

Polyethyleneglycol-Betulinic Acid (PEG-BA) Polymer-Drug Conjugate Induces Apoptosis and Antioxidation in a Biological Model of Pancreatic Cancer

Abstract
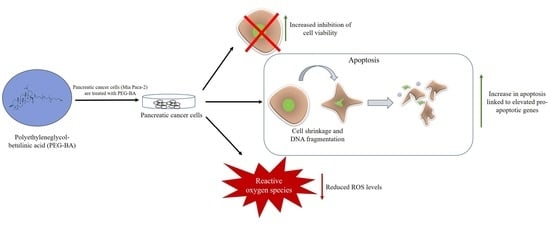
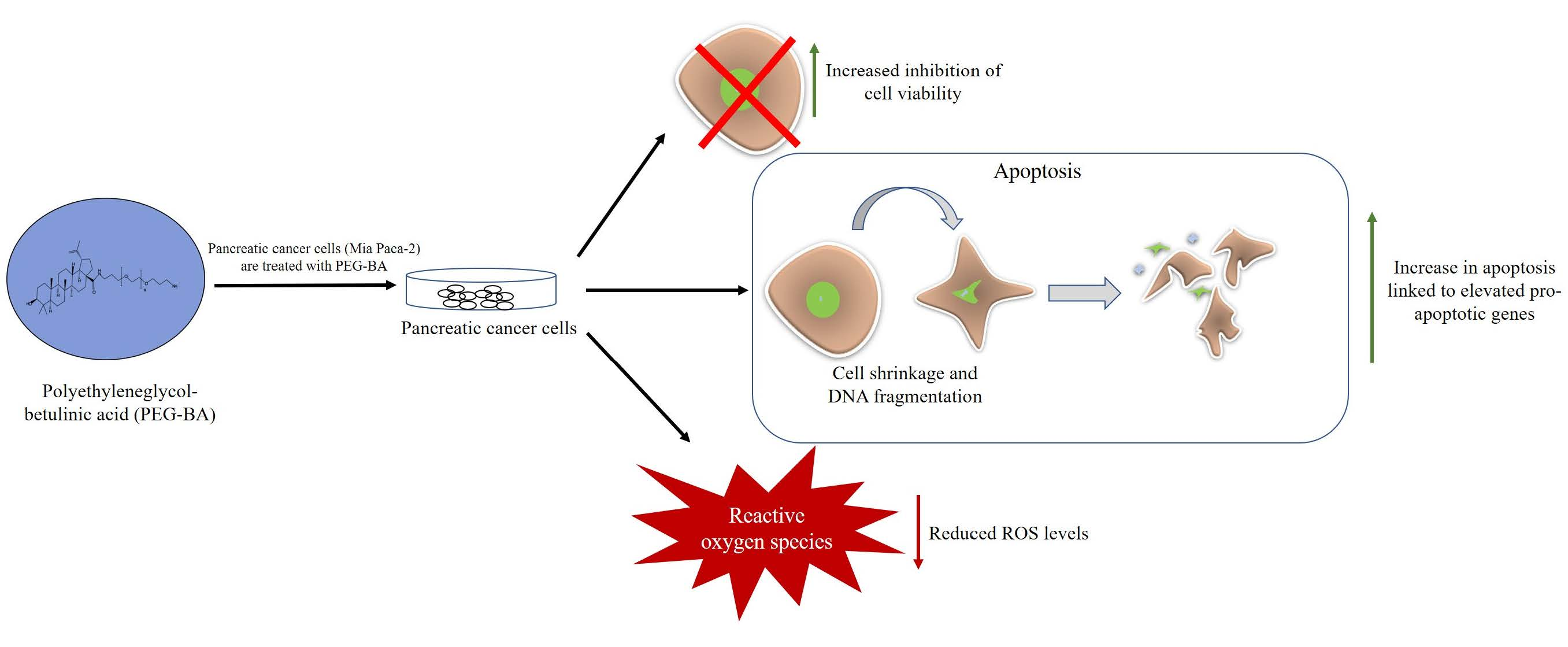
:
1. Introduction
2. Materials and Methods
2.1. Compound Preparation
2.2. Cell Culture Models and Maintenance
2.3. Cell Treatment
2.4. Microscopic Analysis
2.5. Cytotoxicity Analysis
2.6. Cell Cycle Status Determination with Propidium Iodide Dye
2.7. Gene Expression Analysis of Apoptotic Genes
2.7.1. Extraction of Total RNA
2.7.2. Complementary DNA (cDNA) Synthesis
2.7.3. Quantitative Real-Time Polymerase Chain Reaction Analysis
2.8. NF-κB/p65 Transcription Factor Assay
2.9. ROS Reduction and Potential Antioxidant Analysis
2.9.1. DPPH Assay
2.9.2. DEPPD Assay
2.10. Statistical Analysis
3. Results
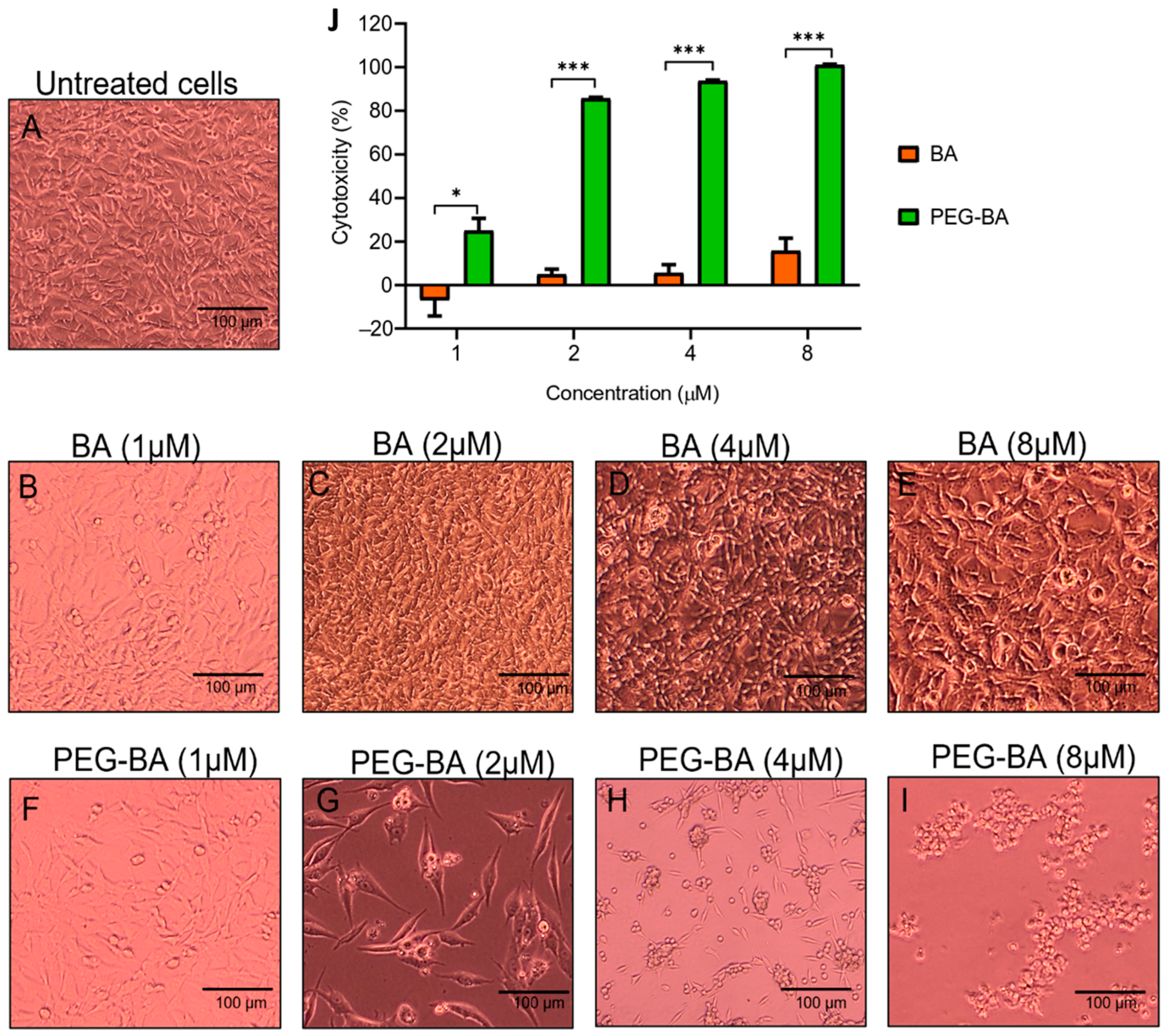
3.1. PEG-BA Causes Cell Rounding in a Dose and Time-Dependent Manner
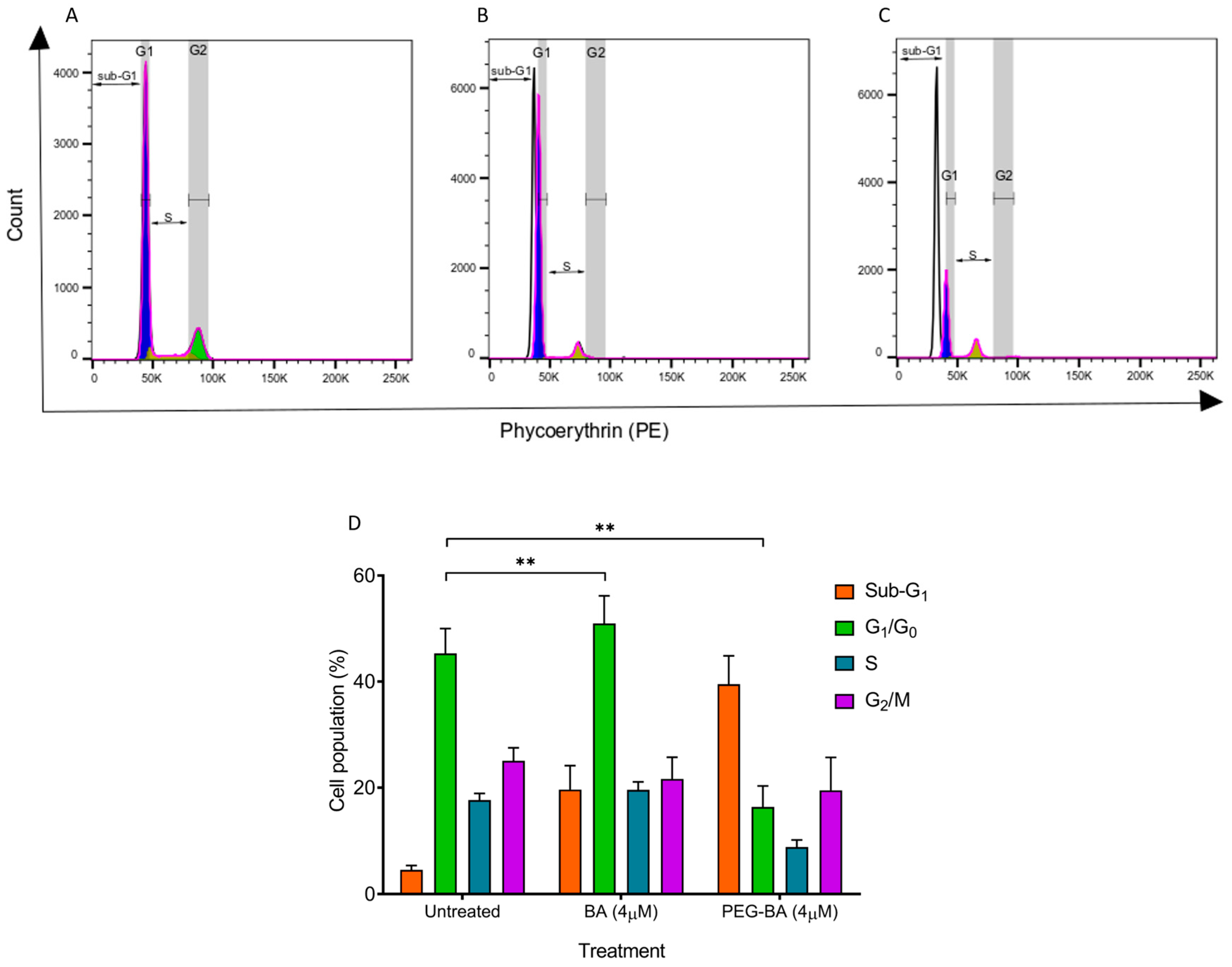
3.2. PEG-BA Induces a Sub-G1 Arrest in Pancreatic Cancer Cells
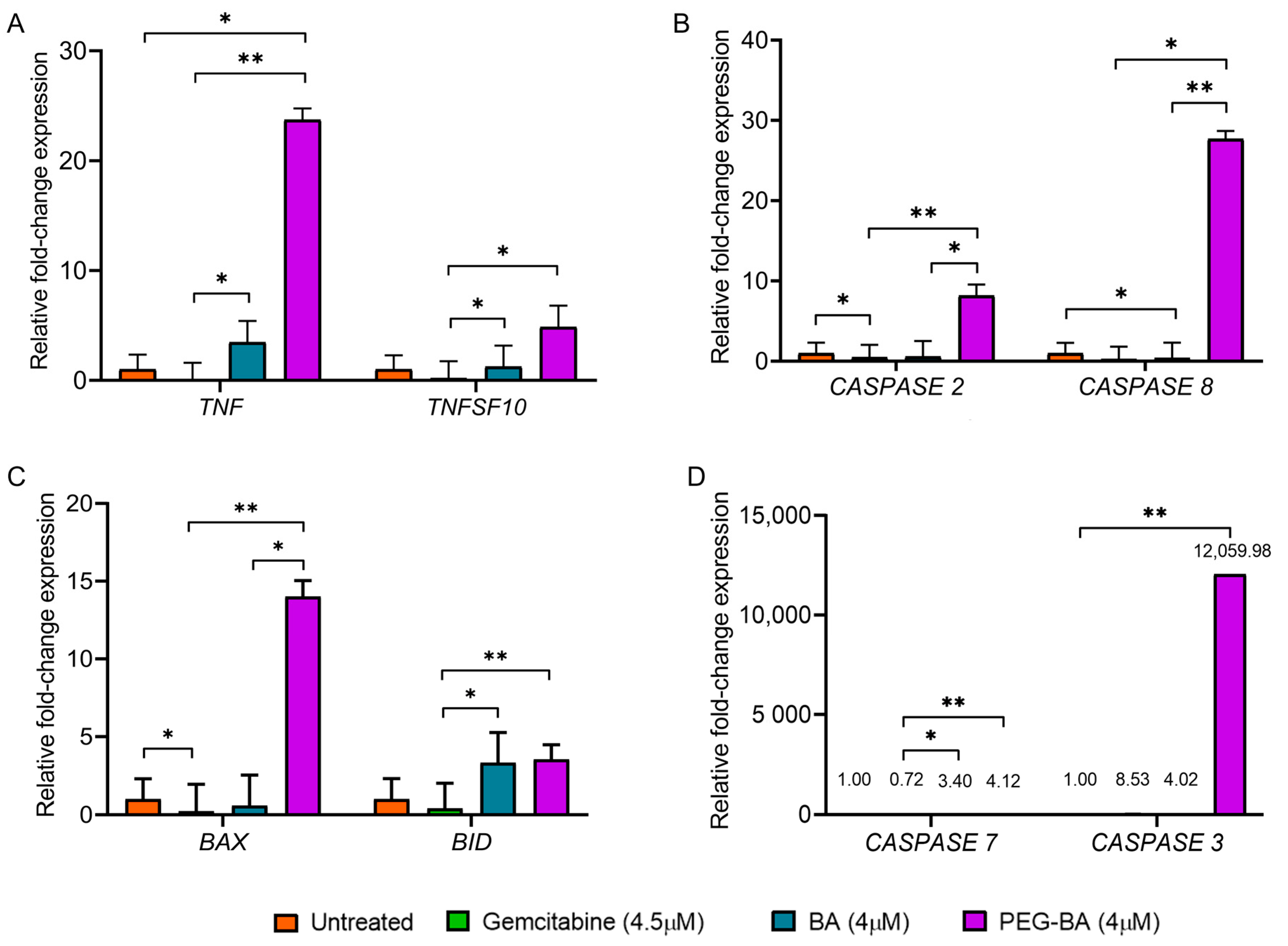
3.3. PEG-BA Elevates the Expression of Proapoptotic Genes
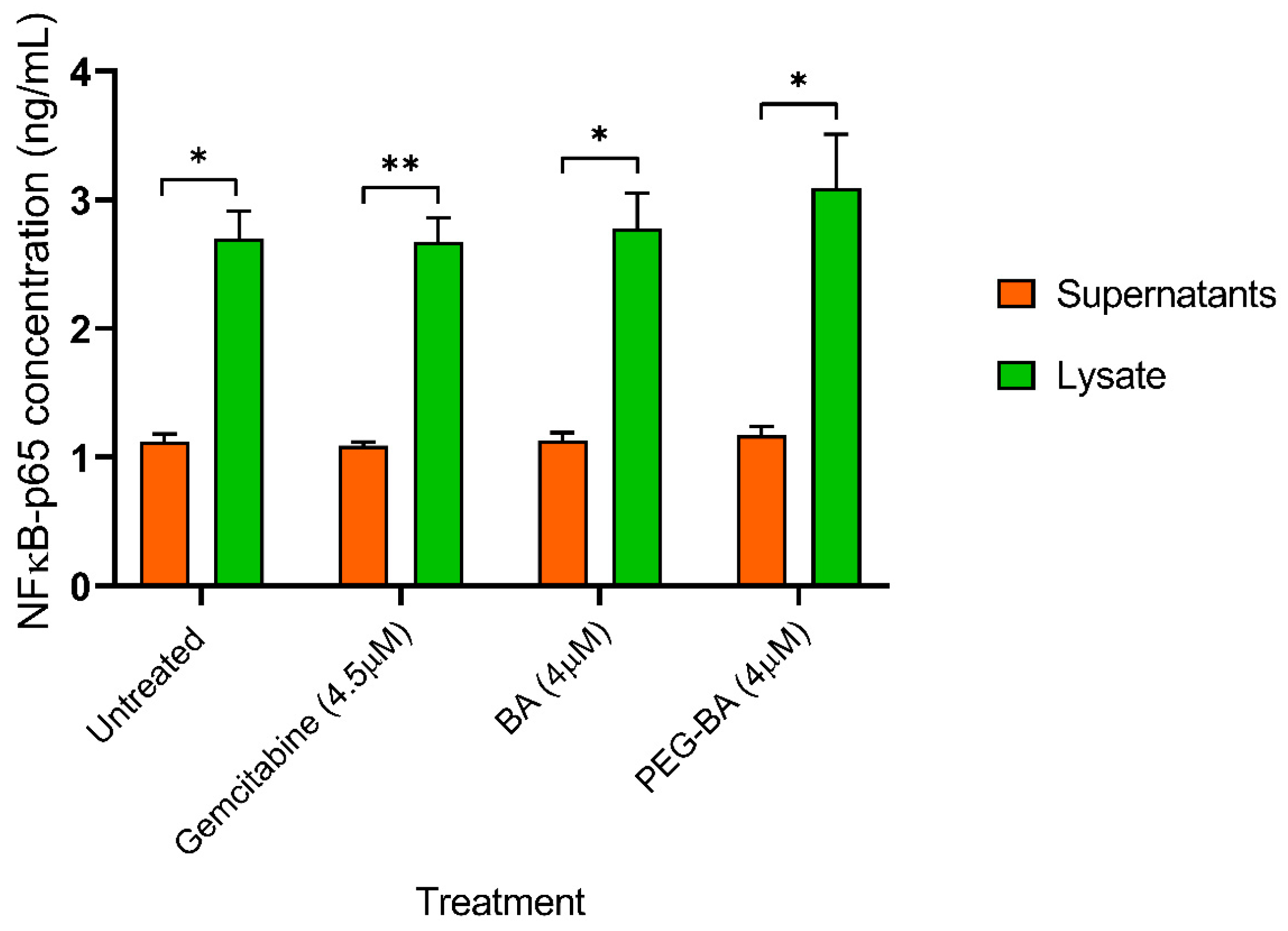
3.4. PEG-BA Treatment May Result in NF-κB Expression in Pancreatic Cancer Cells
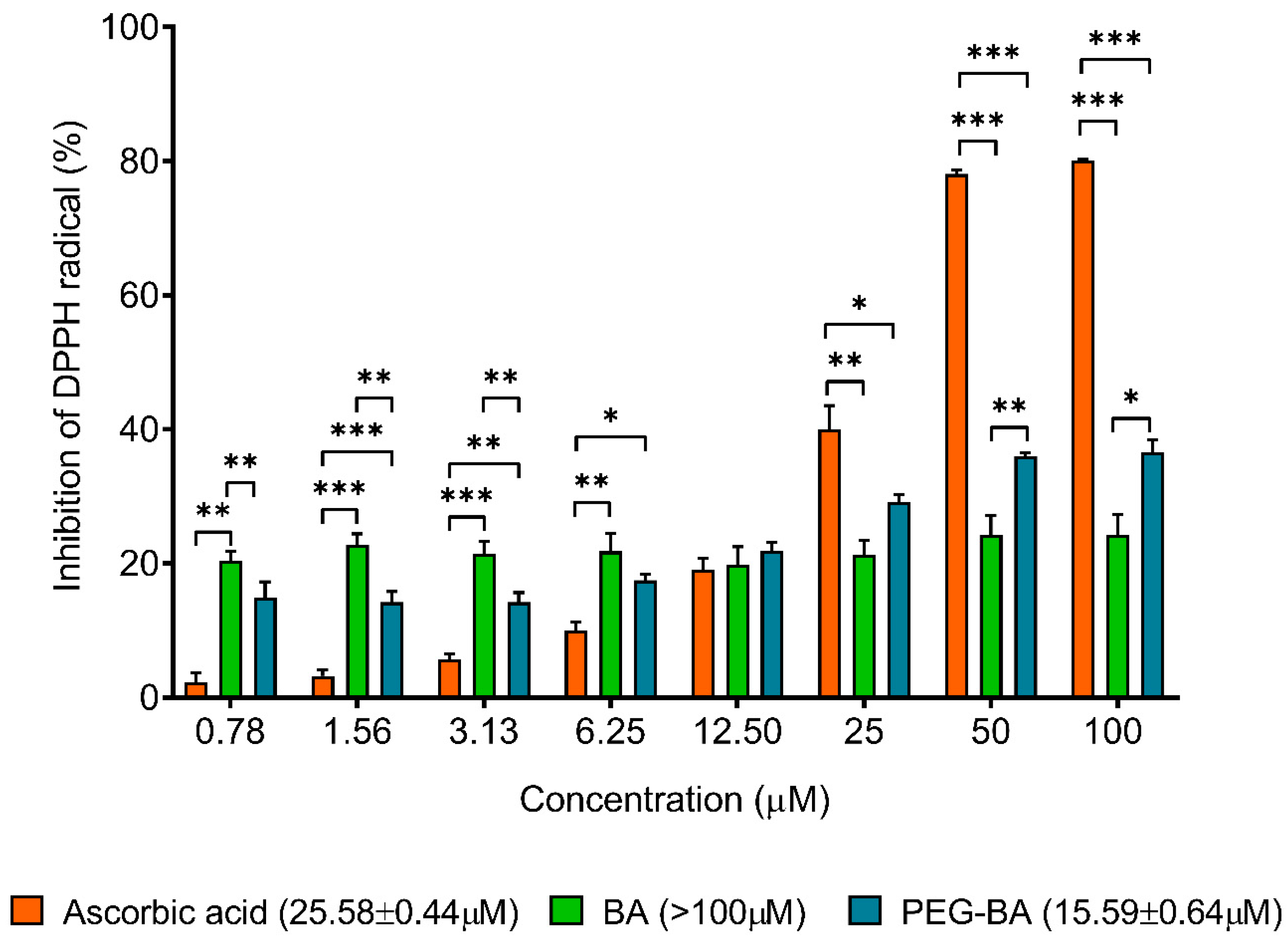
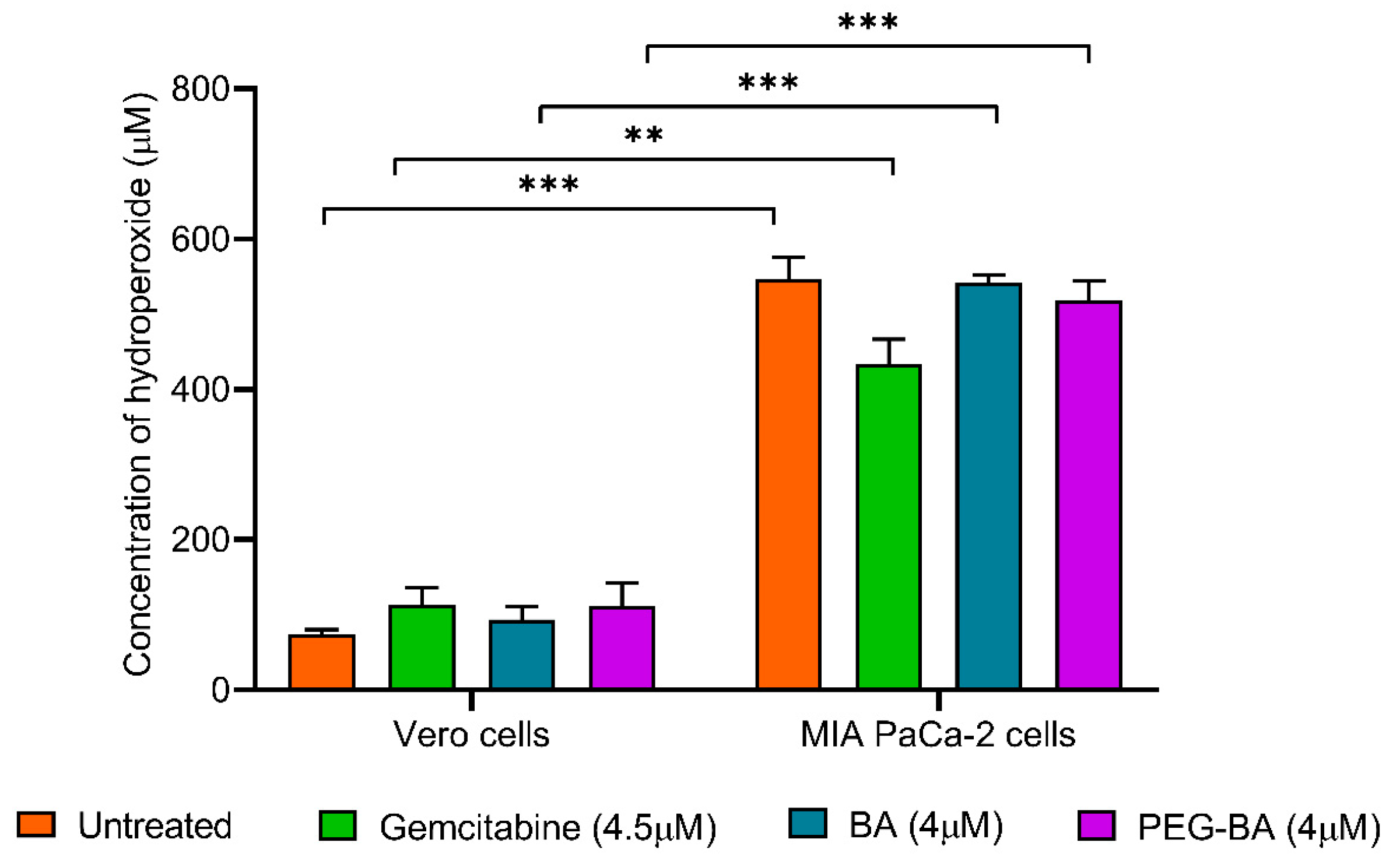
3.5. PEG-BA Treatment Induces Antioxidant Activities in Pancreatic Cancer Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Maisonneuve, P. Epidemiology and burden of pancreatic cancer. Presse Médicale 2019, 48, e113–e123. [Google Scholar] [CrossRef]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primer. 2016, 2, 16022. [Google Scholar] [CrossRef]
- Kim, A.; Ha, J.; Kim, J.; Cho, Y.; Ahn, J.; Cheon, C.; Kim, S.; Ko, S.; Kim, B. Natural products for pancreatic cancer treatment: From traditional medicine to modern drug discovery. Nutrients. 2021, 13, 3801. [Google Scholar] [CrossRef]
- Yogeeswari, P.; Sriram, D. Betulinic acid and its derivatives: A review on their biological properties. Curr. Med. Chem. 2005, 12, 657–666. [Google Scholar] [CrossRef]
- Fulda, S. Betulinic acid for cancer treatment and prevention. Int. J. Mol. Sci. 2008, 9, 1096–1107. [Google Scholar] [CrossRef] [Green Version]
- Ali-Seyed, M.; Jantan, I.; Vijayaraghavan, K.; Bukhari, S.N.A. Betulinic acid: Recent advances in chemical modifications, effective delivery, and molecular mechanisms of a promising anticancer therapy. Chem. Biol. Drug. Des. 2016, 87, 517–536. [Google Scholar] [CrossRef]
- Fru, P.N.; Nweke, E.E.; Mthimkhulu, N.; Mvango, S.; Nel, M.; Pilcher, L.A.; Balogun, M. Anti-cancer and immunomodulatory activity of a polyethylene glycol-betulinic acid conjugate on pancreatic cancer cells. Life 2021, 11, 462. [Google Scholar] [CrossRef] [PubMed]
- Mthimkhulu, N.; Mosiane, K.S.; Nweke, E.E.; Balogun, M.; Fru, P.N. Prospects of delivering natural compounds by polymer-drug conjugates in cancer therapeutics. Anticancer. Agents Med. Chem. 2021, 22, 1699–1713. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, B.; Kulkarni, N.S.; Shukla, S.K.; Parvathaneni, V.; Chauhan, G.; Damon, J.K.; Sarode, A.; Garcia, J.V.; Kunda, N.; Mitragotri, S.; et al. Development of inhalable quinacrine loaded bovine serum albumin modified cationic nanoparticles: Repurposing quinacrine for lung cancer therapeutics. Int. J. Pharm. 2020, 557, 118995. [Google Scholar] [CrossRef] [PubMed]
- Alven, S.; Nqoro, X.; Buyana, B.; Aderibigbe, B.A. Polymer-drug conjugate, a potential therapeutic to combat breast and lung cancer. Pharmaceutics 2020, 12, 406. [Google Scholar] [CrossRef]
- Ekladious, I.; Colson, Y.L.; Grinstaff, M.W. Polymer-drug conjugate therapeutics: Advances, insights and prospects. Nat. Rev. Drug Discov. 2018, 18, 273–294. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Kang, J.; Fu, C. The independence of and associations among apoptosis, autophagy, and necrosis. Signal Transduct. Target Ther. 2018, 3, 18. [Google Scholar] [CrossRef] [Green Version]
- Samm, N.; Werner, K.; Rückert, F.; Saeger, H.D.; Grützmann, R.; Pilarsky, C. The role of apoptosis in the pathology of pancreatic cancer. Cancers 2010, 3, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Lafont, E.; Hartwig, T.; Walczak, H. Paving TRAIL’s path with Ubiquitin. Trends Biochem. Sci. 2018, 43, 44–60. [Google Scholar] [CrossRef] [PubMed]
- Szegezdi, E.; Leverkus, M. Guiding the killer and bringing in accomplices: Bispecific antibody treatment for malignant melanoma. J. Investig. Dermatol. 2016, 136, 362–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.H.; Zhou, H.J.; Wang, W.Q.; Wu, G.D. Antimalarial dihydroartemisinin also inhibits angiogenesis. Cancer Chemother. Pharmacol. 2004, 53, 423–432. [Google Scholar] [CrossRef]
- Zhang, E.; Lu, X.; Yin, S.; Yan, M.; Lu, S.; Fan, L.; Hu, H. The functional role of Bax/Bak in palmitate-induced lipoapoptosis. Food Chem. Toxicol. 2019, 123, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of cancer: New dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.S. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef]
- Hong-Duck, U.M. Bcl-2 family proteins as regulators of cancer cell invasion and metastasis: A review focusing on mitochondrial respiration and reactive oxygen species. Oncotarget 2015, 7, 5193–5203. [Google Scholar]
- Evans, J.D.; Cornford, P.A.; Dodson, A.; Greenhalf, W.; Foster, C.S.; Neoptolemos, J.P. Detailed tissue expression of bcl-2, bax, bak and bcl-x in the normal human pancreas and in chronic pancreatitis, ampullary and pancreatic ductal adenocarcinomas. Pancreatology 2001, 1, 254–262. [Google Scholar] [CrossRef]
- Virkajärvi, N.; Pääkkö, P.; Soini, Y. Apoptotic index and apoptosis influencing proteins bcl-2, mcl-1, bax and caspases 3, 6 and 8 in pancreatic carcinoma: Apoptosis influencing proteins in pancreatic carcinoma. Histopathology 1998, 33, 432–439. [Google Scholar] [CrossRef]
- Campani, D.; Esposito, I.; Boggi, U.; Cecchetti, D.; Menicagli, M.; De Negri, F.; Colizzi, L.; Del Chario, M.; Mosca, F.; Fornaciari, G.; et al. Bcl-2 expression in pancreas development and pancreatic cancer progression. J. Pathol. 2001, 194, 444–450. [Google Scholar] [CrossRef]
- Rajabi, S.; Maresca, M.; Yumashev, A.V.; Choopani, R.; Hajimehdipoor, H. The most competent plant-derived natural products for targeting apoptosis in cancer therapy. Biomolecules 2021, 11, 534. [Google Scholar] [CrossRef]
- Riley, J.S.; Hutchinson, R.; McArt, D.G.; Crawford, N.; Holohan, C.; Paul, I.; Van Schaeybroeck, S.; Salto-Tellez, M.; Johnston, P.G.; Fennell, D.A.; et al. Prognostic and therapeutic relevance of FLIP and procaspase-8 overexpression in non-small cell lung cancer. Cell Death Dis. 2013, 4, e951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenev, T.; Marani, M.; McNeish, I.; Lemoine, N.R. Pro-caspase-3 overexpression sensitises ovarian cancer cells to proteasome inhibitors. Cell Death Differ. 2001, 8, 256–264. [Google Scholar] [CrossRef] [Green Version]
- Jakubowska, K.; Guzińska-Ustymowicz, K.; Famulski, W.; Cepowicz, D.; Jagodzińska, D.; Pryczynicz, A. Reduced expression of caspase-8 and cleaved caspase-3 in pancreatic ductal adenocarcinoma cells. Oncol. Lett. 2016, 11, 1879–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, R.; Dua, J.S.; Prasad, D.N.; Hira, S.; Monika. Advancement in Novel Drug Delivery System: Niosomes. JDDT 2019, 9, 995–1001. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and cancer: Triggers, mechanisms, and consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Milkovic, L.; Cipak Gasparovic, A.; Cindric, M.; Mouthuy, P.A.; Zarkovic, N. Short overview of ROS as cell function regulators and their implications in therapy concepts. Cells 2019, 8, 793. [Google Scholar] [CrossRef]
- Liu, Z.; Ren, Z.; Zhang, J.; Chuang, C.C.; Kandaswamy, E.; Zhou, T.; Zuo, L. Role of ROS and nutritional antioxidants in human diseases. Front. Physiol. 2018, 9, 477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS generation and antioxidant defence systems in normal and malignant cells. Oxid. Med. Cell. Longev. 2019, 2019, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adhami, V.M. Pancreatic cancer and inflammation. Am. J. Biomed. Sci. 2010, 2, 322–328. [Google Scholar] [CrossRef]
- Algül, H.; Treiber, M.; Lesina, M.; Schmid, R.M. Mechanisms of disease: Chronic inflammation and cancer in the pancreas—A potential role for pancreatic stellate cells? Nat. Clin. Pract. Gastroenterol. Hepatol. 2007, 4, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer. 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKay, C.J.; Glen, P.; McMillan, D.C. Chronic inflammation and pancreatic cancer. Best Pract. Res. Clin. Gastroenterol. 2008, 22, 65–73. [Google Scholar] [CrossRef]
- Tak, P.P.; Firestein, G.S. NF-κB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef]
- Mvango, S.; Mthimkhulu, N.; Fru, P.N.; Pilcher, L.A.; Balogun, M.O. Physico-Chemical Characterisation of Polyethylene Glycol-conjugated Betulinic Acid; AIP Publishing: Mallorca, Spain, 2020; p. 020039. Available online: http://aip.scitation.org/doi/abs/10.1063/5.0028479 (accessed on 3 September 2022).
- Deer, E.L.; González-Hernández, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and genotype of pancreatic cancer cell lines. Pancreas 2010, 39, 425–435. [Google Scholar] [CrossRef] [Green Version]
- Strober, W. Trypan blue exclusion test of cell viability. Curr. Protoc. Immunol. 2019, 111, A3.B.1–A3.B.3. [Google Scholar]
- Aykul, S.; Martinez-Hackert, E. Determination of half-maximal inhibitory concentrations using biosensor-based protein interaction analysis. Anal. Biochem. 2016, 508, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Zhang, Q.; Zeng, L.; Chen, Y.; Lian, G.; Qian, C.; Chen, S.; Li, J.; Huang, K. Pancreatic cancer epidemiology, detection, and management. Gastroenterol. Res. Pract. 2016, 2016, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mistry, S.; Shah, S. Antioxidant activity of methanolic extract of Ficus religiosa linn bark by using DPPH ((1, 1-Diphenyl-2-Picrylhydrazyl). Int. J. Phytopharm. 2014, 4, 57–58. [Google Scholar]
- Williams, A.A. Metabonomics Profile and Corresponding Immune Parameters of HIV Infected Individuals. Ph.D. Thesis, University of Pretoria, Pretoria, South Africa, 2012. [Google Scholar]
- Wegert, J.; Bausenwein, S.; Kneitz, S.; Roth, S.; Graf, N.; Geissinger, E.; Gessler, M. Retinoic acid pathway activity in wilms tumors and characterization of biological responses in Vitro. Mol. Cancer 2011, 10, 136–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, J.V.; Chambers, S.H.; Smith, P.J. A pragmatic approach to the analysis of DNA histograms with a definable G1 peak. Cytometry 1987, 8, 1–8. [Google Scholar] [CrossRef]
- Sasaki, N.; Gomi, F.; Hasegawa, F.; Hirano, K.; Fujiwara, M.; Toyoda, M.; Ishiwata, T. Characterization of the metastatic potential of the floating cell component of MIA PaCa-2, a human pancreatic cancer cell line. Biochem. Biophys. Res. Commun. 2020, 522, 881–888. [Google Scholar] [CrossRef]
- Margo, A.; Magro, A.; Shrestha, S.; Brundage, K.; Rankin, G. Metalloproteinase dependent reduction of cell surface cluster determinants upon the induction of apoptosis. Int. J. Oncol. 2014, 44, 1539–1550. [Google Scholar]
- Anaya-Eugenio, G.D.; Eggers, N.A.; Ren, Y.; Rivera-Chavez, J.; Kinghorn, A.D.; De Blanco, E.J.C. Apoptosis induced by (+)-Betulin through NF-ĸb inhibition in MDA-MB-231 breast cancer cells. Anticancer Res. 2020, 40, 6637–6647. [Google Scholar] [CrossRef]
- Vorobjev, I.; Barteneva, N.S. Temporal heterogeneity metrics in apoptosis induced by anticancer drugs. J. Histochem. Cytochem. 2015, 63, 494–510. [Google Scholar] [CrossRef] [Green Version]
- Coricovac, D.; Dehelean, C.A.; Pinzaru, I.; Mioc, A.; Aburel, O.; Macasoi, I.; Draghici, G.A.; Petean, C.; Soica, C.; Boruga, M.; et al. Assessment of betulinic acid cytotoxicity and mitochondrial metabolism impairment in a human melanoma cell line. Int. J. Mol. Sci. 2021, 22, 4870. [Google Scholar] [CrossRef]
- Xu, T.; Pang, Q.; Zhou, D.; Zhang, A.; Luo, S.; Wang, Y.; Yan, X. Proteomic investigation into betulinic acid-induced apoptosis of human cervical cancer HeLa cells. PLoS ONE 2014, 9, e105768. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.M.; Chiu, C.C.; Wu, P.F.; Chen, C.Y. Subamolide E from Cinnamomum subavenium induces sub-G1 cell-cycle arrest and caspase-dependent apoptosis and reduces the migration ability of human melanoma cells. J. Agric. Food Chem. 2011, 59, 8187–8192. [Google Scholar] [CrossRef] [PubMed]
- Crowley, L.C.; Scott, A.P.; Marfell, B.J.; Boughaba, J.A.; Chojnowski, G.; Waterhouse, N.J. Measuring cell death by propidium iodide uptake and flow cytometry. Cold Spring Harb. Protoc. 2016, 2016, pdb.prot087163. [Google Scholar] [CrossRef] [PubMed]
- Ehrhardt, H.; Fulda, S.; Führer, M.; Debatin, K.M.; Jeremias, I. Betulinic acid-induced apoptosis in leukemia cells. Leukemia 2004, 18, 1406–1412. [Google Scholar] [CrossRef]
- Fulda, S.; Scaffidi, C.; Susin, S.A.; Krammer, P.H.; Kroemer, G.; Peter, M.E.; Debatin, K. Activation of mitochondria and release of mitochondrial apoptogenic factors by betulinic acid. J. Biol. Chem. 1998, 273, 33942–33948. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.L.; Kuzmanoff, K.L.; Ling-Indeck, L.; Pezzuto, J.M. Betulinic acid induces apoptosis in human neuroblastoma cell lines. Eur. J. Cancer 1997, 33, 2007–2010. [Google Scholar] [CrossRef]
- Wang, D.; Xu, M.; Li, F.; Gao, Y.; Sun, H. Target identification-based analysis of mechanism of betulinic acid-induced cell apoptosis of cervical cancer SiHa. Nat. Prod. Commun. 2022, 17, 1934578X2211155. [Google Scholar] [CrossRef]
- Dutta, D.; Sarkar, A.; Chakraborty, B.; Chowdhury, C.; Das, P. Induction of apoptosis by a potent betulinic acid derivative in human colon carcinoma HT-29 cells. Int. J. Sci. Res. Publ. 2015, 5, 7. [Google Scholar]
- Shen, M.; Hu, Y.; Yang, Y.; Wang, L.; Yang, X.; Wang, B.; Huang, M. Betulinic acid induces ROS-dependent apoptosis and S-phase arrest by inhibiting the NF- κ B pathway in human multiple myeloma. Oxid. Med. Cell. Longev. 2019, 2019, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saneja, A.; Kuma, R.; Singh, A.; Dhar Dubey, R.; Mintoo, M.J.; Singh, G.; Mondhe, D.M.; Panda, A.M.; Gupta, P.N. Development and evaluation of long-circulating nanoparticles loaded with betulinic acid for improved anti-tumor efficacy. Int. J. Pharm. 2017, 531, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Zuco, V.; Supino, R.; Righetti, S.C.; Cleris, L.; Marchesi, E.; Gambacorti-Passerini, C.; Formelli, F. Selective cytotoxicity of betulinic acid on tumor cell lines, but not on normal cells. Cancer Lett. 2002, 175, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Krishnan, J. mRNA translation and protein synthesis: An analysis of different modelling methodologies and a new PBN based approach. BMC Syst. Biol. 2014, 8, 1–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2013, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Nachmias, B.; Ashhab, Y.; Ben-Yehuda, D. The inhibitor of apoptosis protein family (IAPs): An emerging therapeutic target in cancer. Semin. Cancer Biol. 2004, 14, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Zhao, W.; Tong, P.; Li, P.; Zhao, Y.; Li, H.; Liang, J. Comprehensive molecular characterisation of inhibitors of apoptosis proteins (IAPs) for therapeutic targeting in cancer. BMC Med. Genom. 2020, 13, 7. [Google Scholar] [CrossRef]
- Desagher, S.; Osen-Sand, A.; Nichols, A.; Eskes, R.; Montessuit, S.; Lauper, S.; Maundrell, K.; Antonsson, B.; Martinou, J. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J. Cell Biol. 1999, 144, 891–901. [Google Scholar] [CrossRef]
- Gu, Q.; Wang, J.D.; Xia, H.H.X.; Lin, M.C.M.; He, H.; Zou, B.; Tu, S.P.; Yang, Y.; Liu, X.G.; Lam, S.K.; et al. Activation of the caspase-8/Bid and Bax pathways in aspirin-induced apoptosis in gastric cancer. Carcinogenesis 2005, 26, 541–546. [Google Scholar] [CrossRef] [Green Version]
- Korsmeyer, S.J.; Wei, M.C.; Saito, M.; Weiler, S.; Oh, K.J.; Schlesinger, P.H. Proapoptotic cascade activates BID, which oligomerises BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 2000, 7, 1166–1173. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Fisher, J.C.; Dillon, C.P.; Kriwacki, R.W.; Kuwana, T.; Green, D.R. Mechanism of apoptosis induction by inhibition of the antiapoptotic BCL-2 proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 20327–20332. [Google Scholar] [CrossRef] [Green Version]
- Naseri, M.H.; Mahdavi, M.; Davoodi, J.; Tackallou, S.H.; Goudarzvand, M.; Neishabouri, S.H. Upregulation of Bax and downregulation of Bcl2 during 3-NC mediated apoptosis in human cancer cells. Cancer Cell Int. 2015, 15, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Y.; Shen, S.; Verma, I.M. NF-κB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [Green Version]
- Xia, L.; Tan, S.; Zhou, Y.; Lin, J.; Wang, H.; Oyang, L.; Tian, Y.; Liu, L.; Su, M.; Wang, H.; et al. Role of the NFκB-signaling pathway in cancer. OncoTargets Ther. 2018, 11, 2063–2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinatizadeh, M.R.; Schock, B.; Chalbatani, G.M.; Zarandi, P.K.; Jalali, S.A.; Miri, S.R. The nuclear factor Kappa B (NF-kB) signaling in cancer development and immune diseases. Genes Dis. 2021, 8, 287–297. [Google Scholar] [CrossRef]
- Lin, B.; Williams-Skipp, C.; Tao, Y.; Schleicher, M.S.; Cano, L.L.; Duke, R.C.; Schienman, R.L. NF-kB functions as both a proapoptotic and antiapoptotic regulatory factor within a single cell type. Cell Death Differ. 1999, 6, 570–582. [Google Scholar] [CrossRef]
- Ryan, K.; Ernst, M.; Rice, N.; Vousden, K. Role of NF-kB in p53-mediated programmed cell death. Nature 2000, 404, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, N.; Simpson, J.; Taylor, G.; Rafique, S.; Whitehouse, A.; Hiscox, J.; Stark, L.A. Nucleolar NF-κB/RelA mediates apoptosis by causing cytoplasmic relocalisation of nucleophosmin. Cell Death Differ. 2011, 18, 1889–1903. [Google Scholar] [CrossRef] [Green Version]
- Adesanwo, J.K.; Makinde, O.O.; Obafemi, C.A. Phytochemical analysis and antioxidant activity of methanol extract and betulinic acid isolated from the roots of Tetracera potatoria. J. Pharm. Res. 2013, 6, 903–907. [Google Scholar] [CrossRef]
- Sati, P.; Pandey, A.; Rawat, S.; Rani, A. Phytochemicals and antioxidants in leaf extracts of Ginkgo biloba with reference to location, seasonal variation and solvent system. J. Pharm. Res. 2013, 7, 804–809. [Google Scholar] [CrossRef]
- Anggraini, T.; Wilma, S.; Syukri, D.; Azima, F. Total phenolic, anthocyanin, catechins, DPPH radical scavenging activity, and toxicity of Lepisanthes alata (Blume) Leenh. Int. J. Food Sci. 2019, 2019, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakabayashi, T.; Kawashima, T.; Matsuzawa, Y. Evaluation of reactive oxygen metabolites in patients with non-small cell lung cancer after chemotherapy. MRM 2014, 9, 44. Available online: http://www.mrmjournal.com/content/9/1/44 (accessed on 15 September 2022). [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward Primer Sequence | Reverse Primer Sequence |
|---|---|---|
| TNF | 5′-TGCACTTTGGAGTGATCGGC-3′ | 5′-TTGTCACTCGGGGTTCGAGA-3′ |
| TNFSF10 | 5′-TTGGGACCCCAATGACGAAGA-3′ | 5′-TGGTCCCAGTTATGTGAGCTG-3′ |
| BAX | 5′-CCAGCAAACTGGTGCTCAAGG-3′ | 5′-ACAGGGACATCAGTCGCTTCA-3′ |
| BID | 5′-GACCCTGGGAGAGCTCTGAAGC-3′ | 5′-CTCCGACTCACTCCTGGTTCAC-3′ |
| CASPASE 2 | 5′-TACTCCCACCGTTGAGCTGT-3′ | 5′-TGCCAGCTGGAAGTGTGTTTG-3′ |
| CASPASE 3 | 5′-TTGGAACCAAAGATCATACATGGAA-3′ | 5′-TGAGGTTTGCTGCATCGACA-3′ |
| CASPASE 7 | 5′-AGTGGATGCTAAGCCAGACCG-3′ | 5′-TCGAACGCCCATACCTGTCA-3′ |
| CASPASE 8 | 5′-ATAGGCCTGTGACGAAGGTGC-3′ | 5′-GCGGAATGTAGTCCAGGCTCA-3′ |
| ACTB1 | 5′-CTTCGCGGGCGACGAT-3′ | 5′-CCACATAGGAATCCTTCTGACC-3′ |
| MRPL191 | 5′-CCTAGGCCGGAGTTTCCAA-3′ | 5′-GACTCAAGAACCTGCGTTCC-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mosiane, K.S.; Nweke, E.E.; Balogun, M.; Fru, P.N. Polyethyleneglycol-Betulinic Acid (PEG-BA) Polymer-Drug Conjugate Induces Apoptosis and Antioxidation in a Biological Model of Pancreatic Cancer. Polymers 2023, 15, 448. https://0-doi-org.brum.beds.ac.uk/10.3390/polym15020448
Mosiane KS, Nweke EE, Balogun M, Fru PN. Polyethyleneglycol-Betulinic Acid (PEG-BA) Polymer-Drug Conjugate Induces Apoptosis and Antioxidation in a Biological Model of Pancreatic Cancer. Polymers. 2023; 15(2):448. https://0-doi-org.brum.beds.ac.uk/10.3390/polym15020448
Chicago/Turabian StyleMosiane, Karabo Sekopi, Ekene Emmanuel Nweke, Mohammed Balogun, and Pascaline Nanga Fru. 2023. "Polyethyleneglycol-Betulinic Acid (PEG-BA) Polymer-Drug Conjugate Induces Apoptosis and Antioxidation in a Biological Model of Pancreatic Cancer" Polymers 15, no. 2: 448. https://0-doi-org.brum.beds.ac.uk/10.3390/polym15020448