. J. 2007, 93, 3169–3181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kure, J.L.; Andersen, C.B.; Rasmussen, T.E.; Lagerholm, B.C.; Arnspang, E.C. Defining the diffusion in model membranes using line fluorescence recovery after photobleaching. Membranes 2020, 10, 434. [Google Scholar] [CrossRef] [PubMed]
- Schoenmaker, L.; Witzigmann, D.; Kulkarni, J.A.; Verbeke, R.; Kersten, G.; Jiskoot, W.; Crommelin, D.J.A. mRNA-lipid nanoparticle COVID-19 vaccines: Structure and stability. Int. J. Pharm. 2021, 601, 120586. [Google Scholar] [CrossRef]
- Gettel, D.L.; Sanborn, J.; Patel, M.A.; de Hoog, H.-P.; Liedberg, B.; Nallani, M.; Parikh, A.N.N. Mixing, Diffusion, and percolation in binary supported membranes containing mixtures of lipids and amphiphilic block copolymers. J. Am. Chem. Soc. 2014, 136, 10186–10189. [Google Scholar] [CrossRef]
- Zhang, H.-Y.; Hill, R.J. Concentration dependence of lipopolymer self-diffusion in supported bilayer membranes. J. R. Soc. Interface 2011, 8, 127–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussell, S.J.; Koch, D.L.; Hammer, D.A. Effect of hydrodynamic interactions on the diffusion of integral membrane-proteins—tracer diffusion in organelle and reconstituted membranes. Biophys. J. 1995, 68, 1828–1835. [Google Scholar] [CrossRef] [Green Version]
- Le Roux, A.-L.; Quiroga, X.; Walani, N.; Arroyo, M.; Roca-Cusachs, P. The plasma membrane as a mechanochemical transducer. Philos. Trans. R. Soc. B 2019, 374, 20180221. [Google Scholar] [CrossRef]
- Scalettar, B.A.; Hearst, J.E.; Klein, M.P. FRAP and FCS studies of self-diffusion and mutual diffusion in entangled DNA solutions. Macromolecules 1989, 22, 4550–4559. [Google Scholar] [CrossRef]
- Krishnaswamy, S. The transition of prothrombin to thrombin. J. Thromb. Haemost. 2013, 11, 265–276. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Pearce, K.H.; Thompson, N.L. Translational diffusion of bovine prothrombin fragment 1 weakly bound to supported planar membranes: Measurement by total internal reflection with fluorescence pattern photobleaching recovery. Biophys. J. 1994, 67, 1754–1766. [Google Scholar] [CrossRef] [Green Version]
- Abarca-Cabrera, L.; Fraga-García, P.; Berensmeier, S. Bio-nano interactions: Binding proteins, polysaccharides, lipids and nucleic acids onto magnetic nanoparticles. Biomater. Res. 2021, 25, 12. [Google Scholar] [CrossRef] [PubMed]
- Chan, V.; Fortina, P.; McKenzie, S.E.; Graves, D.J. The effects of electrostatics and hydrophobicity on the adsorption and surface diffusion of DNA oligonucleotides. J. Colloid Interface Sci. 1998, 203, 197–207. [Google Scholar] [CrossRef]
- Mossalam, M.; Dixon, A.S.; Lim, C.S. Controlling subcellular delivery to optimize therapeutic effect. Ther. Deliv. 2010, 1, 169–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayel, M.J.; Hom, E.F.Y.; Verkman, A.S. Diffusion of green fluorescent protein in the aqueous-phase lumen of endoplasmic reticulum. Biophys. J. 1999, 76, 2843–2851. [Google Scholar] [CrossRef] [Green Version]
- Mitrea, D.M.; Kriwacki, R.W. Phase separation in biology; functional organization of a higher order. Cell Commun. Signal. 2016, 14, 1. [Google Scholar] [CrossRef] [Green Version]
- Partikian, A.; Ölveczky, B.; Swaminathan, R.; Li, Y.; Verkman, A.S. Rapid diffusion of green fluorescent protein in the mitochondrial matrix. J. Cell Biol. 1998, 140, 821–829. [Google Scholar] [CrossRef]
- Johnstone, C.P.; Wang, N.B.; Sevier, S.A.; Galloway, K.E. Understanding and engineering chromatin as a dynamical system across length and timescales. Cell Syst. 2020, 11, 424–448. [Google Scholar] [CrossRef]
- Sprague, B.L.; Pego, R.L.; Stavreva, D.A.; McNally, J.G. Analysis of binding reactions by fluorescence recovery after photobleaching. Biophys. J. 2004, 86, 3473–3495. [Google Scholar] [CrossRef] [Green Version]
- Ben-Yishay, R.; Shav-Tal, Y. The dynamic lifecycle of mRNA in the nucleus. Curr. Opin. Cell Biol. 2019, 58, 69–75. [Google Scholar] [CrossRef]
- Kim, S.H.; Vieira, M.; Shim, J.Y.; Choi, H.; Hye Yoon Park, H.Y. Recent progress in single-molecule studies of mRNA localization in vivo. RNA Biol. 2019, 16, 1108–1118. [Google Scholar] [CrossRef] [PubMed]
- Braga, J.; McNally, J.G.; Carmo-Fonseca, M. A reaction-diffusion model to study RNA motion by quantitative fluorescence recovery after photobleaching. Biophys. J. 2007, 92, 2694–2703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makoto Nagano, M.; Toshima, J.Y.; Siekhaus, D.E.; Toshima, J. Rab5-mediated endosome formation is regulated at the trans-Golgi network. Commun. Biol. 2019, 2, 419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.-S.; Lee, S.Y.; Gao, M.; Bourgoin, S.; Randazzo, P.A.; Premont, R.T.; Hsu, V.W. ARFGAP1 promotes the formation of COPI vesicles, suggesting function as a component of the coat. J. Cell Biol. 2002, 159, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Gondré-Lewis, M.C.; Arnaoutova, I.; Loh, Y.P. Dense-core secretory granule biogenesis. Physiology 2006, 21, 124–133. [Google Scholar] [CrossRef] [Green Version]
- Weiss, A.N.; Bittner, M.A.; Holz, R.W.; Axelrod, D. Protein mobility within secretory granules. Biophys. J. 2014, 107, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Weiss, A.N.; Anantharam, A.; Bittner, M.A.; Axelrod, D.; Holz, R.W. Lumenal protein within secretory granules affects fusion pore expansion. Biophys. J. 2014, 107, 26–33. [Google Scholar] [CrossRef] [Green Version]
- Vinothkumar, K.R.; Henderson, R. Structures of membrane proteins. Q. Rev. Biophys. 2010, 43, 65–158. [Google Scholar] [CrossRef] [Green Version]
- Premkumar, L.S. Transient receptor potential channels as targets for phytochemicals. ACS Chem. Neurosci. 2014, 5, 1117–1130. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, D.; Segal, A.; Voets, T. Distinct modes of perimembrane TRP channel turnover revealed by TIR-FRAP. Sci. Rep. 2014, 4, 7111. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Types of Biophysical Techniques | Measurable Processes | Limitations | References |
|---|---|---|---|
| Fluorescence Recovery After Photobleaching | Convection | Require sophisticated models; | [13] |
| Diffusion | require high-powered lasers; | [45] | |
| Reaction/Binding | only valid for large ROI * | [60] | |
| Fluorescence Correlation Spectroscopy | Diffusion | Lack of interpreting models; | [56] |
| Reaction/Binding Concentration | difficult to apply in live cells; require high S/N * ratio | [61] [62] | |
| Single Particle Tracking | Diffusion | Only for dilute species; | [22] |
| Viscosity | measure lower mobility; | [55] | |
| Molecular Binding | requires feedback tracking | [63] | |
| Surface Plasmon Resonance Sensor | Reaction/Binding | Require gold substrate; lack of | [52] |
| Mass transfer | interpreting models; noise from used optoelectronics | [64] [65] | |
| Stochastic Optical Reconstruction | Diffusion | High costs; require photo- | [66] |
| Molecular Binding | switchable dyes as labels; require experts in operation | [67] [68] | |
| Molecular Interaction | Complicated mathematical | [57] | |
| FCCS # | Composition/Fraction Large Complex | modeling; supplicated instrumentation; high cost | [69] [70] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, N.; Lai, A.C.-K.; Liao, K.; Corridon, P.R.; Graves, D.J.; Chan, V. Recent Advances in Fluorescence Recovery after Photobleaching for Decoupling Transport and Kinetics of Biomacromolecules in Cellular Physiology. Polymers 2022, 14, 1913. https://0-doi-org.brum.beds.ac.uk/10.3390/polym14091913
Cai N, Lai AC-K, Liao K, Corridon PR, Graves DJ, Chan V. Recent Advances in Fluorescence Recovery after Photobleaching for Decoupling Transport and Kinetics of Biomacromolecules in Cellular Physiology. Polymers. 2022; 14(9):1913. https://0-doi-org.brum.beds.ac.uk/10.3390/polym14091913
Chicago/Turabian StyleCai, Ning, Alvin Chi-Keung Lai, Kin Liao, Peter R. Corridon, David J. Graves, and Vincent Chan. 2022. "Recent Advances in Fluorescence Recovery after Photobleaching for Decoupling Transport and Kinetics of Biomacromolecules in Cellular Physiology" Polymers 14, no. 9: 1913. https://0-doi-org.brum.beds.ac.uk/10.3390/polym14091913