
Highly Branched Bio-Based Unsaturated Polyesters by Enzymatic Polymerization


Abstract
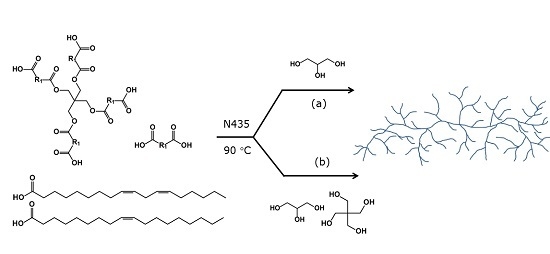
:
1. Introduction
2. Materials and Methods
2.1. Experimental Procedure
2.1.1. General Synthesis Procedure for the Lipase-Catalyzed Preparation of the UBPs
2.1.2. CALB-Catalyzed Bulk One-Pot UBP Synthesis for Preparation of UBP3
2.1.3. Structural Data of UBPs
2.1.4. General Synthesis Procedure for a Classical Alkyd Binder
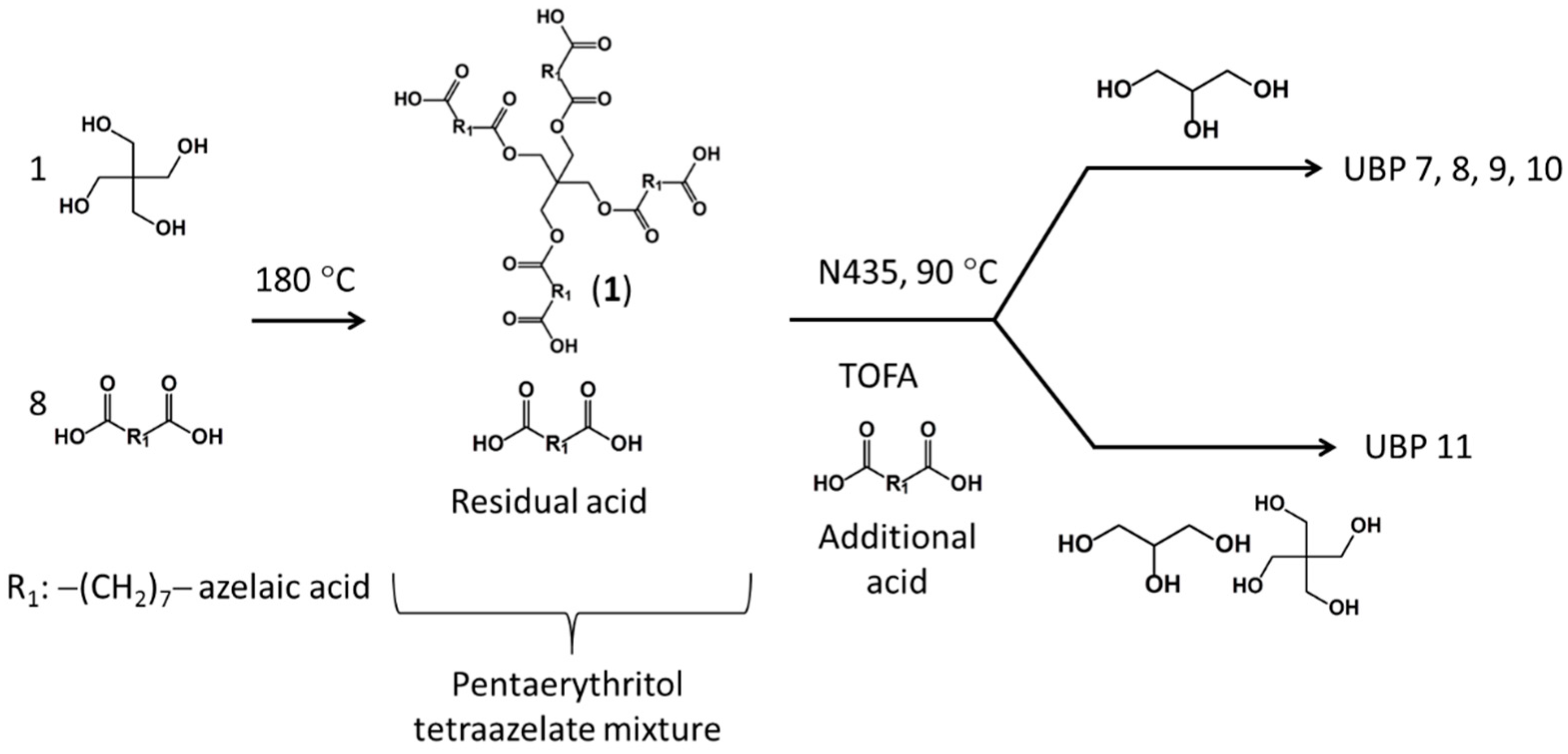
2.1.5. Pentaerythritol Tetraazelate (PTA) Mixture Synthesis
3. Results and Discussion
3.1. One-Pot Enzymatic Polymerization Procedure for Synthesis of Glycerol Based UBPs
3.1.1. Analysis of the Tall Oil Fatty Acid (TOFA) Content
3.1.2. Structural Characterization of Glycerol Based UBPs
3.1.3. Testing Possibilities with the Enzymatic Procedure
3.2. Increased Degree of Branching by Use of Pentaerythritol
3.2.1. Synthesis of Pentaerythritol Tetraazelate (PTA) Mixture
3.2.2. Optimization of Glycerol-Pentaerythritol Based Enzymatic Alkyds
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Evans, P.D. Weathering of wood and wood composites. In Handbook of Wood Chemistry and Wood Composites; Rowell, R.M., Ed.; CRC Press Taylor & Francis Group: Boca Raton, FL, USA, 2013. [Google Scholar]
- Flexner, B. Wood Finishing 101; F+W Media, Inc.: New York, NY, USA, 2011. [Google Scholar]
- Jones, F.N. Alkyd Resins. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2003; pp. 1–31. [Google Scholar]
- Van Gorkum, R.; Bouwman, E. The oxidative drying of alkyd paint catalysed by metal complexes. Coord. Chem. Rev. 2005, 249, 1709–1728. [Google Scholar] [CrossRef]
- Wicks, Z.W. Alkyd resins. In Encyclopedia of Polymer Science and Technology; John Wiley & Sons, Inc.: Weinheim, Germany, 2007. [Google Scholar]
- Mallégol, J.; Gardette, J.-L.; Lemaire, J. Long-term behavior of oil-based varnishes and paints. Photo- and thermooxidation of cured linseed oil. J. Am. Oil Chem. Soc. 2000, 77, 257–263. [Google Scholar] [CrossRef]
- Fourcade, D.; Ritter, B.S.; Walter, P.; Schönfeld, R.; Mülhaupt, R. Renewable resource-based epoxy resins derived from multifunctional poly(4-hydroxybenzoates). Green Chem. 2013, 15, 910–918. [Google Scholar] [CrossRef]
- Holmberg, K. Alkyd Resins. In Coatings Technology Handbook; Tracton, A.A., Ed.; CRC Press: Boca Raton, FL, USA, 2006. [Google Scholar]
- Morrow, C.J.; Wallace, J.S. Synthesis of polyesters by lipase-catalyzed polycondensation in organic media. In Biocatalysis; Abramowicz, D.A., Ed.; Springer: Dordrecht, The Netherlands, 1990. [Google Scholar]
- Linko, Y.Y.; Seppala, J. Producing high molecular weigh biodegradable polyesters. Chemtech 1996, 26, 25–31. [Google Scholar]
- Anderson, E.M.; Larsson, K.M.; Kirk, O. One biocatalyst many applications: The use of Candida antarctica B-lipase in organic synthesis. Biocatal. Biotransform. 1998, 16, 181–204. [Google Scholar] [CrossRef]
- Nayak, P.L. Enzyme-catalyzed polymerization: An opportunity for innovation. Des. Monomers Polym. 1998, 1, 259–284. [Google Scholar] [CrossRef]
- Gross, R.A.; Kalra, B.; Kumar, A. Polyester and polycarbonate synthesis by in vitro enzyme catalysis. Appl. Microbiol. Biotechnol. 2001, 55, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Varma, I.K.; Albertsson, A.-C.; Rajkhowa, R.; Srivastava, R.K. Enzyme catalyzed synthesis of polyesters. Prog. Polym. Sci. 2005, 30, 949–981. [Google Scholar] [CrossRef]
- Kobayashi, S. Recent developments in lipase-catalyzed synthesis of polyesters. Macromol. Rapid Commun. 2009, 30, 237–266. [Google Scholar] [CrossRef] [PubMed]
- Linares, G.; Baldessari, A. Lipases as efficient catalysts in the synthesis of monomers and polymers with biomedical applications. Curr. Org. Chem. 2013, 17, 719–743. [Google Scholar] [CrossRef]
- Miletić, N.; Loos, K.; Gross, R.A. Enzymatic polymerization of polyester. In Biocatalysis in Polymer Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010. [Google Scholar]
- Binns, F.; Harffey, P.; Roberts, S.M.; Taylor, A. Studies of lipase-catalyzed polyesterification of an unactivated diacid/diol system. J. Polym. Sci. Part A Polym. Chem. 1998, 36, 2069–2080. [Google Scholar] [CrossRef]
- Moreno, M.; Lligadas, G.; Ronda, J.C.; Galià, M.; Cádiz, V. Polyketoesters from oleic acid. Synthesis and functionalization. Green Chem. 2014, 16, 1847–1853. [Google Scholar] [CrossRef]
- Eriksson, M.; Hult, K.; Malmström, E.; Johansson, M.; Trey, S.M.; Martinelle, M. One-pot enzymatic polycondensation to telechelic methacrylate-functional oligoesters used for film formation. Polym. Chem. 2011, 2, 714–719. [Google Scholar] [CrossRef]
- Vaida, C.; Keul, H.; Moeller, M. Tailor-made polyesters based on pentadecalactone via enzymatic catalysis. Green Chem. 2011, 13, 889–899. [Google Scholar] [CrossRef]
- Feder, D.; Gross, R.A. Exploring chain length selectivity in HIC-catalyzed polycondensation reactions. Biomacromolecules 2010, 11, 690–697. [Google Scholar] [CrossRef] [PubMed]
- Gubicza, L.; Bélafi-Bakó, K.; Fehér, E.; Fráter, T. Waste-free process for continuous flow enzymatic esterification using a double pervaporation system. Green Chem. 2008, 10, 1284–1287. [Google Scholar] [CrossRef]
- Kline, B.J.; Beckman, E.J.; Russell, A.J. One-step biocatalytic synthesis of linear polyesters with pendant hydroxyl groups. J. Am. Chem. Soc. 1998, 120, 9475–9480. [Google Scholar] [CrossRef]
- Major, B.; Kelemen-Horváth, I.; Csanádi, Z.; Bélafi-Bakó, K.; Gubicza, L. Microwave assisted enzymatic esterification of lactic acid and ethanol in phosphonium type ionic liquids as co-solvents. Green Chem. 2009, 11, 614–616. [Google Scholar] [CrossRef]
- Mahapatro, A.; Kumar, A.; Gross, R.A. Mild, solvent-free omega-hydroxy acid polycondensations catalyzed by Candida antarctica lipase B. Biomacromolecules 2004, 5, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Uyama, H.; Inada, K.; Kobayashi, S. Lipase-Catalyzed Synthesis of Aliphatic Polyesters by Polycondensation of Dicarboxylic Acids and Glycols in Solvent-Free System. Polym. J. 2000, 32, 440–443. [Google Scholar] [CrossRef]
- Jiang, Y.; Van Ekenstein, G.O.R.A.; Woortman, A.J.J.; Loos, K. Fully biobased unsaturated aliphatic polyesters from renewable resources: Enzymatic synthesis, characterization, and properties. Macromol. Chem. Phys. 2014, 215, 2185–2197. [Google Scholar] [CrossRef]
- Corici, L.; Pellis, A.; Ferrario, V.; Ebert, C.; Cantone, S.; Gardossi, L. Understanding potentials and restrictions of solvent-free enzymatic polycondensation of itaconic acid: An experimental and computational analysis. Adv. Synth. Catal. 2015, 357, 1763–1774. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, C.; Stuparu, M.C.; Daugaard, A.E.; Khan, A. Aza-michael addition reaction: Post-polymerization modification and preparation of PEI/PEG-based polyester hydrogels from enzymatically synthesized reactive polymers. J. Polym. Sci. Part A Polym. Chem. 2015, 53, 745–749. [Google Scholar] [CrossRef]
- Tsujimoto, T.; Uyama, H.; Kobayashi, S. Enzymatic synthesis of cross-linkable polyesters from renewable resources. Biomacromolecules 2001, 2, 29–31. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, T.; Uyama, H.; Kobayashi, S. Enzymatic synthesis and curing of biodegradable crosslinkable polyesters. Macromol. Biosci. 2002, 2, 329–335. [Google Scholar] [CrossRef]
- Zhang, Y.R.; Spinella, S.; Xie, W.; Cai, J.; Yang, Y.; Wang, Y.Z.; Gross, R.A. Polymeric triglyceride analogs prepared by enzyme-catalyzed condensation polymerization. Eur. Polym. J. 2013, 49, 793–803. [Google Scholar] [CrossRef]
- Köckritz, A.; Martin, A. Synthesis of azelaic acid from vegetable oil-based feedstocks. Eur. J. Lipid Sci. Technol. 2011, 113, 83–91. [Google Scholar] [CrossRef]
- Bravi, E.; Benedetti, P.; Marconi, O.; Perretti, G. Determination of free fatty acids in beer wort. Food Chem. 2014, 151, 374–378. [Google Scholar] [CrossRef] [PubMed]
- Rabiller, C.; Maze, F. Quantitative analysis and determination of the enantiomeric purity of glycerides by 13C-NMR spectroscopy. Application to the lipase-catalysed transesterification of triacylglycerides. Magn. Reson. Chem. 1989, 27, 582–584. [Google Scholar] [CrossRef]
- Stamatov, S.D.; Stawinski, J. Regioselective opening of an oxirane system with trifluoroacetic anhydride. A general method for the synthesis of 2-monoacyl- and 1,3-symmetrical triacylglycerols. Tetrahedron 2005, 61, 3659–3669. [Google Scholar] [CrossRef]
- Jie, L.K.; Marcel, S.F.; Lam, C.C. 1H-Nuclear magnetic resonance spectroscopic studies of saturated, acetylenic and ethylenic triacylglycerols. Chem. Phys. Lipids 1995, 77, 155–171. [Google Scholar]
- Hatzakis, E.; Agiomyrgianaki, A.; Kostidis, S.; Dais, P. High-resolution NMR spectroscopy: An alternative fast tool for qualitative and quantitative analysis of diacylglycerol (DAG) oil. J. Am. Oil Chem. Soc. 2011, 88, 1695–1708. [Google Scholar] [CrossRef]
- Duclos, R.I.; Johnston, M.; Vadivel, S.K.; Makriyannis, A.; Glaser, S.T.; Gatley, S.J. A methodology for radiolabeling of the endocannabinoid 2-arachidonoylglycerol (2-AG). J. Org. Chem. 2011, 76, 2049–2055. [Google Scholar] [CrossRef] [PubMed]
- Compton, D.L.; Vermillion, K.E.; Laszlo, J.A. Acyl migration kinetics of 2-monoacylglycerols from soybean oil via 1H NMR. J. Am. Oil Chem. Soc. 2007, 84, 343–348. [Google Scholar] [CrossRef]
- Yao, D.; Li, G.; Kuila, T.; Li, P.; Kim, N.H.; Kim, S.-I.; Lee, J.H. Lipase-catalyzed synthesis and characterization of biodegradable polyester containing l-malic acid unit in solvent system. J. Appl. Polym. Sci. 2011, 120, 1114–1120. [Google Scholar] [CrossRef]
- Kumar, A.; Kulshrestha, A.S.; Gao, W.; Gross, R.A. Versatile route to polyol polyesters by lipase catalysis. Macromolecules 2003, 36, 8219–8221. [Google Scholar] [CrossRef]
- Kulshrestha, A.S.; Gao, W.; Gross, R.A. Glycerol copolyesters: Control of branching and molecular weight using a lipase catalyst. Macromolecules 2005, 38, 3193–3204. [Google Scholar] [CrossRef]
- Yang, Y.; Lu, W.; Cai, J.; Hou, Y.; Ouyang, S.; Xie, W.; Gross, R.A. Poly(oleic diacid-co-glycerol): Comparison of polymer structure resulting from chemical and lipase catalysis. Macromolecules 2011, 44, 1977–1985. [Google Scholar] [CrossRef]
- Andrianov, K.A.; Emelyanov, V.N. Synthesis of polyesters and polyester-amides of cycloreticular structures. Izvestiya Akademii Nauk SSSR Seriya Khimicheskaya 1963, 7, 1267–1272. [Google Scholar]
- Happe, M.; Kouadio, M.; Treanor, C.; Sawall, J.-P.; Fornage, A.; Sugnaux, M.; Fischer, F. Size selectivity in lipase catalysed tetrol acylation. J. Mol. Catal. B Enzym. 2014, 109, 40–46. [Google Scholar] [CrossRef]
- Poojari, Y.; Beemat, J.S.; Clarson, S.J. Enzymatic synthesis of poly(ε-caprolactone): Thermal properties, recovery, and reuse of lipase B from Candida antarctica immobilized on macroporous acrylic resin particles. Polym. Bull. 2013, 70, 1543–1552. [Google Scholar] [CrossRef]




{kind=link}
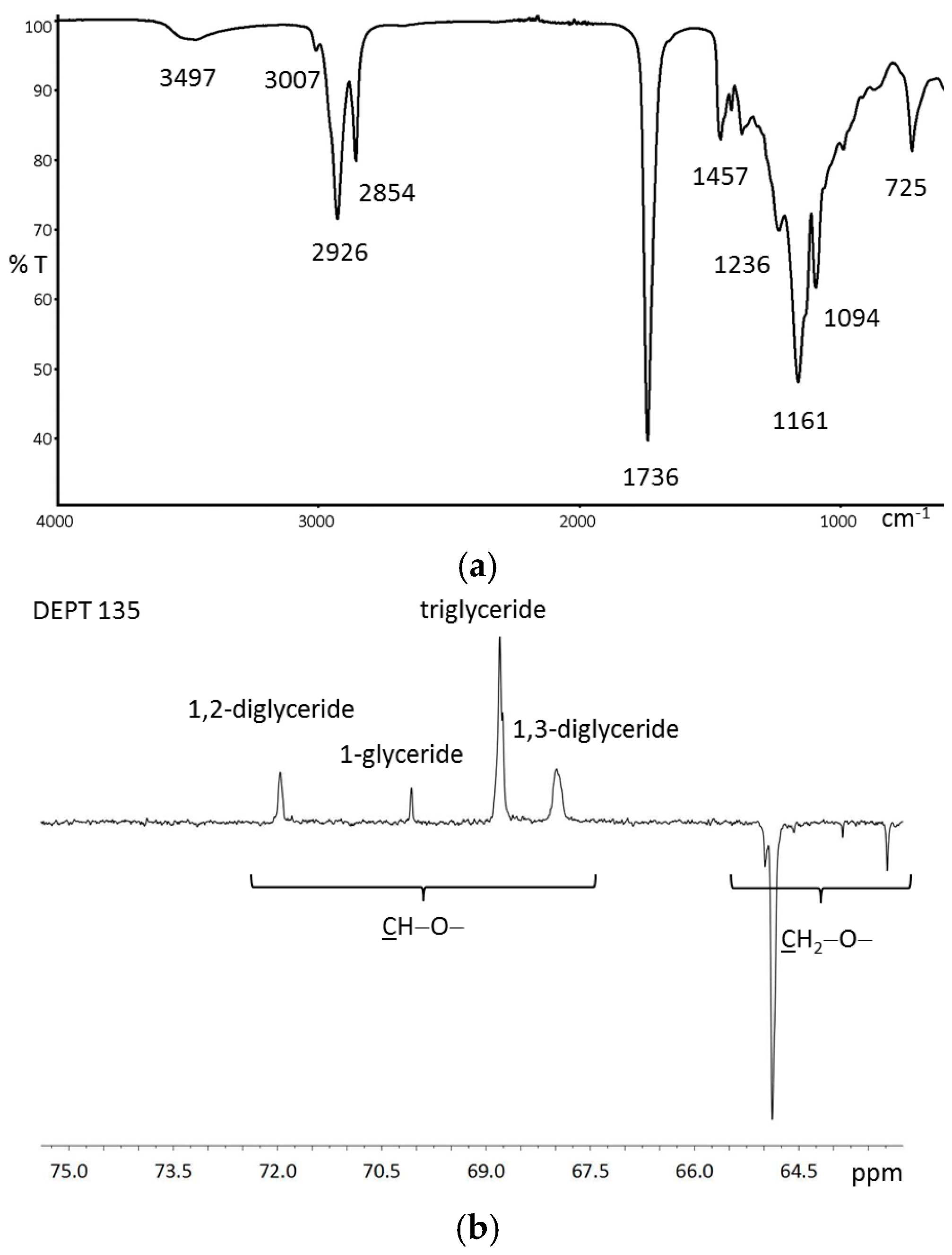{kind=link}
{kind=link}
{kind=link}
| UBP | Molar ratio 1 | React. time (h) | Mw (g/mol) | PDI | Tg (°C) | Film properties | ||
|---|---|---|---|---|---|---|---|---|
| Azelaic acid | TOFA | Tg (°C) | Adv. WCA (°) | |||||
| 1 | 1 | 1.13 | 25 | 37,400 | 5 | −72 | −21 | 104 ± 1 |
| 2 | 1 | 0.85 | 25 | 27,400 | 3.6 | −66 | −27 | 104 ± 2 |
| 3 | 1 | 0.57 | 25 | 28,100 | 3.6 | −60 | −30 | 109 ± 1 |
| 4 | 1 | 0.29 | 25 | 30,400 | 3.5 | −54 | −37 | 117 ± 2 |
| 5 | 1.16 | 0.27 | 25 | 20,900 | 3.2 | −42 | −34 | 141 ± 1 |
| 6 | 1 | 0.29 | 84 | 39,700 | 3.8 | −52 | −35 | 111 ± 1 |
| UBP | React. time (h) | Mw (g/mol) | PDI | Tg (°C) | Acid number | Film properties | |
|---|---|---|---|---|---|---|---|
| Tg (°C) | Adv. WCA (°) | ||||||
| 7 | 25 | 34,200 | 3.3 | −51 | 31.7 ± 0.4 | −27 | 104 ± 2 |
| 8 | 48 | 40,600 | 3.4 | −51 | 26.0 ± 0.6 | −27 | 104 ± 2 |
| 9 | 110 | 56,700 | 4.5 | −55 | 20.1 ± 0.9 | −25 | 114 ± 3 |
| 10 1 | 25 | 16,800 | 2.8 | −54 | 87.3 ± 1.7 | −31 | 132 ± 3 |
| 11 2 | 25 | 26,800 | 2.9 | −53 | 28.3 ± 0.6 | −28 | 114 ± 1 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, H.D.; Löf, D.; Hvilsted, S.; Daugaard, A.E. Highly Branched Bio-Based Unsaturated Polyesters by Enzymatic Polymerization. Polymers 2016, 8, 363. https://0-doi-org.brum.beds.ac.uk/10.3390/polym8100363
Nguyen HD, Löf D, Hvilsted S, Daugaard AE. Highly Branched Bio-Based Unsaturated Polyesters by Enzymatic Polymerization. Polymers. 2016; 8(10):363. https://0-doi-org.brum.beds.ac.uk/10.3390/polym8100363
Chicago/Turabian StyleNguyen, Hiep Dinh, David Löf, Søren Hvilsted, and Anders Egede Daugaard. 2016. "Highly Branched Bio-Based Unsaturated Polyesters by Enzymatic Polymerization" Polymers 8, no. 10: 363. https://0-doi-org.brum.beds.ac.uk/10.3390/polym8100363