Mapping of the Waxy Bloom Gene in ‘Black Jewel’ in a Parental Linkage Map of ‘Black Jewel’ × ‘Glen Ample’ (Rubus) Interspecific Population

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and Phenotyping
2.2. DNA Isolation and SSR Marker Analysis
2.3. Tunable Genotyping-by-Sequencing (tGBS)
2.4. Trimming of Sequencing Reads and Alignment to the Reference Genome
2.5. Discovery of Polymorphic Sites
2.6. Criteria for Homozygous and Heterozygous Calls
2.7. Rubus Linkage Group 2 (LG2) SSR Genotyping
2.8. Genetic Map Construction
2.9. Predicted Gene Search in the Rubus Reference Genome
3. Results
3.1. Waxy Bloom Phenotyping
3.2. SSR Marker Analyses
3.3. tGBS SNPs Identification and Genotyping
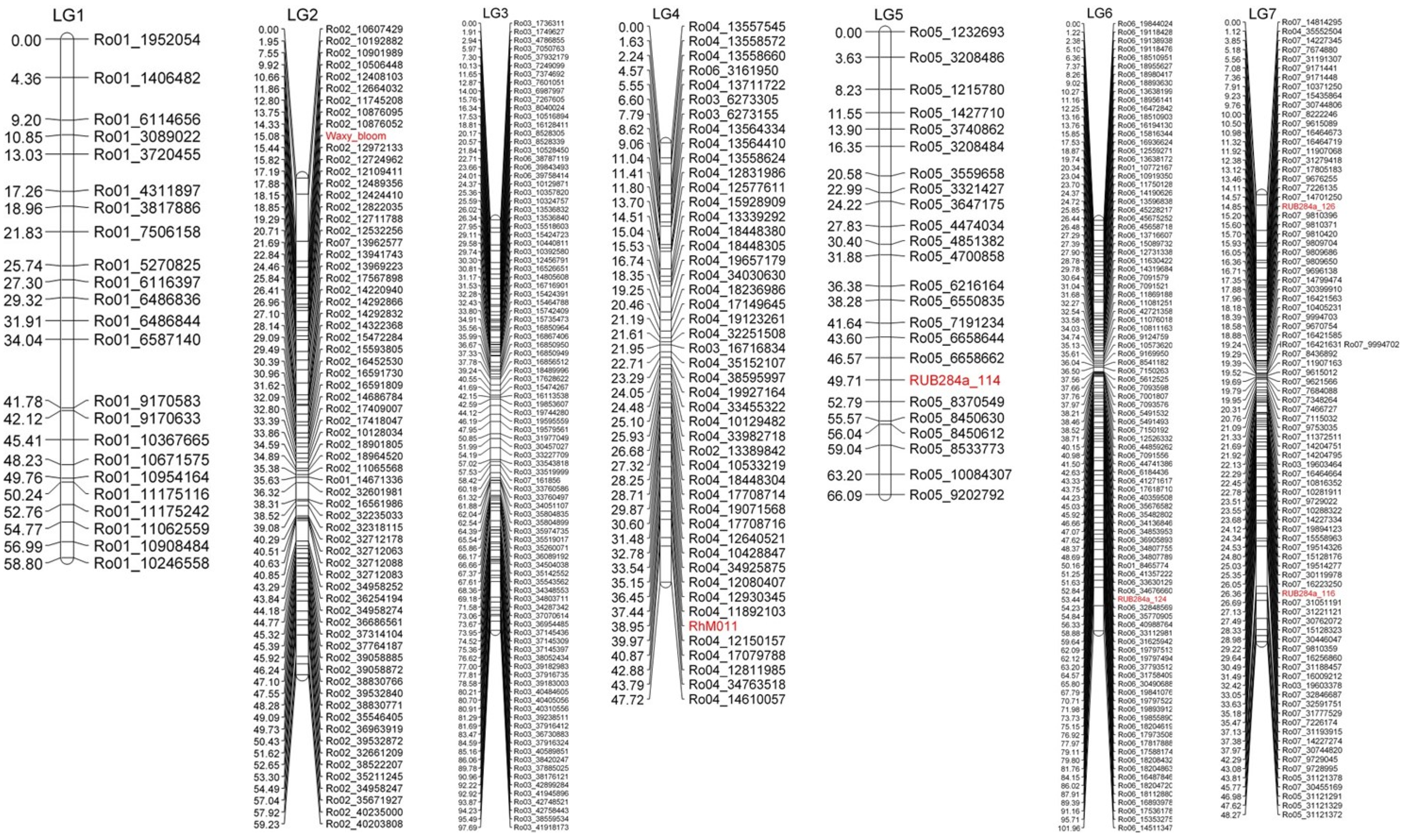
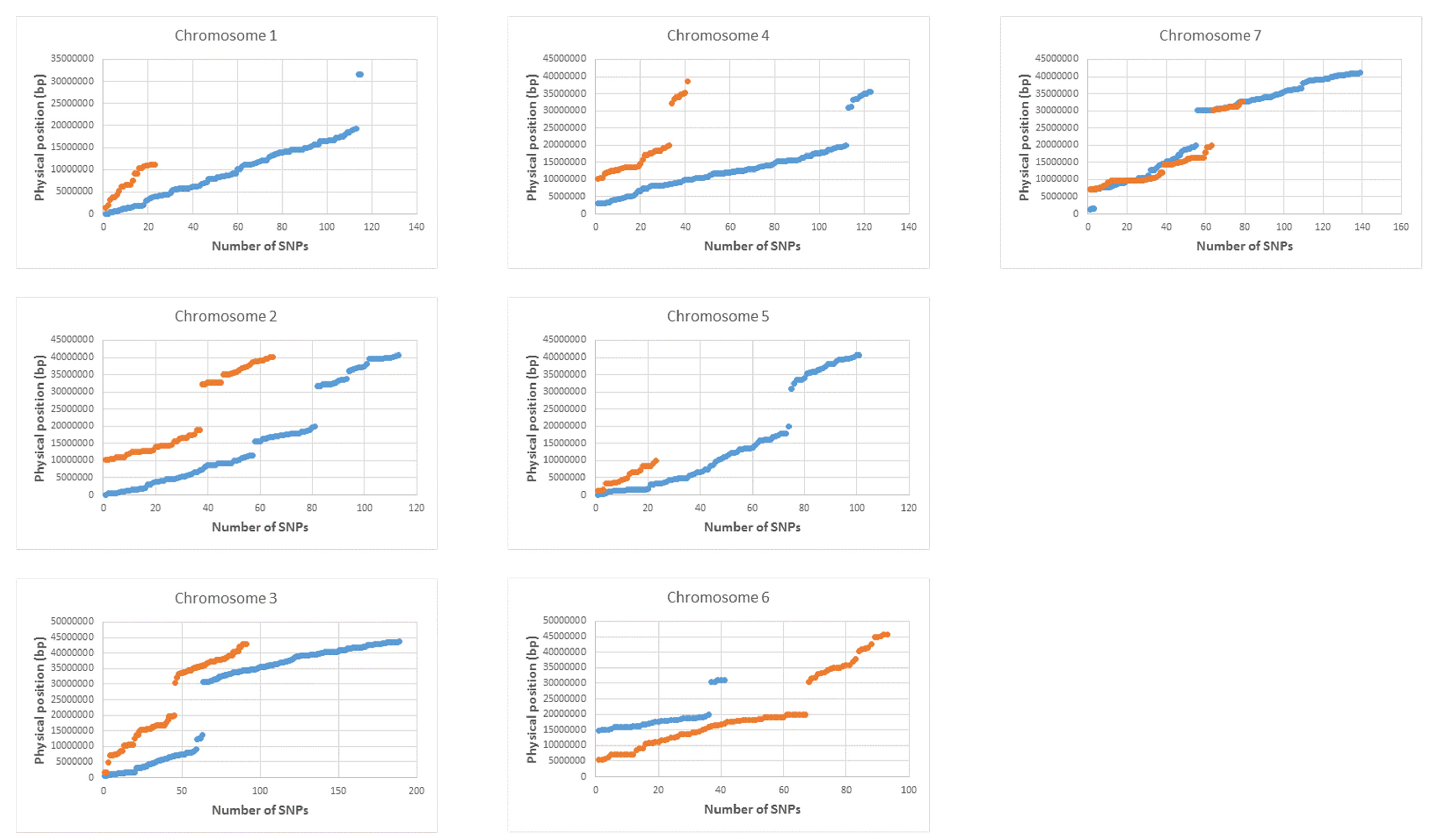
3.4. Linkage Map Construction
3.5. Mapping of the Waxy Bloom Gene
3.6. Annotated Genes in the Rubus V3.0 Reference Genome within the Physical Interval of the Waxy Bloom Gene
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Graham, J.; Hein, I.; Powell, W. “Raspberry.” Fruits and Nuts; Springer: Berlin/Heidelberg, Germany, 2007; pp. 207–216. [Google Scholar]
- Foster, T.M.; Bassil, N.V.; Dossett, M.; Worthington, M.L.; Graham, J. Genetic and genomic resources for Rubus breeding: A roadmap for the future. Hortic. Res. 2019, 6, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinonen, I.M.; Meyer, A.S.; Frankel, E.N. Antioxidant Activity of Berry Phenolics on Human Low-Density Lipoprotein and Liposome Oxidation. J. Agric. Food Chem. 1998, 46, 4107–4112. [Google Scholar] [CrossRef]
- FAOSTAT; Agriculture Organization of the United Nations: Rome, Italy, 2020.
- Graham, J.; Smith, K.; Woodhead, M.; Russell, J. Development and use of SSR markers in Rubus species. Mol. Ecol. Notes 2002, 2, 250–252. [Google Scholar] [CrossRef]
- Graham, J.; Smith, K.; MacKenzie, K.; Jorgenson, L.; Hackett, C.; Powell, W. The construction of a genetic linkage map of red raspberry (Rubus idaeus subsp. idaeus) based on AFLPs, genomic-SSR and EST-SSR markers. Theor. Appl. Genet. 2004, 109, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Graham, J.; Smith, K.; Tierney, I.; MacKenzie, K.; Hackett, C.A. Mapping gene H controlling cane pubescence in raspberry and its association with resistance to cane botrytis and spur blight, rust and cane spot. Theor. Appl. Genet. 2006, 112, 818–831. [Google Scholar] [CrossRef] [PubMed]
- Sargent, D.; Fernández-Fernández, F.; Rys, A.; Knight, V.; Simpson, D.; Tobutt, K. Mapping of A1 conferring resistance to the aphid Amphorophora idaei and dw (dwarfing habit) in red raspberry (Rubus idaeus L.) using AFLP and microsatellite markers. BMC Plant Biol. 2007, 7, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattison, J.A.; Samuelian, S.K.; Weber, C.A. Inheritance of Phytophthora root rot resistance in red raspberry determined by generation means and molecular linkage analysis. Theor. Appl. Genet. 2007, 115, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Ward, J.A.; Bhangoo, J.; Fernández-Fernández, F.; Moore, P.; Swanson, J.D.; Viola, R.; Velasco, R.; Bassil, N.; Weber, C.A.; Sargent, D. Saturated linkage map construction in Rubus idaeus using genotyping by sequencing and genome-independent imputation. BMC Genom. 2013, 14, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Bushakra, J.M.; Bryant, D.W.; Dossett, M.; Vining, K.J.; VanBuren, R.; Gilmore, B.S.; Lee, J.; Mockler, T.C.; Finn, C.E.; Bassil, N.V. A genetic linkage map of black raspberry (Rubus occidentalis) and the mapping of Ag 4 conferring resistance to the aphid Amphorophora agathonica. Theor. Appl. Genet. 2015, 128, 1631–1646. [Google Scholar] [CrossRef] [Green Version]
- Bushakra, J.M.; Stephens, M.J.; Atmadjaja, A.N.; Lewers, K.S.; Symonds, V.V.; Udall, J.A.; Chagné, D.; Buck, E.J.; Gardiner, S.E. Construction of black (Rubus occidentalis) and red (R. idaeus) raspberry linkage maps and their comparison to the genomes of strawberry, apple, and peach. Theor. Appl. Genet. 2012, 125, 311–327. [Google Scholar] [CrossRef]
- Hackett, C.A.; Milne, L.; Smith, K.; Hedley, P.; Morris, J.; Simpson, C.G.; Preedy, K.; Graham, J. Enhancement of Glen Moy x Latham raspberry linkage map using GbS to further understand control of developmental processes leading to fruit ripening. BMC Genet. 2018, 19, 59. [Google Scholar] [CrossRef] [PubMed]
- McCallum, S.; Simpson, C.; Graham, J. QTL Mapping and Marker Assisted Breeding in Rubus spp. In Raspberry; Springer International Publishing: Cham, Switzerland, 2018; pp. 121–144. [Google Scholar]
- Jennings, D. Some evidence on the influence of the morphology of raspberry canes upon their ability to be attacked by certain fungi. Hortic. Res. 1962, 1, 100–111. [Google Scholar]
- MacKenzie, K.; Williamson, S.; Smith, K.; Woodhead, M.; McCallum, S.; Graham, J. Characterisation of Gene H in Red Raspberry: Explaining its Role in Cane Morphology, Disease Resistance and Timing of Fruit Ripening. J. Hortic. 2015, 2, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Lewis, D. Genetical studies in cultivated raspberries. J. Genet. 1939, 38, 367–379. [Google Scholar] [CrossRef]
- Baker, E.A.; Batt, R.F.; Silva Fernandes, A.M.S.; Martin, J.T. Cuticular waxes of plant species and varieties. In Proceedings of the Annual Report of the Agricultural and Horticultural Research Station, Long Ashton; University of Bristol: Bristol, UK, 1963; pp. 106–110. [Google Scholar]
- Jeffree, C.E.; Baker, E.A.; Holloway, P.J. Ultrastructure and Recrystallization of Plant Epicuticular Waxes. New Phytol. 1975, 75, 539–549. [Google Scholar] [CrossRef]
- Jennings, D.L. Some evidence on the genetic structure of present-day raspberry varieties and some possible implications for further breeding. Euphytica 1963, 12, 229–243. [Google Scholar] [CrossRef]
- Dossett, M.; Finn, C.E. Performance and phenology of wild black raspberry (Rubus occidentalis L.) germplasm in a common garden. Genet. Resour. Crop. Evol. 2015, 63, 653–673. [Google Scholar] [CrossRef]
- Jennings, D.L. Balanced lethals and polymorphism in Rubus idæeus. Heredity 1967, 22, 465–479. [Google Scholar] [CrossRef] [Green Version]
- Jennings, D.L.; Tulloch, B.M.M. Studies on factors which promote germination of raspberry seeds. J. Exp. Bot. 1965, 16, 329–340. [Google Scholar] [CrossRef]
- Castillo, N.R.F.; Reed, B.M.; Graham, J.; Fernández-Fernández, F.; Bassil, N.V. Microsatellite Markers for Raspberry and Blackberry. J. Am. Soc. Hortic. Sci. 2010, 135, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Fernández, F.; Antanaviciute, L.; Govan, C.L.; Sargent, D.J. Development of a multiplexed microsatellite set for fingerprinting red raspberry (Rubus idaeus) germplasm and its transferability to other Rubus species. J. Berry Res. 2011, 1, 177–187. [Google Scholar] [CrossRef] [Green Version]
- Ewing, B.; Green, P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998, 8, 186–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewing, B.; Hillier, L.; Wendl, M.C.; Green, P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998, 8, 175–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Chou, S.S. LUCY2: An interactive DNA sequence quality trimming and vector removal tool. Bioinformatics 2004, 20, 2865–2866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VanBuren, R.; Wai, C.M.; Colle, M.; Wang, J.; Sullivan, S.; Bushakra, J.M.; Liachko, I.; Vining, K.J.; Dossett, M.; Finn, C.E.; et al. A near complete, chromosome-scale assembly of the black raspberry (Rubus occidentalis) genome. GigaScience 2018, 7, giy094. [Google Scholar] [CrossRef]
- Wu, T.D.; Nacu, S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 2010, 26, 873–881. [Google Scholar] [CrossRef] [Green Version]
- Schuelke, M. An economic method for the fluorescent labeling of PCR fragments. Nat. Biotechnol. 2000, 18, 233–234. [Google Scholar] [CrossRef]
- Van Oojien, J. JoinMap 4, software for the calculation of genetic linkage maps in experimental populations. Kyzama Wagening. 2006. [Google Scholar]
- VanBuren, R.; Bryant, D.; Bushakra, J.M.; Vining, K.J.; Edger, P.P.; Rowley, E.R.; Priest, H.D.; Michael, T.P.; Lyons, E.; Filichkin, S.A.; et al. The genome of black raspberry (Rubus occidentalis). Plant J. 2016, 87, 535–547. [Google Scholar] [CrossRef] [Green Version]
- Jibran, R.; Dzierzon, H.; Bassil, N.; Bushakra, J.M.; Edger, P.P.; Sullivan, S.; Finn, C.E.; Dossett, M.; Vining, K.J.; VanBuren, R.; et al. Chromosome-scale scaffolding of the black raspberry (Rubus occidentalis L.) genome based on chromatin interaction data. Hortic. Res. 2018, 5, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [Green Version]
- Poland, J.A.; Brown, P.J.; Sorrells, M.E.; Jannink, J.-L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 2012, 7, e32253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emeriewen, O.F.; Richter, K.; Berner, T.; Keilwagen, J.; Schnable, P.S.; Malnoy, M.; Peil, A. Construction of a dense genetic map of the Malus fusca fire blight resistant accession MAL0045 using tunable genotyping-by-sequencing SNPs and microsatellites. Sci. Rep. 2020, 10, 16358. [Google Scholar] [CrossRef]
- Woodhead, M.; McCallum, S.; Smith, K.; Cardle, L.; Mazzitelli, L.; Graham, J. Identification, characterisation and mapping of simple sequence repeat (SSR) markers from raspberry root and bud ESTs. Mol. Breed. 2008, 22, 555–563. [Google Scholar] [CrossRef]
- Wight, H.; Zhou, J.; Li, M.; Hannenhalli, S.; Mount, S.; Liu, Z. Draft genome assembly and annotation of red raspberry Rubus Idaeus. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Dossett, M. Variation and Heritability of Vegetative, Reproductive and Fruit Chemistry Traits in Black Raspberry (Rubus occidentalis L.). Master’s Thesis, Oregon State University, Corvallis, OR, USA, 23 March 2007. [Google Scholar]
- Bassil, N.V.; Nyberg, A.; Hummer, K.E.; Graham, J.; Dossett, M.; Finn, C.E. A Universal Fingerprinting Set for Red Raspberry. Acta Hortic. 2012, 946, 83–87. [Google Scholar] [CrossRef] [Green Version]
- Dossett, M.; Bassil, N.V.; Finn, C.E. SSR Fingerprinting of Black Raspberry Cultivars Shows Discrepancies in Identification. Acta Hortic. 2012, 946, 49–53. [Google Scholar] [CrossRef]
- Pinczinger, D.; von Reth, M.; Hanke, M.-V.; Flachowsky, H. SSR fingerprinting of raspberry cultivars traded in Germany clearly showed that certainty about the genotype authenticity is a prerequisite for any horticultural experiment. Eur. J. Hortic. Sci. 2020, 85, 79–85. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| SSR | LG | Source | ‘Black Jewel’ Alleles | ‘Glen Ample’ Alleles |
|---|---|---|---|---|
| ‡ RhM011 1 | LG7 | Castillo et al. [24] | 280, 282 | 288, 292 |
| ‡ RhM043 2 | LG4 | Castillo et al. [24] | – | 374, 377 |
| ‡ RiM017 3 | LG7 | Castillo et al. [24] | 192, 194 | 196 |
| ‡ Rub123a 2 | LG6 | Graham et al. [7] | 139 | 148, 162 |
| ‡ Rub285a 2 | LG1 | Graham et al. [7] | 167 | 172, 174 |
| † Rub107a 2 | LG2 | Graham et al. [7] | 173 | 166, 168 |
| † Rub210a 2 | LG1 | Graham et al. [7] | 103 | 117, 123 |
| † Rub124a 4 | LG1 | Graham et al. [7] | – | 163 |
| † Rub270a 2 | LG2 | Graham et al. [7] | 183 | 175, 183 |
| † Rub56a 5 | LG2 | Graham et al. [7] | – | – |
| † Rub76b 4 | LG2 | Graham et al. [7] | 217 | 211, 217 |
| † Rub4a 4 | LG2 | Graham et al. [7] | – | 154 6 |
| † Rub163a 5 | LG2 | Graham et al. [7] | – | – |
| † Rub293b 2 | LG2 | Graham et al. [7] | – | 162, 164, 200, 202 |
| † Rub284a 1 | LG2 | Graham et al. [7] | 114, 116, 122 6, 124, 126 | 156, null |
| † Rubnebp2O23 4 | LG2 | Graham et al. [7] | 234 | 238, 240 |
| ‘Black Jewel’ | ‘Glen Ample’ | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LG | No. of Markers LOD15 | No. of Markers Mapped | Length of Map cM | Maximum Gap cM | Minimum Gap cM | Average Gap cM | LG | No. of Markers LOD15 | No. of Markers Mapped | Length of Map cM | Maximum Gap cM | Minimum Gap cM | Average Gap cM |
| 1 | 30 | 23 | 58.80 | 7.74 | 0.34 | 2.67 | 1 | 312 | 123 | 103.75 | 4.78 | 0.25 | 0.85 |
| 2 | 81 | 68 | 59.23 | 5.61 | 0.07 | 0.88 | 2 | 384 | 120 | 103.76 | 2.79 | 0.08 | 0.87 |
| 3 | 123 | 96 | 97.69 | 3.72 | 0.15 | 1.03 | 3 | 586 | 200 | 112.91 | 1.59 | 0.06 | 0.57 |
| 4 | 74 | 47 | 47.72 | 3.92 | 0.35 | 1.04 | 4 | 320 | 140 | 96.03 | 3.82 | 0.01 | 0.69 |
| 5 | 25 | 25 | 66.09 | 4.59 | 0.47 | 2.87 | 5 | 295 | 106 | 98.87 | 3.54 | 0.08 | 0.94 |
| 6 | 113 | 96 | 101.96 | 6.25 | 0.04 | 1.07 | 6 | 58 | 42 | 47.80 | 4.74 | 0.02 | 1.17 |
| 7 | 120 | 88 | 48.27 | 4.32 | 0.01 | 0.55 | 7 | 364 | 146 | 97.41 | 3.57 | 0.02 | 0.67 |
| Total | 566 | 443 | 479.76 | Total | 2319 | 877 | 660.53 | ||||||
| R. occidentalis Genomes | Map Locations in This Study in Comparison to Previous Studies | ||||||
|---|---|---|---|---|---|---|---|
| Chromosomes Located | This Study | Graham et al. [7] | Castillo et al. [24] | ||||
| Rubus SSRs | Rubus V3 | Rubus V1 | ‘Black Jewel’ | ‘Glen Ample’ | ‘Glen Moy’ × ‘Latham’ | ‘Glen Moy’ × ‘Latham’ | ‘Autumn Bliss’ × ‘Malling’ |
| Rub284a | – (F/R) | chr7 (F) | LG7 | LG3 | LG2 | N/A | N/A |
| chr6 (F) | LG6 | ||||||
| chr5 (F) | LG5 | ||||||
| chr4 (F) | |||||||
| – (R) | |||||||
| RiM017 | chr4 (F/R) | chr4 (F/R) | LG4 ‡ | NP | N/A | LG7 | LG7 |
| RhM011 | chr4 (R) | chr4 (R) | LG4 | LG4 | N/A | LG7 | LG7 |
| chr3 (R) | chr3 (R) | ||||||
| – (F) | – (F) | ||||||
| Rub123a | chr1 (F) | chr1 (F) | NP | LG1 | LG6 | N/A | N/A |
| – (R) | – (R) | ||||||
| Rub285a | – (F/R) | chr1 (F) | NP | LG1 | LG1 | N/A | N/A |
| – (R) | |||||||
| RhM043 | – (F/R) | chr7 (F) | NP | LG2 | N/A | NP | NP |
| chr5 (F) | LG4 (secondary locus) | ||||||
| – (R) | |||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinczinger, D.; Reth, M.v.; Keilwagen, J.; Berner, T.; Peil, A.; Flachowsky, H.; Emeriewen, O.F. Mapping of the Waxy Bloom Gene in ‘Black Jewel’ in a Parental Linkage Map of ‘Black Jewel’ × ‘Glen Ample’ (Rubus) Interspecific Population. Agronomy 2020, 10, 1579. https://0-doi-org.brum.beds.ac.uk/10.3390/agronomy10101579
Pinczinger D, Reth Mv, Keilwagen J, Berner T, Peil A, Flachowsky H, Emeriewen OF. Mapping of the Waxy Bloom Gene in ‘Black Jewel’ in a Parental Linkage Map of ‘Black Jewel’ × ‘Glen Ample’ (Rubus) Interspecific Population. Agronomy. 2020; 10(10):1579. https://0-doi-org.brum.beds.ac.uk/10.3390/agronomy10101579
Chicago/Turabian StylePinczinger, Dora, Marcel von Reth, Jens Keilwagen, Thomas Berner, Andreas Peil, Henryk Flachowsky, and Ofere Francis Emeriewen. 2020. "Mapping of the Waxy Bloom Gene in ‘Black Jewel’ in a Parental Linkage Map of ‘Black Jewel’ × ‘Glen Ample’ (Rubus) Interspecific Population" Agronomy 10, no. 10: 1579. https://0-doi-org.brum.beds.ac.uk/10.3390/agronomy10101579