
Temperature Dependences of IR Spectra of Humic Substances of Brown Coal


1
Department of Chemistry, Lomonosov Moscow State University, Leninskie Gory, 1-3, GSP-1, 119991 Moscow, Russia
2
Department of Chemistry and Physical Chemistry of Soils, V.V. Dokuchaev Soil Science Institute, Pyzhevsky per., 7/2, 119017 Moscow, Russia
*
Author to whom correspondence should be addressed.
Agronomy 2021, 11(9), 1822; https://0-doi-org.brum.beds.ac.uk/10.3390/agronomy11091822
Submission received: 2 August 2021
/
Revised: 5 September 2021
/
Accepted: 9 September 2021
/
Published: 11 September 2021
(This article belongs to the Topic Frontier Studies in Composition of Humic Substances and Soil Organic Matter)
Abstract
:The capabilities of temperature-monitored IR spectroscopy for studying the organic matter and mineral composition of humic substances (HS) were tested. Temperature dependences of the mid-IR spectra of humic substances heated in the air in the range 25–215 °C (298–488 K, with a step of 2.5 °C)—for three commercially available samples isolated from brown coal (leonardite)—were performed. The characteristic bands were identified, and their changes in band maxima positions and intensities were compared. From the viewpoint of interpretation of HS components, the spectra were divided into regions of quartz lattice region (800–260 cm−1), quartz overtone region (1270–800 cm−1), humic substance organic matter region (1780–1270 cm−1), quartz combination region (2800–1780 cm−1), CH-speciation region (3100–2800 cm−1), and hydrogen-speciation region (4000–3100 cm−1) thus selected to contain the dominating type of bands. For the first time, a reversible change in the frequencies of the band maxima in IR spectra upon heating was observed, which can be interpreted as forming structures with a particular order in the studied humic substances in the dry state. For a single sample, both the band-shift scale and the functional dependence of the various bands on temperature differ significantly. The approach differentiates crystalline quartz bands, amorphous silica, and HSOM/surface groups experiencing a different temperature behavior of the band maxima and their intensities. Band-maximum temperature dependence can be considered more stable to changes in experimental conditions than band maxima at a single temperature, thus providing a more detailed HS structure analysis without HS decomposition or destruction.
1. Introduction
Humic substances (HS) are a predominant species of organic matter in natural (soil and waters) and human-made environments such as sewage and landfills [1,2,3,4]. Many studies deal with HS as antibacterial/antiviral, antioxidant, metabolic, immunomodulating, and other medicines [5,6,7]. Additionally, HS are demanded in various branches of soil science and agronomy: fertilizers, plant growth regulators, and mediators and detoxifiers in the polluted ecosystems [6,7,8,9,10,11]. HS of various origins—soil, sediments, peat, lignites, brown coal—are relevant resources [1,3] involved in soil [9,12], chemical, and biochemical processes of brown coal treatment [13,14] and biomass treatment [15]. These processes require a large volume of information of HS systems, which requires the development of corresponding analysis and characterization methods.
IR spectroscopy is a developed method to study HS and is used to characterize and assess functional (mainly carboxylic and hydroxylic) groups and whole HS organic matter (HSOM) [16,17,18,19,20,21]. However, like in many other multicomponent samples, the bands of different HS components and functional groups overlap, and a resulting HS IR spectrum shows a small number of relatively uninformative broad overlapping bands. Thus, despite the simplicity and versatility of conventional IR spectroscopy, the information on HS by this method is somewhat limited and usually constrained by qualitative comparison of the functional group composition [17,18,21,22,23,24,25]. Nevertheless, with the potential of IR spectroscopy, it is far from being exhausted for HS and can be seriously enhanced using temperature-dependent IR studies.
Changes in the vibrational spectra of solid samples upon heating or cooling provide information on polymorphic transformations, structural phase transitions, intramolecular interactions, and second-order phase transitions. However, even if such changes are absent, temperature-dependent IR spectra may reveal alterations in the band intensities, positions, shape, and width [26]. Structural deformation by pressure or temperature usually results in small changes in interatomic distances, significantly altering molecular and lattice vibrations that can be monitored using FTIR and Raman spectroscopies [27]. In addition, changes in the crystal structure usually shift the molecule center symmetry and may also lead to changes in intermolecular forces and thus band frequencies [28].
According to Hooke’s law, the decreased nearest neighbor distances imply larger force constants and higher frequencies. For crystalline forms, lattice potentials are partially anharmonic; the mean interatomic distances and forces depend on the level of vibrational excitation [29]. Thus, transitions between higher levels are less energetic than transitions between lower levels, which causes a redshift of band maxima with temperature and band broadening [29]. As a rule, the stronger the intermolecular interactions, the more significant the shift. Combined internal and external mode bands move faster with temperature [26]. According to Tielens and Allamandola [30], absorption features sharpen with temperature from the crystal lattice thermal shrinkage, but the integrated absorption strength stays approximately constant [31]. The study of the temperature dependences of vibrational spectra provides information on the structure of a solid sample and, on a decrease in temperature, bands hidden or inactive at room temperature may be revealed. Different modes (e.g., stretching versus bending vibrations) in a material may behave differently upon cooldown since they may have different anharmonicity constants and interact with different phonons, which may account for the observed behavior of amorphous magnesium silicates [32].
Thus, the temperature dependence of band parameters in the IR spectrum of a solid has a fundamental nature that is mainly associated with the structural deformation of its crystal lattice. This effect has long been observed and studied in various minerals such as silicates, analogs of interstellar dust forsterite, hydrous silicates at 4 K and 300 K [33], and olivine and enstatite at 80 K and 300 K [32]. Mennella et al. [34] investigated the absorption coefficient per unit mass for amorphous and crystalline fayalite, crystalline forsterite, and two kinds of disordered carbon grains over the temperature range 24–295 K. They found that the shift magnitude was 1–5 cm−1 at the maxima in the region of 100–500 cm−1. The temperature effects on the IR modes in silicates are mainly due to the contraction of the material structure at low temperatures [34]. The reduced interatomic distance induces a more vital constant and a progressive shift to coupling to higher frequencies. Bowey et al. [35] studied some silicates (olivine, orthopyroxene, clinopyroxene) at 295 K and 3.5 K. A shift of the bands with temperature was also found. Of particular note is the work by Johnston et al. [36], in which low-temperature FTIR spectra were used in resolving dickite- and nacrite-like features present in the spectra of kaolin clays when cooled to <30 K. These features were not resolved at room temperature and only partially resolved at liquid nitrogen temperature (77 K). The room-temperature and low-temperature positions of the n(OH) bands of kaolinite, dickite, and nacrite were linearly correlated with the interatomic OH–O distances; this relationship served for polytype/disorder identification. An increase in the thermal energy of a solid can provide information on the degree of anharmonicity of the interaction potentials of atoms. For example, significant shifts in band positions for a given temperature change indicate a softer, more anharmonic interatomic potential. These types of effects have been studied in temperature-dependent IR bands and width trends in apatite and kaolinite [37].
Thus, temperature dependences of IR spectra are essential for many reasons. First, the spectra in different phase states of a substance can be compared, thereby clarifying the role of intermolecular forces. Second, the bandwidth and spectral resolvability are temperature-dependent; hence temperature measurements improve the chemical analysis. Finally, frequency correlations require temperature-dependent band shifts and intensity changes to solve some theoretical problems [38].
For HS, the temperature dependence of IR spectra has hardly been studied, especially in the region where there are no significant molecular structure changes. The early paper [25] studied the moisture effects on IR spectra using a KBr transmission technique. There are studies devoted to the change in the molecular structure of HS during heating to simulate a fire [39]. HS IR spectra are measured upon heating from 325 °C [40]. Decarboxylation was found as the primary dehydration reaction from 150 to 400 °C, and anhydride was identified in HS from the FT-IR spectra at a higher temperature region, from 200 to 400 °C [41]. Along with other methods, IR spectra at 20–800 °C were obtained to study the temperature effects on the stability of soil humic acids with main features only (OH stretching at 3450 cm−1, methylene bands C–H at 2920 cm−1 and 2850 cm−1, the C=O stretching vibrations due to carboxylic groups at 1720 cm−1, the band at 1620 cm−1 due to C–C bonds) [42].
This work aims to demonstrate the capabilities of temperature-monitored IR spectroscopy for studying HS structure, both organic matter and mineral component composition. The IR spectra of several HS of leonardite were studied upon continuous heating from 25 to 215 °C; all the experiments (temperature dependence, centrifugation, and matrix assignment) were used previously but never combined.
2. Materials and Methods
2.1. Samples
Humic acid sodium salt (technical grade, Sigma-Aldrich Inc., St. Louis, MO, USA, CAS Number: 68131-04-4) was used. Malcolm and MacCarthy [43] showed that this HS is similar to leonardite humic acid and Wyoming dopplerite. Other samples are Powhumus (potassium humate) from German leonardite (HUMINTECH, GmbH, Grevenbroich, Germany) and Potassium humate from Sakhalin leonardite. These samples are further referred to as Sigma-Aldrich, Powhumus, and Sakhalin from this point on. Kaolin came from the Polog deposit (Ukraine, 97% of kaolinite in a purified sample) was used for comparison. The samples were not purified and used in their native state. All the samples were measured in five replicates. Bulk Si and Al in the studied samples were previously estimated by ICP-AES [22].
2.2. FTIR Measurements
The analysis by IR spectroscopy was carried out on a Vertex 70 IR Fourier spectrometer (Bruker Optik GmbH, Ettlingen, Germany) with a room temperature DLaTGS detector. A GladiATR attachment of a single attenuated total internal reflection (ATR) with a software-controlled diamond crystal heated in the range from room temperature to 215 °C (Pike Technologies, Madison, WI, USA) was used. The parameters of measurements are summarized in Table 1. The spectrometer and the attachment were purged with air with a dew point of −70 °C (produced by a PG28L Purge Gas Generator, PEAK Scientific, Glasgow, United Kingdom) at a rate of 500 L/h. The ambient temperature was maintained at 23 °C with an allowable variation of ± 1 °C using an air conditioner. The post-registration process was performed using OPUS 8.2 software (Bruker Optik GmbH, Ettlingen, Germany).
Before recording the spectra with heating, an empty ATR crystal spectrum was recorded at 25 °C as a background. Then, a small amount of the sample was placed on its surface, pressed with a screw, and a program of controlled heating was started at a rate of 0.25 °C/min from 25 °C to 215 °C; spectra registration step is 2.5 °C. After heating to 215 °C, the sample was cooled to 25 °C in the same manner at a 0.25 °C/min rate. The resulting heating and cooling spectra were combined, a corresponding array of spectra of an empty ATR crystal was subtracted from it, an ATR correction was performed, and the maxima positions were determined. The sample was in the ambient atmosphere during the measurement.
Extended ATR correction [44] was performed using the following conditions: ATR crystal diamond, radiation incidence angle 45 degrees, number of ATR reflections, 1. After ATR correction, a 13-point smoothing procedure was performed. Baseline correction was not performed. Band maxima were determined using the standard band search method using the OPUS software. The standard method is finding the x-position of the interpolated maximum or minimum. The intensity is the corresponding y-value of the interpolated maximum or minimum. The sensitivity parameter was 5% (in some cases 1%) to 20%. The higher the threshold is, the fewer bands are displayed.
3. Results
The IR spectra of the studied HS samples are shown in Figure 1; more details are given in the Supplementary Materials (Figures S1–S11, see below). The band frequency shifts are visible, discussed below. The spectral series also show a rather complicated and multidirectional change in the signal amplitude, i.e., the total absorbance of the spectral bands; however, in the case of ATR measurements, it depends in a complicated way on the refractive index of the sample and crystal, which also complicatedly depend on temperature and vary nonlinearly. Therefore, the reasonable use of frequency changes and amplitude requires separate measurements beyond this work scope.
To assess the reversibility of the observed changes, we recorded not only the spectra upon heating but also cooling the samples and compared the spectra before heating and after cooling back to 25 °C (Supplementary Materials, Figures S9–S11). The main differences are water absorption; its content has significantly decreased after the heating–cooling cycle, as expected. The most significant change in all three samples upon heating is the disappearance of the broad band in the region of 2400–3800 cm−1 due to water evaporation. No new bands appeared in the spectra.
Other band–intensity ratios showed insignificant changes during heating and cooling, which indicates the absence or insignificance of changes in the molecular structure of the studied samples. This behavior agrees with thermogravimetric data: dehydration (temperature interval I), from 70 to 110 °C; degradation of aliphatic structures and polysaccharides (interval II), from 210 to 380 °C [45,46]; DSC under oxidative conditions in combination with IR analysis of samples after heating [47,48]; and combinations of TGA, DTA, and DSC [49,50,51]. Some authors point to the possibility of hydroxyl elimination in the temperature range up to 200 °C under air conditions for potassium humates extracted from coal [52,53]. However, even in this case, the major changes in the HS molecular skeleton begin at significantly higher temperatures. Hoffmann et al. [54] used TGA, DTA, and DSC to study, in particular, a Sigma-Aldrich sample. They noted that the decomposition of polysaccharides, the elimination of functional groups, and the subsequent destruction of phenolic compounds occur in the range of 200–400 °C. Boguta et al. [55] mentioned that the primary process at temperatures below 200 °C is dehydration. However, a small contribution from the organic-carbon decomposition processes is also possible in the 40–220 °C range. Thus, it was worth noting that exothermic processes could slightly overlap endothermic effects.
3.1. Post-Registration Processing of IR Spectra
The post-registration processing algorithm may considerably change the spectrum. Therefore, all samples were processed in a strictly identical way to minimize the effect of mathematical processing on the results. For clarity, all post-registration processing stages are illustrated with spectra in the Supplementary Materials (Figures S1–S8). The spectra of the samples after processing are shown in the main text.
Extended ATR correction was used. However, since the ATR crystal absorption is close to 100% in the region 2400–1900 cm−1, this region is significantly noisy (Figure S1, Supplementary Materials). Additionally, it does not contain absorption bands for the studied HS samples. Therefore, this region is replaced by a straight line to apply the extended ATR correction procedure correctly.
The ATR refractive index is determined by the crystal material and the accessory design (the angle and the number of reflections). Additionally, the average refractive index of the sample must be known, and this value, on the one hand, significantly affects the corrected spectrum shape; on the other hand, it is not known precisely for the test sample. There are no data of the sample refractive index used for ATR correction in humic substances in the literature.
For most organic materials, a sample refractive index of ca. 1.5, being a good starting point, is recommended as an ATR-correction base value and generally provides excellent correction performance [44]. Some materials, such as samples rich in amorphous carbon such as high carbon-black rubbers, have higher indices, and a value of 2.0 is recommended for these samples [56]. Thus, it is logical to expect an optimum refractive index for HS between these values. According to the literature, the HS refractive index in the visible region is 1.6 [57], 1.60–1.65 [58], or 1.63–1.70 [59]. Therefore, we varied the sample refractive index for ATR correction in the range of 1.5–2.0 on the example of the Powhumus sample (Supplementary Materials, Figures S3 and S4; the initial spectrum is shown in Figure S2). Figure S2 shows that the corrected spectra at n = 1.5, 1.6, 1.8, and 1.9 are identical. At n = 2.0, the spectrum undergoes significant distortions. At n = 1.7, the spectrum also differs from other corrections, mainly in the band intensities. Besides, in regions of 4000–3800 cm−1 and 1900–1700 cm−1, where a strong distortion band caused by light scattering is observed, a value of 1.7 provides the highest degree of its compensation. Based on these experiments and the literature data, we have chosen n = 1.7 for the ATR correction of all HS spectra in this study.
The spectrum of kaolinite in the region of 3720–3600 cm−1 recorded under the same conditions and on the same equipment as the HS samples compared with the HS spectra is shown in Supplementary Materials, Figures S5–S8. In this case, the initial ATR spectrum of kaolinite was corrected using two refractive indices, 1.5, which is more justified for this mineral, and 1.7 for a correct comparison with HS spectra. For n = 1.5, the obtained values of the band maxima at 3691.0, 3669.5, 3652.3, and 3620.0 cm−1 coincide entirely with the position of the kaolinite bands at a temperature of 305 K [36]. For n = 1.7, the difference is observed only for the first band and produces a shift to 3692.0 cm−1.
All spectra after ATR correction are presented as absorption vs. wavenumber. In this case, the absolute value of absorption in ATR depends on many factors, including the particle size, and is not of fundamental importance. After ATR correction, a smoothing procedure was performed. Such processing turned out to be necessary since small noises and, especially, weak water vapor bands introduced an error in some cases in determining the band maximum and were not cut off by the maximum-finder algorithm. The band maxima were determined before and after the smoothing procedure to assess its effect on the band position and general trends. The fluctuations of the maxima (and, thus, the relative band displacements) decreased significantly upon smoothing, but all the change tendencies were preserved. The temperature dependence of the maximum frequency of bands is nonlinear. Baseline correction was not performed since this operation introduces an additional difficult-to-control distortion in determining the exact maximum position.
3.2. General Description of IR Spectra
The exact positions of the maxima, the wavenumber shifts in band maxima caused by heating, and the band assignment are given in Table 2. It sums up the maxima at 25 °C, shifts when heated to 215 °C, and the difference between maxima at 215 and 25 °C. A positive difference indicates a redshift (towards higher wavenumbers); a negative, a blueshift (towards lower wavenumbers).
The mid-IR region bands commonly associated with characteristic HS and soil attributes drop into three regions: 1200–400 cm−1, 2000–1400 cm−1, and 4000–2000 cm−1 [71] corresponding to inorganic soil matrix, organic matter, and hydrogen-bond bands, respectively. In our previous paper [72], we divided the whole mid-IR region for silicate soils in a slightly different way: matrix fingerprint region, 1100–400 cm−1; matrix overtone/combination band region, 2500–1100 cm−1; and OH-region, 4000–2500 cm−1. Both divisions roughly serve the same purpose, but the chemical speciation of various samples shows that the region selection should be more detailed [72].
In this paper, we divided the mid-IR region 4000–260 cm−1 into even in more detail—six regions to divide inorganic components and organic matter. Namely, these regions are the quartz lattice region (800–260 cm−1); quartz overtone region (1270–800 cm−1); humic substance organic matter (HSOM) region (1780–1270 cm−1); quartz combination region (2800–1780 cm−1); CH-speciation region (3100–2800 cm−1); and hydrogen-speciation region (4000–3100 cm−1). These regions were selected to contain the dominating type of bands and be readily and reproducibly dividable in the same manner for all the similar samples, and although some boundaries (e.g., 1260 and 1780 cm−1) may be provisional, they are still generally reliable.
3.3. Primary Band Assignment
In the following description, we use the direction from low to high energies (low to high wavenumbers). The dominating calibrations in each region go from fundamental matrix vibrations to variable amorphous and organic components and then to speciation due to surface groups and coordination (Figure 1).
In the quartz lattice region (800–260 cm−1), most bands belong to quartz lattice, 797 cm−1, O–Si–O stretch, 697 cm−1 (probably, overtone ≅ 2 × 398 cm−1), 535 cm−1 (probably, overtone ≅ 2 × 265 cm−1), 510 cm−1, Si–O–Si bend; 460 cm−1, 450 (shoulder) cm−1, 430 cm−1, 420 cm−1, and 410 cm−1, O–Si–O bend [61,70]. Bands at 750 cm−1 and 455 cm−1 can be attributed to amorphous silica [61]. Broad bands with maxima at 640 cm−1 and 610 cm−1 are probably the manifestations of water librations [73]; they are present in kaolinite and quartz (noisy in the latter case) or AlO bending [68]. Bands at 775 cm−1 and 715 cm−1 are absent in kaolinite spectra. The band at 715 cm−1 cannot be reliably attributed and may manifest in-phase rock vibrations of AlO vibrations [68] or C4+ alkanes [69]. The band at 775 cm−1 may belong to aromatic CH out-of-plane bending [20].
The quartz overtone band region (1270–800 cm−1) contains a broad, weak band at 1260 cm−1, which could be a quartz combination band (≅460 + 797 cm−1) or Aryl–O stretching of SOM [74]; broadband at 1120–1070 cm−1, O–Si–O stretch in crystalline and amorphous SiO2 species; bands at 1035 cm−1 and 1010 cm−1, quartz lattice O–Si–O stretch; 1115 cm−1, 937 cm−1, and 915 cm−1 (OH deformation of inner-surface hydroxyl groups of kaolinite [61]). The latter band is also the manifestation of the O–H bend of carboxylic species [61].
Other SiO vibrations are amorphous SiO2 O–Si–O stretch (not present in quartz); 915–912 cm−1, amorphous silica, probably overtone ≅2 × 460 cm−1 [75] or (O–H bend [76]); 890 cm−1, Si–O– [64], Si–O–Si bridge [65], or probably polyaromatic bend vibrations. Weak bands at 855 cm−1 and 845 cm−1 may belong to Al–OH [77]. Comparing the spectra of HS samples with kaolinite spectra in 1270–260 cm−1 (Supplementary Materials, Figures S5–S8) reveals that bands at 1115 cm−1, 937 cm−1, 915 cm−1, 797 cm−1, 750 cm−1, 697 cm−1, 535 cm−1, 460 cm−1, and 430 cm−1 correspond to kaolinite, as well as bands at 1035 cm−1 and 1010 cm−1, although they are displaced more strongly in HS samples than other bands in kaolinite. Bands at 1164 cm−1, 1085 (1089) cm−1, and 875 cm−1 are absent in kaolinite spectra. The 1164 cm−1 band corresponds to C–OH stretching of aliphatic O–H, 1085 cm−1, to alcoholic or polysaccharide C–O stretch and O–H deformation, or an Si–OH bend in silicate impurities [62].
Bands at 875 cm−1 and 840 cm−1 (weak shoulder companion of the previous band; in all three samples studied, the intensities of these two bands are correlated) are attributed to silicate species by some authors (Si–O– [64] or Si–O–Si bridge [65]), characteristic of large amounts of defects in crystalline quartz, usually in artificial samples [78,79,80]. However, these band intensities do not correlate with bulk Si and Al in the studied samples by ICP-AES [22], which may prove that they are not related to silicates and aluminosilicates. Therefore, this band may also be attributed to carbonate [60] or calcite [66], as previously reported for various silica-containing samples. An alternative to this attribution is HSOM, most probably polyaromatic bend vibrations. However, the band at 875 cm−1 is significantly more intense than those attributed to aromatic compounds [67].
In the HSOM region (1780–1270 cm−1), the bands at 1730 cm−1, 1720 cm−1, 1700 cm−1, 1695 cm−1, 1685 cm−1, 1650 cm−1, 1640 cm−1, 1620 cm−1, and 1550 cm−1 correspond to an aromatic C–C stretch. Bands at 1750 cm−1, 1730 cm−1, and 1720 cm−1 (shoulder) are attributed to carbonyl/carboxyl C=O [42]. The band at 1645–1640 cm−1, the shoulder band, is the bend (v2) of the covalent bonds of liquid absorbed water [81]. The complex, broad band at 1574/1569 cm−1 is an antisymmetric carboxylate stretch, with contributions from hydrogen-bonded SiOH…H2O HO–H stretch (amorphous) [82,83,84,85]. A broad band at 1395/1380 cm−1 is the symmetric carboxylate stretch [20]. Other bands are 1470 cm−1, 1450 cm−1 (shoulder), O–H, C–H scissoring, antisymmetric methyl bend [20,65]; 1370 cm−1, symmetric methyl bend; and 1310 cm−1 (shoulder), probably carboxyl C–O stretch [20].
In the quartz-combination-band region (2800–1780 cm−1), primary bands are 2650 cm−1, carboxylic O–H stretch [86], and 2430 cm−1, probably, amines. In addition, residual bands of CO2 at 2370 cm−1 and 2350 cm−1 are visible.
The CH-speciation region (3100–2800 cm−1) is comprised of bands at 3020 cm−1 (3020–3100 cm−1), alkene/aromatic sp2 =CH2 stretch; 2970 cm−1 and 2880 cm−1, the antisymmetric and symmetric stretch of methyl groups; 2925 cm−1 and 2850 cm−1, the antisymmetric and symmetric stretch of methylene groups [18,62]. Bands of CHx groups are on the shoulder of the OH continuum band. A broad band of 2400–3700 cm−1 with a maximum of about 3370 cm−1 is a signature band of OH vibrations of water with different hydrogen bonds. Additionally, in this region, vibrations of the OH group in –COOH (3400–3200 cm−1) and –C–OH are present [16]. Thus, we can consider the bands at 2925/2919 cm−1, 2854/2850 cm−1, 1574/1569 cm−1, 1395/1378 cm−1, 1164 cm−1, and 1085/1089 cm−1 and probably 875 cm−1, 775 cm−1, and 715 cm−1 mainly assigned to the organic matter of HS samples.
The hydrogen-speciation region (4000–3100 cm−1) shows several bands. The bands at 3665 cm−1 and 3650 cm−1 (Figure 1) are weak in Sigma-Aldrich and Powhumus samples, absent in the Sakhalin spectrum. Although they are identified in the spectra at 25 °C, upon heating, their intensities become comparable to the noise, and it is difficult to determine the exact maximum position. A comparison with kaolinite (Supplementary Materials, Figures S5–S8) shows that all four bands in 3720–3600 cm−1 in the Sigma-Aldrich and Powhumus samples refer to kaolinite fragments; the spectra coincide in the series of bands, the ratio of their intensities, and the maximum wavenumbers. Johnston, Kogel, Bish, Kogure, and Murray [36] showed that a more in-depth study of the structure of these fragments requires cryogenic conditions, which was beyond the scope of this work. In this region, the bands are much more broad and less intense in the Sakhalin spectrum than kaolinite bands, and two of the four bands are visible. Other bands in the hydrogen-speciation region are 3710 cm−1 (unbonded SiO–H stretch, tilted (kaolinite, clay) [82]; 3700–3680 cm−1, hydrogen-bonded SiO–H…H2O stretch (amorphous species); 3665 cm−1, 3655 cm−1, 3645 cm−1, 3635 cm−1, and 3610 cm−1 non-silicate OH; 3650 cm−1 and 3630 cm−1, isolated SiO–H in quartz [60,61]; and 3400 cm−1 and 3290 cm−1, condensed-phase antisymmetric and symmetric hydrogen-bond ensembles.
3.4. Temperature Changes
As a review of existing data shows, a shift in the frequency of the bands with temperature arises if we exclude the cases of phase transitions or chemical changes when the length of bonds changes, which is usually caused by deformation of the crystal lattice (including deformation due to a temperature change) [26,27,28,29,30,31]. As a multicomponent mixture, HS does not form a crystal lattice, which is consistent with the X-ray diffraction spectra, in which there are no HS reflections. HS is predominantly in solid form under natural conditions and is associated with clay minerals [87]. Thus, the behavior of HS either indicates the formation of local structured fragments, which nevertheless are not found in the XRD spectra, or remnants of mineral fragments, the bands of which are hidden by broad bands of HS. The situation is complicated by the lack of a theoretical model, even for simple crystalline phases, which would make it possible to predict such a frequency shift or relate it to the lattice parameters. The work by Bronnikov et al. [88] deserves special attention. Several polymers were studied, and a change (decrease) in the band frequency is interpreted as stretching of the macromolecule skeleton (an increase in the C–C–C bond angles and the lengths of the valence bonds in the skeleton). The magnitude of the band displacement is related to the relative thermal elongation of the skeleton.
Thus, with a high degree of confidence, temperature-induced changes in the frequency position in the HS spectra indicate the presence of ordered fragments in which phonon vibrations can arise.
3.4.1. Quartz Lattice Region (800–260 cm−1)
Most bands in this region are fundamental lattice vibrations of quartz and other structured components of the inorganic matrix. The lattice bend vibrations of Si–O–Si (510 cm−1) and O–Si–O (460 cm−1, 450 cm−1, and 430 cm−1) do not show any shifts (Figure 2) following their behavior of first-order fundamental vibrations [89]. On the contrary, lattice O–Si–O stretch vibrations at 797 cm−1 (Figure 3) exhibit a redshift of 3 cm−1, smaller than shifts of 1–2 cm−1 of bands in the region 1250–800 cm−1.
The band at 535 cm−1 experiences a redshift, probably indicating a second-order (overtone) band of the primary lattice vibration at 263 cm−1. The same can be assumed for the intense signature band at 697 cm−1 as it has a counterpart lattice vibration at 398–400 cm−1 [80]. Bands of 535 cm−1 and 460 cm−1 are red-shifted compared to kaolinite spectra, while the band of 455 cm−1, the principal band in kaolinite, is either insignificant or much lower in intensity in HS.
The bands at 775 and 750 cm−1 do not belong to quartz lattice frequencies, and they do not shift, which is clear from comparing with neighboring quartz bands. Likewise, the band at 715 cm−1 is not attributed to any lattice bands of quartz or silicate minerals, and its position does not change with temperature so that it can be the manifestation of OM, in-phase rock CH2 vibrations of C4+ alkanes [69], or polyaromatic entities [67].
A rather broad band at 700–500 cm−1 with main components at 640 cm−1 and 610 cm−1 is attributed to water librations [73]. Its intensities decrease with temperature (Figure 3), which contrasts with the behavior of other bands in this region. Both bands disappear at temperatures of 75–80 °C. This behavior is in league with water bands at 1640 cm−1 and 3690 cm−1 and the hydrogen-bond ensemble (see Section 3.4.3 and Section 3.4.5 below) attributed to loosely bound water. Water absorption decrease with temperature reveals a weak band at 670–665 cm−1, which does not shift with the temperature and may be attributed to CO2. A band at 425 cm−1 can be attributed to C–C vibrations, but this is rather spurious due to its low intensity and only reliable appearance in the Sigma-Aldrich sample.
3.4.2. Quartz-Overtone-Band Region (1270–800 cm−1)
All bands in this region experience a redshift with similar parameters (Figure 4) except for the carbonate band at 875 cm−1 (Figure 5). Following the literature [32], these are overtone lattice bands in α-quartz shifting due to changing lattice parameters [90]. Broad bands at around 1030 cm−1 (Figure 6) and 1120–1070 cm−1 (Figure 7) can be considered the total SiO2 components as they are characteristic of both amorphous and crystalline species [32,68,80,90,91]. However, the band behavior at 1030 cm−1 is similar to that of 1010 cm−1.
However, the band at 1120–1070 cm−1 is absent in kaolinite, so it may be partially attributed to HSOM (in-plane C–H bending of non-aromatic and cellulose species [92]). Still, if we follow the concept of temperature-independent non-quartz vibrations, no such components are found in this broad band. It can be deconvoluted into two components at 1110 cm−1 and 1070 cm−1 that experience similar and synchronous redshifts, so they must be attributed to quartz overtone components. This band is roughly the same as the dual structure of temperature experiments for quartz samples. The carbonate band at 875 cm−1 and its companion at 840 cm−1 is visible at low temperatures (Figure 4), less affected by water bands, and does not decrease at the end of the temperature cycle.
3.4.3. HSOM Region (1780–1270 cm−1)
All bands except for 1380 cm−1 (Figure 8) and 1580 cm−1 (Figure 9) are temperature-independent, which is evidence that they are not overtone counterparts of matrix vibrations of quartz and belong to isolated group vibrations. The pair of 1580 cm−1 and 1380 cm−1 (antisymmetric and symmetric carboxylate stretches, respectively) is dominated by combination vibrations [93,94] and behaves as expected (such as hydrogen-bond ensembles), Figure 10. The antisymmetric band exhibits a blueshift, while the counterpart symmetric band shows a redshift. The antisymmetric band blueshift is significantly higher than the redshift in the symmetric vibration, which may be accounted for the effect on the high-wavenumber edge of this band from decreasing water amount with temperature. The latter fact is confirmed by the change in the slope of the temperature change of 120 °C.
The absence of temperature-dependent vibrations around 1700 cm−1 at low temperatures shows that HS carboxylic groups are mainly carboxylate species. Such significant shifts in carboxylate bands show some similarity with Si–OH bands in the hydrogen-speciation region (see below, Section 3.4.5). The series increasing bands at 1750 cm−1 and 1730–1720 cm−1 (carboxylic C=O) with the temperature (Figure 9) may be evidence of increasing amounts of the carboxylic acid species, which is confirmed by the correlated increase in the band at 1260 cm−1 characteristic to carboxylic C–OH. Additionally, other small increasing bands at 1700 cm−1, 1695 cm−1, 1685 cm−1, 1650 cm−1, 1640 cm−1, and 1620 cm−1, which are present for all HS samples, could be different carboxyl anions appearing at high temperatures as well.
The water band position at 1640 cm−1 at the shoulder does not prevent the analysis of the principal band. The position of the center of the broad band of water around 1645–1640 cm−1 may be evidence of the loosely absorbed water [95]. The temperatures at which the band at 1640 cm−1 decreases are not synchronous with other changes in this area, whereas they are similar to the behavior of the bands in the water libration area (700–500 cm−1, Figure 3), hydrogen-bond ensemble, and Si–OH bands in the speciation region (see below).
3.4.4. Quartz-Combination-Band and CH-Speciation Regions
All bands are temperature-independent in the quartz-combination-band region (2800–1780 cm−1), Figure 11. All four signature bands in the CH-speciation region are stable as expected for first-order isolated vibrations [74]. The estimation ratio of methylene-to-methyl shows 2:1, provided the absorption coefficient is the same [96]. Alkene/aromatic CH2 bands at 3100–3030 appear clearly in the whole region, but the whole structure is more resolved at temperatures over 85 °C. The decrease of the hydrogen-bond ensemble can account for this behavior. The intensities of these bands start to disappear at temperatures over 150 °C due to the volatilization of aromatic components. Small blue and redshifts, respectively, are observed for asymmetric and symmetric C–H stretching of aliphatic groups as expected (Table 2).
3.4.5. Hydrogen-Speciation Region (4000–3100 cm−1)
The band at 3692 cm−1 (3700–3680 cm−1) shows a complex behavior with a general trend in the energy increase (Figure 12). This band experiences the most significant redshift by modulus (by 10 cm−1 for Powhumus and Sigma-Aldrich) among all the spectrum bands. The trend can be divided into three parts, a nearly stable band position (from 20 to 55 °C), a sharp twofold decrease in intensity with a redshift by ca. 2 cm−1 from 55 to 105 °C, and a monotonous redshift in the band maximum from 105 to 250 °C, without a change in the band intensity. In addition, the shift in the band at 3692 cm−1 is accompanied by the appearance of red-shifting weak bands at 3710 cm−1 and 3715 cm−1 that can be attributed to unbonded SiO–H stretch tilted vibrations in amorphous silica [82].
A band at 3620 cm−1 experiences a mirror-like blueshift behavior synchronous with the band at 3692 cm−1, although the whole shift is 5 cm−1. Both bands at 3700–3680 cm−1 and 3620 cm−1 are observed in kaolinite and attributed to amorphous and quartz SiO–H bands [82]. Noteworthy is that the data on the bulk species of these bands (crystalline or amorphous) are contradictory [82], although all the authors claim these bands belong to hydrogen-bonded vibrations (Figure 13). According to the behavior of these bands, they are interconnected, and the initial part of the plots shows that they are attributed to loosely bound molecular water, which results in the initial slow shift followed by the more tightly bound water, which results in its loss from the amorphous silica matrix and more substantial shifts at higher temperatures.
The band at 3695 cm−1 seems unusual as dehydration results in the redshift despite expected loss in coordination and a blueshift [97]. This feature may be accounted for a change in coordination rather than isolation due to hydration. Another reason for such a behavior can be the ionic character of the related species so that the dehydration results in a more organized character of these bonds.
Bands at 3655 cm−1, 3645 cm−1, 3665 cm−1, 3635 cm−1, and 3610 cm−1 are assigned as OH of alcohol/phenolic species [74]; the latter three are equal in intensities, which slightly increase with temperature and do not shift. Therefore, they are not coincident with the bands attributed to pure kaolin samples. On the contrary, a change in the region of 3660–3640 cm−1 cannot be attributed to a simple shift. This behavior is different for all three samples due to the formation of isolated OH species from the neighboring species, resulting in a rather acute redshift by 5 cm−1.
Condensed-phase antisymmetric (3400 cm−1) and symmetric (3290 cm−1) hydrogen-bond ensembles behave as predicted: the energy of antisymmetric vibrations increases and the energy of symmetric vibrations decreases [98] monotonously.
3.5. Differences between HS Samples
The samples have almost the same set of prominent bands, differing mainly in their intensity ratios. The behavior of all quartz and silica bands for all the samples is the same; all the signature bands of quartz and kaolinite are attributable; the difference is only the relative intensity or the shape (band or shoulder). No significant differences between the samples were found in the quartz-combination-band (2800–1780 cm−1) and CH-speciation (3100–2800 cm−1) regions.
The bands at 715 cm−1 and 425 cm−1 are reliable only in Sigma-Aldrich; the CO2 band at 667 cm−1 is much broader in Powhumus and is very weak in the Sakhalin sample. In the Sakhalin sample, the band with a maximum at 875 cm−1 is absent; in the rest of the samples, it differs in intensity much more strongly than the other bands. The bands at 875 cm−1 and 840 cm−1 in Sigma-Aldrich are more intense than in Powhumus and Sakhalin (barely identified in the latter sample).
All the primary and minor bands in the HSOM region are the same for all three samples, but the intensity ratio for carboxylate bands of Powhumus and Sakhalin is different from Sigma-Aldrich. This difference may result from different HA counterions, potassium for the former two, and sodium for Sigma-Aldrich. This ratio inversely correlates with the band at 875 cm−1. The symmetric carboxylate stretch band for Sigma-Aldrich does not experience a significant shift with temperature.
The bands at 3665 cm−1, 3655 cm−1, 3645 cm−1, 3635 cm−1, and 3610 cm−1 are the same for all the samples; however, the behavior of the broad band at 3700–3680 cm−1 is somewhat different. Comparing the three HS samples shows that this band is a poorly resolved or unresolved band with maxima at 3685 cm−1 and 3695 cm−1. It is not apparent in Sigma-Aldrich and Powhumus but is seen in the Sakhalin sample and confirmed (though with a high overall error) by centrifuged samples.
Thus, the overall anomalously large redshift is apparent. A decrease in the band at 3695 cm−1 and an increase in the band at 3685 cm−1, along with an intensity decrease in the hydrogen-bond ensemble band edge, visually shifts this band to lower wavenumbers.
3.6. Uncentrifuged and Centrifuged Samples
The decrease in intensities of most quartz lattice bend vibrations, which are removed upon centrifugation (see Figure 1 and Figure 14), is shown, while the bands corresponding to the generic SiO2 vibrations or amorphous silica (1030 cm−1, 915 cm−1, 797 cm−1, 697 cm−1, 535 cm−1, and 465 cm−1) remain. This decrease is confirmed by changes in intensities of other bands attributed to crystalline/amorphous SiO2 (see below, Section 4.1.3). As well, in the quartz-overtone-band region, most bands attributed to crystalline quartz disappear after centrifugation, while SiO2 bands characteristic of amorphous species remain. The intensity of bands that may be attributed to polyaromatic compounds significantly decreases after centrifugation (reliably confirmed for Powhumus HS only).
Centrifugation does not change the overall picture but reveals the band complex structure at 1590 cm−1. Thus, probably partly, this band contains hydrogen-bonded amorphous SiOH…H2O HO–H stretch. The broad carboxylic O–H stretch is present in both uncentrifuged and centrifuged samples, which may be proof of its nature. The band is weak, but its intensity increases along with other carboxylic bands in increasing temperature. A broad and weak band at 2430 cm−1 is also present in both samples, but its assignment to amine vibrations is not proven due to its low intensity.
No significant differences except for the spectra of centrifuged samples at high temperature reveal fine structures of bands (3020–3100 cm−1) that may be attributed to aromatic stretch and confirm other bands attributed to aromatic species. Hydrogen-speciation region (4000–3100 cm−1): all SiO–H bands significantly decrease in intensity; the bands at 3665 cm−1, 3655 cm−1, 3645 cm−1, and 3635 cm−1 remain approximately the same.
As a whole, the comparison of the temperature behavior of centrifuged and uncentrifuged samples shows that the effect of hydrogen-bond ensemble affects the region up to 3700 cm−1 more or less monotonically (a constant decrease with the temperature, while the edge of this continuum decreases significantly, which may affect the shape and behavior of the bands at 3700 cm−1.
4. Discussion
4.1. Components
4.1.1. HSOM
Thus, the comparison of spectra of different samples and in the studied temperature range shows that aliphatic compounds/groups are represented by all signature bands. They are methylene and methyl stretch at 3000–2800 cm−1 (Figure 11), CH2 scissoring at 1460 cm−1 (Figure 8), probably CH2 wagging/twisting at 1310 cm−1 (Figure 8), and in-plane bend vibrations at 715 cm−1 (Figure 3). All bands attributed to aliphatic compounds/groups are temperature-independent.
The presence of aromatic compounds is possible due to the existence of almost all characteristic bands: (1) –C=C– stretch [99] at 1540–1560 cm−1 (Figure 9); (2) substituted/polyaromatic bands at 1660–1700 cm−1 (Figure 9), and (3) the band of =C–H stretch at 3020 cm−1 (Figure 11). In addition, aromatic in-plane =C–H bend in 1090–1060 cm−1 may contribute to the broad complex band at 1110–1070 cm−1 (Figure 7). All bands attributed to aromatic compounds are temperature-independent. An exciting feature of aromatic bands is their appearance after eliminating the loosely bound molecular water (3400–3000 cm−1, 1660–1630 cm−1: Figure 9, and 650–600 cm−1: Figure 3.) followed by a synchronous decrease (though relatively small) in the band intensities upon temperatures of 150 °C. As aromatic compounds are stable at the used range of temperatures, such behavior may evidence the volatilization of aromatic compounds, i.e., low-molecular entities containing aromatic compounds. Such behavior was shown for all three HS samples for 3400–3000 cm−1, while changes at 1660–1630 cm−1 were only reliably found for Powhumus.
Concerning functional groups, as expected, HS is dominated by the carboxylate bands, which form an antisymmetric/symmetric pair. According to the existing studies [100], the maxima of HSOM carboxylate bands correspond to α-substituted aliphatic carboxylic acids and possibly halogen-substituted and aromatic carboxylic acids. The temperature behavior shows the appearance and synchronous growth of a series of nearly located bands at 1740–1300 cm−1 (Figure 8 and Figure 9) that can be considered isolated unbonded carboxyl species, though they also manifest aliphatic and aromatic group vibrations. No other functional groups can be reliably attributed.
The absence of the carboxylic acid band at 1700 cm−1 and two broad dominating carboxylate bands shows that the latter is the major carboxylic species in these samples. The presence of carboxylic O–H fully confirms the low-energy-range bands. The aliphatic-to-aromatic ratio in the CH-speciation region (3100–2800 cm−1) can be estimated in principle, but this would result in relatively high uncertainty due to overlapping bands of aromatic bands with other constituents. At least four kinds of Si–OH groups and three types of water can be attributed in HS, summed up below (Section 4.1.3).
4.1.2. Water
Three types of water can be attributed to HS. First is loosely bound molecular water that dehydrates below 200 °C and is a minor component in the studied samples. The bands are 3400 cm−1 and 3290 cm−1 (Figure 11), the bend at 1640 cm−1 (Figure 9) [101], and, though with some doubts, librations at 640 cm−1. The bands decrease synchronously, insignificantly affect other bands, and do not depend on sample centrifugation.
Another type of water is tightly bound molecular water that dehydrates above 350 °C. This kind of water is bonded to surface –Si–OH groups. The 3740 cm−1 and 3660 cm−1 bands due to surface Si–OH appeared upon the dehydration of this type of water. These bands are absent in centrifugated samples. Finally, there is water tightly bound to HSOM, according to the behavior of weak bands at 3660–3640 cm−1; this type of water is not bound to OH groups and instead provides the coordination of the carboxylic groups. The same type of water is also expected for temperature changes in carboxylate bands and causes the appearance of C=O and C–OH bands. Significant shifts in carboxylate bands could result from two simultaneously acting factors: an intrinsic change in vibrations and the blueshift due to the decrease in the hydrogen bond coordination effect of carboxylate anion with water molecules [28].
4.1.3. Quartz Bulk
The behavior of SiO2 in HS is as expected [32], and all the bands attributed to quartz fall into three categories. First are fundamental frequencies of α-quartz, which are stable, temperature-independent, and coincident with natural α-quartz samples at 700–400 cm−1. Second, lattice O–Si–O stretch vibrations at 797 cm−1 exhibit a small temperature redshift due to changes in the lattice parameters. Finally, most of the intense combination and overtone bands of bulk quartz in the region 1100–600 cm−1 experience a significant redshift due to the changes in the lattice parameters and the theory of the second-order bands’ temperature behavior [90]. Along with a decrease in the bond length, the energy of the associated phonon increases. Frequency increases as pressure increases or temperature decreases, and therefore, the wavelength of the band must decrease. Tetrahedral Si–O bond lengths and bond angles are relatively incompressible compared to the spacings between the oxygen and metal ions and inter-tetrahedral oxygen atoms, so there is slight variation in the wavelength corresponding to the 10 mm stretching and 20 mm bending features with temperature (e.g., [32]). In contrast, the volumes of the divalent cation sites are more compressible. Therefore, one would expect to see a more significant change in the wavelengths of bands associated with the cation sites. Under the conditions of ATR experiments, most bands are fundamental or overtones, while the intensity of combination bands at 2000–1800 cm−1 seen in DRIFT [102] and PAS–FTIR [72] are not seen (Figure 1).
The reliably stable fundamental frequencies of quartz matrix and characteristic redshifts of the combination and overtone bands make it possible to distinguish signature bands of HS organic matter even at relatively low temperatures at such a heating experiment, meaning no severe sample degradation.
4.1.4. Quartz Surface (Si–OH) Groups
At least four kinds of Si–OH groups can be attributed: (1) Si–OH groups are located at the silica surface, and open structural defects in quartz are bound to water [97]. The band is at 3620 cm−1 at 20 °C and shifts to 3625 cm−1 with increasing temperature (Figure 10). Dehydration is the easiest among the three hydroxyl groups. (2) Si–OH groups close together to be hydrogen-bonded to each other. This group probably represents the absorption band at 3660 cm−1, which does not shift significantly with temperature. The organic OH groups can also cause this band. (3) Non-water coordinated Si–OH groups (overlapped band at 3695–3685 cm−1 with an anomalous behavior). The coordination of water most probably causes this band, and the changes with temperature result from the change in coordination. (4) Isolated surface Si–OH groups free of hydrogen bonding (the absorption band at 3715 cm−1). At 25 °C and on the increase to 215 °C and dehydration of loosely attached water molecules, the band grows gradually with a slight redshift to 3712 cm−1.
4.2. Comparison of HS Samples
The three samples are remarkably similar, and the majority of conclusions and assumptions are made by all the three samples and their comparison as a group with either quartz or kaolinite samples. Sigma-Aldrich, Powhumus, and Sakhalin samples have almost identical bands, differing mainly in intensity ratios. Mostly identical are quartz lattice fundamental vibrations and confirmed or probable overtones and bands corresponding to total SiO2 or amorphous species. In the HSOM region, all the samples show the same degree of ionization of HSOM acidic groups (carboxylate bands) and minor series of bands corresponding to carboxylic or aromatic bands and their temperature behavior. Additionally, the spectra show many similarities in (i) the types of bound water and the corresponding temperature behavior; (ii) methyl/methylene ratio and alkyl/aryl CH2 groups; and (iii) the behavior of the complex hydrogen-bonded bands at 3695–3685 cm−1 and 3620 cm−1 and series of minor bands corresponding to OH-groups in the hydrogen-speciation region. With the blueshift and the intensity decrease in the band at 3620 cm−1 (Section 3.5), we concluded that a single mechanism could cause this area’s behavior. In our opinion, a possible mechanism can be a change in the character of bonding groups upon heating, when they do not shift to unbound groups with a well-defined, strong blueshift but change from water-bonded to HSOM-bonded or neighboring-group bonded.
Still, these three HS samples have different features. The Sigma-Aldrich sample is different from two other HS samples by positions and the ratio of carboxylate anion bands at 1580 cm−1 and 1380 cm−1, a more pronounced band at 2650 cm−1, a stronger carbonate band at 875 cm−1, and fewer bands in the region below 450 cm−1. The intensity difference at 875 cm−1 and 840 cm−1 in Sigma-Aldrich compared to Powhumus and Sakhalin may be due to the counterion (Na+ and K+, respectively).
The Sakhalin sample is different from two other HS samples by (i) the maximum number of bands in all the regions, (ii) a different ratio of bands at 3690–3680 cm−1 (surface SiO–H groups), (iii) almost insignificant band at 875 cm−1, (iv) a different ratio and resolved character of the complex band at 3695–3685 cm−1, and (v) the resolved character of the carboxylate band at 1580 cm−1. Powhumus sample shows some similarities with either Sigma-Aldrich or Sakhalin samples. In the quartz and hydrogen-speciation regions, this sample is similar to Sigma-Aldrich, while in the HSOM region, to the Sakhalin sample.
4.3. Summary
Thus, the features of this approach can be summed up as follows. First, it provides a more detailed HS (and on its basis, probably soils) analysis without total HS decomposition or HSOM destruction. The temperature experiment agrees with the existing literature on quartz species [32,90,91,97,103,104,105] and provides the differentiation of crystalline quartz bands, amorphous silica, and HSOM/surface groups. As a bonus, some similarities in the behavior of bands attributed to quartz species may suggest attributing them to overtone or fundamental frequencies of quartz. Second, the temperature experiment combined with centrifugation may help differentiate crystalline/amorphous matrix species and HS species connected to them. Additionally, the used time-resolved heating mode similar to TGA differentiates water elimination, volatilization of HS, and coordination changes, though for the remaining solid samples.
The disadvantages of the proposed can be summed up as follows. First of all, there are possible deficits in the approach that other studies can overcome. There are many relatively spurious band assignments with double and triple choices of HSOM, matrix, and inorganic surface groups, and all these assignments may be doubted (except quartz and major bands) and thus result in incorrect statements regarding the sample composition. For instance, aluminosilicate and silicate species cannot be reliably distinguished due to spurious data. In this study, we double-checked them using both the general spectrum interpretation and heating behavior and bulk composition of corresponding elements (Si, Al). In our opinion, in all the cases, other methods such as XRD, NMR, AES, or chemical speciation (titration, ion exchange) should be used to correct the band attribution. The models other than quartz are needed to account separately for silicate surface groups, purified HSOM, and their potential interaction. No objective evidence of the chemical interaction of HSOM and matrix is found in the present state. As well, non-HSOM acidic surface groups are not revealed, although they may be present. Finally, almost all aryl groups are due to highly molecular HS or polyaromatics, or some aromatics are volatilized (along with water vaporization or afterward), resulting in relatively ambiguous data.
Apart from this, some more ‘technical’ drawbacks are due to the limited number of sample origins used in this study. Thus, a more detailed study is needed to distinguish quartz and other silicate minerals; the most challenging band at 875 cm−1 reveals too many candidates, and more samples are needed to conclude that this is undoubtedly the carbonate band. Furthermore, more evidence is needed in the hydrogen-speciation region and probably in NIR, as band assignment is now ambiguous. Additionally, relative to HS samples, the relevance of potassium and sodium effects on FTIR spectra is left unanswered, as well as other elements, including Ca, Fe, or Al. This could be the subject of the following study. Finally, the approach has another ‘technical’ drawback associated with the relative duration of measurements. However, it is comparable to the duration of thermogravimetric measurements and, in principle, can be further reduced by a factor of 2–4 due to a decrease in the number of scans and an increase in the heating rate. However, optimization of measurement conditions is a challenge for future research.
Additionally, low temperatures could be used to prove that the existing temperature behavior is not connected to water, while higher temperatures compared to the used range can be applied to account for irreversible changes in HSOM. These effects and other uncertainties in spectra would require more statistically wise studies with due account to the reproducibility of the changes upon temperature.
Apart from the concept of HSOM analysis by the proposed approach, the quartz lattice region (800–260 cm−1) can be used as a basis for estimating the amounts of crystalline forms of SiO2 and its ratio with amorphous Si-containing forms. The drawback is that all the bands are temperature-independent, so matrix vibrations are hard to distinguish from organic matter and other components. In the quartz-overtone-band region (1270–800 cm−1), the comparison of fundamental and overtone regions may be used to attribute the bands in the quartz spectrum more finely and compare the crystalline and amorphous species. The bands in this region that do not experience a redshift may be assigned to HSOM. The band at 1120–1070 cm−1 is most problematic as its structure is not revealed fully, either from temperature-based or centrifugation experiments. In HS, it is the least informative region; the only organic constituent may be ambiguous; the characteristic bands of structured quartz 2015 cm−1, 1970 cm−1, and 1880 cm−1 [102,105,106] are not seen, which correlate well with data on the low sensitivity of ATR to quartz overtones [72].
5. Conclusions
Thus, temperature-monitored IR spectroscopy of humic substances provides a more detailed analysis without sample decomposition or HSOM destruction due to different temperature behavior of the maxima and intensities of bands of different components. Therefore, it may be considered a basis for a procedure capable of differentiation of crystalline quartz and amorphous inorganic bands and organic and functional groups of HS systems. Still, some studies are required to expand and strengthen the approach, which should involve the more certain assignment of bands, first of all, complex bands in 1700–1100 cm−1 region, and the use of independent physical and chemical methods along with temperature-based IR measurements.
Still, even in the present state, band-maximum temperature dependence can be considered more stable to changes in experimental conditions than band maxima at a single temperature and serve as a new quantitative parameter characterizing HS using IR spectroscopy. Furthermore, as the main findings of this paper show independence on the HS origin, they can be used in studies involving other types of HS, especially for soils and polluted environments. Additionally, this approach can be used for chemical weathering in soils. The outlooks of this approach may also include studying the changes in HS composition and structure in processed samples used in biotechnology [2,14,15], medicine, and eco-chemistry [4]. Finally, correlation spectroscopy seems expedient for temperature variation to account more correctly for synchronous effects.
Supplementary Materials
The following are available online at www.mdpi.com/article/10.3390/agronomy11091822/s1, Figure S1: A single-channel spectrum of Powhumus humate sample. Figure S2: FTIR spectrum of Powhumus after the registration. Figure S3: Spectra of the Powhumus sample after ATR correction using various refractive indices. Figure S4: Spectra of the Powhumus sample after ATR correction using various refractive indices. Figure S5: IR Absorption spectra in the region 3740–3560 cm−1 after ATR correction. Figure S6: IR Absorption spectra in the region 1900–1200 cm−1 after ATR. Figure S7: IR Absorption spectra in the region 1200–820 cm−1 after ATR correction. Figure S8: IR Absorption spectra in the region 820–400 cm−1 after ATR correction. Figure S9: IR Absorption spectra in the region 4000–400 cm−1 after ATR correction of the Powhumus sample before and after heating and cooling. Figure S10: IR absorption spectra in the region 4000–400 cm−1 after ATR correction. Figure S11: IR absorption spectra in the region 4000–400 cm−1 after ATR correction of the Sakhalin sample before heating after heating and cooling.
Author Contributions
Conceptualization, D.S.V. and O.B.R.; methodology, D.S.V. and O.B.R.; formal analysis, M.A.P., O.B.R., and D.S.V.; investigation, D.S.V. and O.B.R.; resources, D.S.V.; data curation, D.S.V.; writing—original draft preparation, M.A.P. and D.S.V.; writing—review and editing, M.A.P., D.S.V., and O.B.R.; visualization, D.S.V.; supervision, M.A.P.; project administration, M.A.P.; funding acquisition, M.A.P. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by The Russian Science Foundation, project number 19-13-00117.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
This research was performed according to the Development program of the Interdisciplinary Scientific and Educational School of Lomonosov Moscow State University, ‘The future of the planet and global environmental change’.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Malcolm, R.L. The uniqueness of humic substances in each of soil, stream and marine environments. Anal. Chim. Acta 1990, 232, 19–30. [Google Scholar] [CrossRef]
- Kulikova, N.A.; Perminova, I.V. Interactions between Humic Substances and Microorganisms and Their Implications for Nature-like Bioremediation Technologies. Molecules 2021, 26, 2706. [Google Scholar] [CrossRef]
- Stevenson, F.J. Humus Chemistry: Genesis, Composition, Reactions; Wiley: Hoboken, NJ, USA, 1994. [Google Scholar]
- Perminova, I.V.; Garcia-Mina, J.-M.; Podgorski, D.C.; Cervantes, F.J.; Efremenko, E.N.; Domingo, J.L. Humic substances and living systems: Impact on environmental and human health. Environ. Res. 2021, 194, 110726. [Google Scholar] [CrossRef]
- Winkler, J.; Ghosh, S. Therapeutic Potential of Fulvic Acid in Chronic Inflammatory Diseases and Diabetes. J. Diabetes Res. 2018, 2018, 5391014. [Google Scholar] [CrossRef] [Green Version]
- Peña-Méndez, E.M.; Havel, J.; Patočka, J. Humic substances-compounds of still unknown structure: Applications in agriculture, industry, environment, and biomedicine. J. Appl. Biomed. 2005, 3, 13–24. [Google Scholar] [CrossRef] [Green Version]
- de Melo, B.A.; Motta, F.L.; Santana, M.H. Humic acids: Structural properties and multiple functionalities for novel technological developments. Mater. Sci. Eng. C Mater. Biol. Appl. 2016, 62, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Trckova, M.; Matlova, L.; Hudcova, H.; Martin, F.; Zraly, Z.; Dvorska, L.; Beran, V.; Pavlik, I. Peat as a feed supplement for animals: A review. Vet. Med.Czech. 2005, 50, 361–377. [Google Scholar] [CrossRef] [Green Version]
- Pukalchik, M.; Kydralieva, K.; Yakimenko, O.; Fedoseeva, E.; Terekhova, V. Outlining the Potential Role of Humic Products in Modifying Biological Properties of the Soil—A Review. Front. Environ. Sci. 2019, 7, 10. [Google Scholar] [CrossRef]
- Gašparovič, M.; Hrnčár, C.; Gálik, B. The effect of feed additives in pheasants fattening: A review. J. Cent. Eur. Agric. 2017, 18, 749–761. [Google Scholar] [CrossRef]
- Pandey, A.K.; Pandey, S.D.; Misra, V.; Devi, S. Role of humic acid entrapped calcium alginate beads in removal of heavy metals. J. Hazard. Mater. 2003, 98, 177–181. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, Z.; Yin, X.; Wang, N.; Chen, D. Influences of Nitrogen Application Levels on Properties of Humic Acids in Chernozem Amended with Different Types of Organic Materials. Sustainability 2019, 11, 5405. [Google Scholar] [CrossRef] [Green Version]
- Plyatsuk, L.; Chernysh, Y.; Ablieieva, I.; Bataltsev, Y.; Vaskin, R.; Roy, I.; Yakhnenko, E.; Roubík, H. Modelling and development of technological processes for low rank coal bio-utilization on the example of brown coal. Fuel 2020, 267, 117298. [Google Scholar] [CrossRef]
- Skripkina, T.S.; Bychkov, A.L.; Tikhova, V.D.; Lomovsky, O.I. Mechanochemical Solid-Phase Reactions of Humic Acids from Brown Coal with Sodium Percarbonate. Solid Fuel Chem. 2018, 52, 356–360. [Google Scholar] [CrossRef]
- Acharya, B.; Sule, I.; Dutta, A. A review on advances of torrefaction technologies for biomass processing. Biomass Convers. Biorefin. 2012, 2, 349–369. [Google Scholar] [CrossRef]
- Davis, W.M.; Erickson, C.L.; Johnston, C.T.; Delfino, J.J.; Porter, J.E. Quantitative Fourier Transform Infrared spectroscopic investigation humic substance functional group composition. Chemosphere 1999, 38, 2913–2928. [Google Scholar] [CrossRef]
- Tanaka, T.; Nagao, S.; Ogawa, H. Attenuated Total Reflection Fourier Transform Infrared (ATR-FTIR) Spectroscopy of Functional Groups of Humic Acid Dissolving in Aqueous Solution. Anal. Sci. Suppl. 2002, 17icas, i1081–i1084. [Google Scholar] [CrossRef]
- Tatzber, M.; Stemmer, M.; Spiegel, H.; Katzlberger, C.; Haberhauer, G.; Mentler, A.; Gerzabek, M.H. FTIR-spectroscopic characterization of humic acids and humin fractions obtained by advanced NaOH, Na4P2O7, and Na2CO3 extraction procedures. J. Plant. Nutr. Soil Sci. 2007, 170, 522–529. [Google Scholar] [CrossRef]
- Yuan, Y.; Cai, X.; Tan, B.; Zhou, S.; Xing, B. Molecular insights into reversible redox sites in solid-phase humic substances as examined by electrochemical in situ FTIR and two-dimensional correlation spectroscopy. Chem. Geol. 2018, 494, 136–143. [Google Scholar] [CrossRef]
- Baes, A.U.; Bloom, P.R. Diffuse Reflectance and Transmission Fourier Transform Infrared (DRIFT) Spectroscopy of Humic and Fulvic Acids. Soil Sci. Soc. Am. J. 1989, 53, 695–700. [Google Scholar] [CrossRef]
- Vergnoux, A.; Guiliano, M.; Di Rocco, R.; Domeizel, M.; Théraulaz, F.; Doumenq, P. Quantitative and mid-infrared changes of humic substances from burned soils. Environ. Res. 2011, 111, 205–214. [Google Scholar] [CrossRef]
- Karpukhina, E.; Mikheev, I.; Perminova, I.; Volkov, D.; Proskurnin, M. Rapid quantification of humic components in concentrated humate fertilizer solutions by FTIR spectroscopy. J. Soils Sed. 2018, 19, 2729–2739. [Google Scholar] [CrossRef]
- Liu, X.Y.; Chen, W.; Qian, C.; Yu, H.Q. Interaction between Dissolved Organic Matter and Long-Chain Ionic Liquids: A Microstructural and Spectroscopic Correlation Study. Environ. Sci Technol. 2017, 51, 4812–4820. [Google Scholar] [CrossRef] [PubMed]
- MacCarthy, P.; Mark, H.B.; Griffiths, P.R. Direct measurement of the infrared spectra of humic substances in water by Fourier transform infrared spectroscopy. J. Agric. Food Chem. 1975, 23, 600–602. [Google Scholar] [CrossRef]
- Stevenson, F.J.; Goh, K.M. Infrared spectra of humic acids: Elimination of interference due to hygroscopic moisture and structural changes accompanying heating with KBr. Soil Sci. 1974, 117, 34–41. [Google Scholar] [CrossRef]
- Katon, J.E.; Phillips, D.B. Infrared Spectroscopy at Subambient Temperatures. Appl. Spectrosc. Rev. 1973, 7, 1–45. [Google Scholar] [CrossRef]
- Xu, B.; Mei, Y.; Xiao, Z.; Kang, Z.; Wang, R.; Sun, D. Monitoring thermally induced structural deformation and framework decomposition of ZIF-8 through in situ temperature dependent measurements. Phys. Chem. Chem. Phys. 2017, 19, 27178–27183. [Google Scholar] [CrossRef]
- Verma, P.K.; Kundu, A.; Puretz, M.S.; Dhoonmoon, C.; Chegwidden, O.S.; Londergan, C.H.; Cho, M. The Bend+Libration Combination Band Is an Intrinsic, Collective, and Strongly Solute-Dependent Reporter on the Hydrogen Bonding Network of Liquid Water. J. Phys. Chem. B 2018, 122, 2587–2599. [Google Scholar] [CrossRef]
- Koike, C.; Mutschke, H.; Suto, H.; Naoi, T.; Chihara, H.; Henning, T.; Jäger, C.; Tsuchiyama, A.; Dorschner, J.; Okuda, H. Temperature effects on the mid-and far-infrared spectra of olivine particles. Astron. Astrophys. 2006, 449, 583–596. [Google Scholar] [CrossRef] [Green Version]
- Tielens, A.G.G.M.; Allamandola, L.J. Composition, Structure, and Chemistry of Interstellar Dust; Springer: Dordrecht, The Netherlands; pp. 397–470.
- Plendl, J.N. Characteristic Energy Absorption Spectra of Solids; Springer: Boston, MA, USA, 1970; pp. 387–450. [Google Scholar]
- Day, K.L. Temperature Dependence of Mid-Infrared Silicate Absorption. Astrophys. J. 1976, 203, L99. [Google Scholar] [CrossRef]
- Zaikowski, A.; Knacke, R.F.; Porco, C.C. On the presence of phyllosilicate minerals in the interstellar grains. Astrophys. Space Sci. 1975, 35, 97–115. [Google Scholar] [CrossRef]
- Mennella, V.; Brucato, J.R.; Colangeli, L.; Palumbo, P.; Rotundi, A.; Bussoletti, E. Temperature Dependence of the Absorption Coefficient of Cosmic Analog Grains in the Wavelength Range 20 Microns to 2 Millimeters. Astrophys. J. 1998, 496, 1058–1066. [Google Scholar] [CrossRef] [Green Version]
- Bowey, J.E.; Lee, C.; Tucker, C.; Hofmeister, A.M.; Ade, P.A.R.; Barlow, M.J. Temperature effects on the 15–85 μm spectra of olivines and pyroxenes. Mon. Not. R. Astron. Soc. 2001, 325, 886–896. [Google Scholar] [CrossRef] [Green Version]
- Johnston, C.T.; Kogel, J.E.; Bish, D.L.; Kogure, T.; Murray, H.H. Low-temperature Ftir Study of Kaolin-Group Minerals. Clays Clay Miner. 2008, 56, 470–485. [Google Scholar] [CrossRef]
- Balan, E.; Delattre, S.; Roche, D.; Segalen, L.; Morin, G.; Guillaumet, M.; Blanchard, M.; Lazzeri, M.; Brouder, C.; Salje, E.K.H. Line-broadening effects in the powder infrared spectrum of apatite. Phys. Chem. Miner. 2011, 38, 111–122. [Google Scholar] [CrossRef]
- Petrov, A.K.; Sechkarev, A.V. Temperature dependence of infrared spectra. Sov. Phys. J. 1965, 8, 62–64. [Google Scholar] [CrossRef]
- Sirotiak, M.; Bartošová, A. Changes in Structure and Content Humic Substances in Soil During the Laboratory Simulated Fires. Trans. VSB 2016, 11, 42. [Google Scholar] [CrossRef] [Green Version]
- Nanda, S.; Reddy, S.N.; Hunter, H.N.; Butler, I.S.; Kozinski, J.A. Supercritical Water Gasification of Lactose as a Model Compound for Valorization of Dairy Industry Effluents. Ind. Eng. Chem. Res. 2015, 54, 9296–9306. [Google Scholar] [CrossRef]
- Zuyi, T.; Shifang, L.; Jianjun, Z.; Fenling, S. Studies of Thermal Transformations of Humic and Fulvic Acids in Soils I. Infrared Spectroscopy and Temperature-Programmed Pyrolysis Mass Spectrometry. Chem. Ecol. 1997, 13, 237–248. [Google Scholar] [CrossRef]
- Kolokassidou, C.; Pashalidis, I.; Costa, C.N.; Efstathiou, A.M.; Buckau, G. Thermal stability of solid and aqueous solutions of humic acid. Thermochim. Acta 2007, 454, 78–83. [Google Scholar] [CrossRef]
- Malcolm, R.L.; MacCarthy, P. Limitations in the use of commercial humic acids in water and soil research. Environ. Sci. Technol. 1986, 20, 904–911. [Google Scholar] [CrossRef]
- Nunn, S.; Nishikida, K. Advanced ATR Correction Algorithm. In Thermo Fisher Scientific Application Note 50581; Thermo Fisher Scientific: Madison WI, USA, 2008. [Google Scholar]
- Baigorri, R.; Fuentes, M.; González-Gaitano, G.; García-Mina, J.M.; Almendros, G.; González-Vila, F.J. Complementary Multianalytical Approach To Study the Distinctive Structural Features of the Main Humic Fractions in Solution: Gray Humic Acid, Brown Humic Acid, and Fulvic Acid. J. Agric. Food Chem. 2009, 57, 3266–3272. [Google Scholar] [CrossRef] [Green Version]
- Campanella, L.; Tomassetti, M.; Piccolo, A. Thermogravimetric and IR analysis of different extracts of humic substances. Thermochim. Acta 1990, 170, 67–80. [Google Scholar] [CrossRef]
- Provenzano, M.R.; Senesi, N. Thermal Properties of Standard and Reference Humic Substances by Differential Scanning Calorimetry. J. Therm. Anal. Calorim. 1999, 57, 517–526. [Google Scholar] [CrossRef]
- Provenzano, M.R.; Senesi, N.; Miikki, V. Characterization of Composts and Humic Acids from Pulp and Paper Mill Biosludges by DSC in Association with FT-IR Spectroscopy. J. Therm. Anal. Calorim. 1998, 52, 1037–1046. [Google Scholar] [CrossRef]
- Janoš, P.; Kozler, J. Thermal stability of humic acids and some of their derivatives. Fuel 1995, 74, 708–713. [Google Scholar] [CrossRef]
- Cihlář, Z.; Kucerik, J. Regenerated humic acids obtained by the air oxidation of south moravian lignite. part. 2. thermoanalytical characterization of products. Pet. Coal 2010, 52, 254–260. [Google Scholar]
- Giovanela, M.; Parlanti, E.; Soriano-Sierra, E.J.; Soldi, M.S.; Sierra, M.M.D. Elemental compositions, FT-IR spectra and thermal behavior of sedimentary fulvic and humic acids from aquatic and terrestrial environments. Geochem. J. 2004, 38, 255–264. [Google Scholar] [CrossRef]
- Rotaru, A.; Nicolaescu, I.; Rotaru, P.; Neaga, C. Thermal characterization of humic acids and other components of raw coal. J. Therm. Anal. Calorim. 2008, 92, 297–300. [Google Scholar] [CrossRef]
- Sirbu, C.; Cioroianu, T.; Rotaru, P. About the humic acids and thermal behaviour of some humic acids. Ann. Univ. Craiova Phys. 2010, 20, 120–126. [Google Scholar]
- Hoffmann, K.; Huculak-Maczka, M.; Grek, E. INVESTIGATION OF THE PROPERTY OF HUMIC ACIDS BY THERMAL ANALYSIS METHOD. Ecol. Chem. Eng. A 2013, 20, 261–269. [Google Scholar] [CrossRef]
- Boguta, P.; Sokołowska, Z.; Skic, K. Use of thermal analysis coupled with differential scanning calorimetry, quadrupole mass spectrometry and infrared spectroscopy (TG-DSC-QMS-FTIR) to monitor chemical properties and thermal stability of fulvic and humic acids. PLoS ONE 2017, 12, e0189653. [Google Scholar] [CrossRef] [Green Version]
- Larkin, P. Instrumentation and Sampling Methods. In Infrared and Raman Spectroscopy; Larkin, P., Ed.; Elsevier: Oxford, JD, USA, 2011; pp. 27–54. [Google Scholar] [CrossRef]
- Laskina, O.; Young, M.A.; Kleiber, P.D.; Grassian, V.H. Infrared Optical Constants of Organic Aerosols: Organic Acids and Model Humic-Like Substances (HULIS). Aerosol Sci. Technol. 2014, 48, 630–637. [Google Scholar] [CrossRef] [Green Version]
- Dinar, E.; Abo Riziq, A.; Spindler, C.; Erlick, C.; Kiss, G.; Rudich, Y. The complex refractive index of atmospheric and model humic-like substances (HULIS) retrieved by a cavity ring down aerosol spectrometer (CRD-AS). Faraday Discuss. 2008, 137, 279–295. [Google Scholar] [CrossRef]
- Kwon, D.; Sovers, M.J.; Grassian, V.H.; Kleiber, P.D.; Young, M.A. Optical Properties of Humic Material Standards: Solution Phase and Aerosol Measurements. ACS Earth Space Chem. 2018, 2, 1102–1111. [Google Scholar] [CrossRef]
- Russell, J.D.; Fraser, A.R. Infrared methods. In Clay Mineralogy: Spectroscopic and Chemical Determinative Methods; Wilson, M.J., Ed.; Springer: Dordrecht, The Netherlands, 1994; pp. 11–67. [Google Scholar]
- Madejová, J.; Komadel, P. Baseline studies of the clay minerals society source clays: Infrared methods. Clays Clay Miner. 2001, 49, 410–432. [Google Scholar] [CrossRef]
- Senesi, N.; D’Orazio, V.; Ricca, G. Humic acids in the first generation of EUROSOILS. Geoderma 2003, 116, 325–344. [Google Scholar] [CrossRef]
- Dhillon, G.S.; Gillespie, A.; Peak, D.; Van Rees, K.C.J. Spectroscopic investigation of soil organic matter composition for shelterbelt agroforestry systems. Geoderma 2017, 298, 1–13. [Google Scholar] [CrossRef]
- Lucas, S.; Tognonvi, M.T.; Gelet, J.L.; Soro, J.; Rossignol, S. Interactions between silica sand and sodium silicate solution during consolidation process. J. Non-Cryst. Solids 2011, 357, 1310–1318. [Google Scholar] [CrossRef]
- Lønstad Bleken, B.-T.; Mino, L.; Giordanino, F.; Beato, P.; Svelle, S.; Lillerud, K.P.; Bordiga, S. Probing the surface of nanosheet H-ZSM-5 with FTIR spectroscopy. Phys. Chem. Chem. Phys. 2013, 15, 13363–13370. [Google Scholar] [CrossRef] [PubMed]
- De Benedetto, G.E.; Laviano, R.; Sabbatini, L.; Zambonin, P.G. Infrared spectroscopy in the mineralogical characterization of ancient pottery. J. Cult. Herit. 2002, 3, 177–186. [Google Scholar] [CrossRef]
- Roeges, N.P.G. A Guide to the Complete Interpretation of Infrared Spectral of Organic Structures; Wiley: Hoboken, NJ, USA, 1994. [Google Scholar]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part A: Theory and Applications in Inorganic Chemistry; Wiley: Hoboken, NJ, USA, 2008. [Google Scholar]
- Workman, J. The Handbook of Organic Compounds, Three-Volume Set: NIR, IR, R, and UV-Vis Spectra Featuring Polymers and Surfactants; Elsevier Science: Amsterdam, The Netherlands, 2000. [Google Scholar]
- Frost, R.L.; Vassallo, A.M. The Dehydroxylation of the Kaolinite Clay Minerals using Infrared Emission Spectroscopy. Clays Clay Miner. 1996, 44, 635–651. [Google Scholar] [CrossRef]
- Ma, F.; Zeng, Y.; Du, C.; Shen, Y.; Ma, H.; Xu, S.; Zhou, J. Soil variability description using Fourier transform mid-infrared photoacoustic spectroscopy coupling with RGB method. Catena 2017, 152, 190–197. [Google Scholar] [CrossRef]
- Krivoshein, P.K.; Volkov, D.S.; Rogova, O.B.; Proskurnin, M.A. FTIR photoacoustic spectroscopy for identification and assessment of soil components: Chernozems and their size fractions. Photoacoustics 2020, 18, 100162. [Google Scholar] [CrossRef]
- Maréchal, Y. The molecular structure of liquid water delivered by absorption spectroscopy in the whole IR region completed with thermodynamics data. J. Mol. Struct. 2011, 1004, 146–155. [Google Scholar] [CrossRef]
- Coates, J. Interpretation of Infrared Spectra, A Practical Approach. In Encyclopedia of Analytical Chemistry; Meyers, R.A., McKelvy, M.L., Eds.; Wiley: Hoboken, NJ, USA, 2006; p. a5606. [Google Scholar] [CrossRef]
- Bock, J.; Su, G.-J. Interpretation of the Infrared Spectra of Fused Silica. J. Am. Ceram. Soc. 1970, 53, 69–73. [Google Scholar] [CrossRef]
- Vaculíková, L.; Plevová, E.; Vallová, S.; Koutník, I. Characterization and differentiation of kaolinites from selected czech deposits using infrared spectroscopy and differential thermal analysis. Acta Geodyn. Et Geomater. 2011, 8, 59–67. [Google Scholar]
- Inoue, A.; Watanabe, T. Infrared Spectra of Interstratified Illite/Smectite from Hydrothermally Altered Tuffs (Shinzan, Japan) and Diagenetic Bentonites (Kinnekulle, Sweden). Clay Sci. 1989, 7, 263–275. [Google Scholar] [CrossRef]
- Wilson, M.J. Dissolution and formation of quartz in soil environments: A review. Soil Sci. Annu. 2020, 71, 99–110. [Google Scholar] [CrossRef]
- Vettegren, V.I.; Sobolev, G.A.; Kireenkova, S.M.; Morozov, Y.A.; Smul’skaya, A.I.; Mamalimov, R.I.; Kulik, V.B. Effect of water on the α-β phase transition in a surface quartz layer. Phys. Sol. State 2014, 56, 1228–1233. [Google Scholar] [CrossRef]
- Spitzer, W.G.; Kleinman, D.A. Infrared Lattice Bands of Quartz. Phys. Rev. 1961, 121, 1324–1335. [Google Scholar] [CrossRef]
- Max, J.J.; Chapados, C. Isotope effects in liquid water by infrared spectroscopy. III. H2O and D2O spectra from 6000 to 0 cm−1. J. Chem. Phys. 2009, 131, 184505. [Google Scholar] [CrossRef]
- Heaney, P.J.; Kronenberg, A.K.; Prewitt, C.T.; Gibbs, G.V. Chapter 4. Hydrogen Speciation and Chemical Weakening of Quartz. In Silica; De Gruyter: Berlin, Germany, 1994; pp. 123–176. [Google Scholar] [CrossRef]
- Cabaniss, S.E.; McVey, I.F. Aqueous infrared carboxylate absorbances: Aliphatic monocarboxylates. Spectrochim. Acta Part. A Mol. Biomol. Spectrosc. 1995, 51, 2385–2395. [Google Scholar] [CrossRef]
- Cabaniss, S.E.; Leenheer, J.A.; McVey, I.F. Aqueous infrared carboxylate absorbances: Aliphatic di-acids. Spectrochim. Acta Part. A Mol. Biomol. Spectrosc. 1998, 54, 449–458. [Google Scholar] [CrossRef]
- Dunn, G.E.; McDonald, R.S. Infrared spectra of aqueous sodium benzoates and salicylates in the carboxyl-stretching region: Chelation in aqueous sodium salicylates. Can. J. Chem. 1969, 47, 4577–4588. [Google Scholar] [CrossRef]
- Calderón, F.J.; Mikha, M.M.; Vigil, M.F.; Nielsen, D.C.; Benjamin, J.G.; Reeves, J.B. Diffuse-Reflectance Mid-infrared Spectral Properties of Soils under Alternative Crop Rotations in a Semi-arid Climate. Commun. Soil Sci. Plant. Anal. 2011, 42, 2143–2159. [Google Scholar] [CrossRef]
- Nardi, S.; Ertani, A.; Francioso, O. Soil-root cross-talking: The role of humic substances. J. Plant. Nutr. Soil Sci. 2017, 180, 5–13. [Google Scholar] [CrossRef]
- Bronnikov, S.V.; Vettegren, V.I.; Frenkel, S.Y. Description of thermal and mechanical properties of drawn polymers over a wide temperature range. Polym. Eng. Sci. 1992, 32, 1204–1208. [Google Scholar] [CrossRef]
- Poulet, H.; Mathieu, J.P. Vibration Spectra and Symmetry of Crystals; Gordon and Breach: New York, NY, USA; London, UK; Paris, France, 1976. [Google Scholar]
- Scott, J.F.; Porto, S.P.S. Longitudinal and Transverse Optical Lattice Vibrations in Quartz. Phys. Rev. 1967, 161, 903–910. [Google Scholar] [CrossRef]
- Tan, C.Z. Optical interference in overtones and combination bands in α-quartz. J. Phys. Chem. Solids 2003, 64, 121–125. [Google Scholar] [CrossRef]
- Zhuang, J.; Li, M.; Pu, Y.; Ragauskas, A.; Yoo, C. Observation of Potential Contaminants in Processed Biomass Using Fourier Transform Infrared Spectroscopy. Appl. Sci. 2020, 10, 4345. [Google Scholar] [CrossRef]
- Palacios, E.G.; Juárez-López, G.; Monhemius, A.J. Infrared spectroscopy of metal carboxylates: II. Analysis of Fe(III), Ni and Zn carboxylate solutions. Hydrometallurgy 2004, 72, 139–148. [Google Scholar] [CrossRef]
- Palacios, E.G.; Monhemius, A.J. Infrared spectroscopy of metal carboxylates: I. Determination of free acid in solution. Hydrometallurgy 2001, 62, 135–143. [Google Scholar] [CrossRef]
- Herrera-Gómez, A.; Velázquez-Cruz, G.; Martín-Polo, M.O. Analysis of the water bound to a polymer matrix by infrared spectroscopy. J. Appl. Phys. 2001, 89, 5431–5437. [Google Scholar] [CrossRef]
- Honghong, H.; Meifang, G.; Juan, L.; Ortín, A.; Yau, W.W. Analysis of Propylene-1-Butene Copolymer Composition by GPC with Online Detectors. Macromol. Symp. 2015, 356, 110–121. [Google Scholar] [CrossRef]
- Yamagishi, H.; Nakashima, S.; Ito, Y. High temperature infrared spectra of hydrous microcrystalline quartz. Phys. Chem. Miner. 1997, 24, 66–74. [Google Scholar] [CrossRef]
- Mikhaylova, Y.; Adam, G.; Häussler, L.; Eichhorn, K.J.; Voit, B. Temperature-dependent FTIR spectroscopic and thermoanalytic studies of hydrogen bonding of hydroxyl (phenolic group) terminated hyperbranched aromatic polyesters. J. Mol. Struct. 2006, 788, 80–88. [Google Scholar] [CrossRef]
- Du, C.; Goyne, K.W.; Miles, R.J.; Zhou, J. A 1915–2011 microscale record of soil organic matter under wheat cultivation using FTIR-PAS depth-profiling. Agron. Sustain. Dev. 2013, 34, 803–811. [Google Scholar] [CrossRef]
- Kar, S.; Maity, J.P.; Jean, J.-S.; Liu, C.-C.; Nath, B.; Lee, Y.-C.; Bundschuh, J.; Chen, C.-Y.; Li, Z. Role of organic matter and humic substances in the binding and mobility of arsenic in a Gangetic aquifer. J. Environ. Sci. Health Part. A 2011, 46, 1231–1238. [Google Scholar] [CrossRef]
- Volkov, D.S.; Krivoshein, P.K.; Mikheev, I.V.; Proskurnin, M.A. Pristine detonation nanodiamonds as regenerable adsorbents for metal cations. Diam. Relat. Mater. 2020, 110, 108121. [Google Scholar] [CrossRef]
- Nguyen, T.; Janik, L.; Raupach, M. Diffuse reflectance infrared fourier transform (DRIFT) spectroscopy in soil studies. Soil Res. 1991, 29, 49–67. [Google Scholar] [CrossRef]
- Fukuda, J.-i.; Shimizu, I. Water distribution in quartz schists of the Sanbagawa Metamorphic Belt, Japan: Infrared spectroscopic mapping and comparison of the calibrations proposed for determining water contents. Earth Planets Space 2019, 71, 136. [Google Scholar] [CrossRef]
- Jiang, H.; Chen, B.-X.; Fu, C.-S.; Sui, G.-R.; Iso, M. gamma-irradiation damage of quartz fiber and its effects on near-infrared transmission characteristics. Acta Phys. Sin. 2010, 59, 7782–7787. [Google Scholar]
- Hofmeister, A.M.; Bowey, J.E. Quantitative Infrared Spectra of Hydrosilicates and Related Minerals. Mon. Not. R. Astron. Soc. 2006, 367, 577–591. [Google Scholar] [CrossRef]
- Shinoda, K.; Aikawa, N. Absorption coefficients of overtone and combination modes of quartz. Mineral. Mag. 1994, 58, 601–606. [Google Scholar] [CrossRef]
Figure 1.
IR absorption spectra in the range 4000–400 cm−1 after ATR correction of (A) Sigma-Aldrich, (B) Powhumus, and (C) Sakhalin humic substance samples. Temperature increases from 25 to 215 °C from blue to orange lines.
Figure 1.
IR absorption spectra in the range 4000–400 cm−1 after ATR correction of (A) Sigma-Aldrich, (B) Powhumus, and (C) Sakhalin humic substance samples. Temperature increases from 25 to 215 °C from blue to orange lines.


Figure 2.
IR absorption spectra in the region 600–420 cm−1 after ATR correction of Sigma-Aldrich (left) and Powhumus (right) without centrifugation. Temperature increases from 25 to 215 °C from blue to orange lines.
Figure 2.
IR absorption spectra in the region 600–420 cm−1 after ATR correction of Sigma-Aldrich (left) and Powhumus (right) without centrifugation. Temperature increases from 25 to 215 °C from blue to orange lines.

Figure 3.
IR absorption spectra in the region 850–600 cm−1 after ATR correction of Sigma-Aldrich (left) and Powhumus (right) without centrifugation. Temperature increases from 25 to 215 °C from blue to orange lines.
Figure 3.
IR absorption spectra in the region 850–600 cm−1 after ATR correction of Sigma-Aldrich (left) and Powhumus (right) without centrifugation. Temperature increases from 25 to 215 °C from blue to orange lines.

Figure 4.
Redshifts of the smoothed bands at 1032 cm−1 and 797 cm−1 relative to 25 °C temperature for HS samples.
Figure 4.
Redshifts of the smoothed bands at 1032 cm−1 and 797 cm−1 relative to 25 °C temperature for HS samples.

Figure 5.
IR absorption spectra in the region 950–850 cm−1 after ATR correction of Sigma-Aldrich (left) and Powhumus (right) without centrifugation. Temperature increases from 25 to 215 °C from blue to orange lines.
Figure 5.
IR absorption spectra in the region 950–850 cm−1 after ATR correction of Sigma-Aldrich (left) and Powhumus (right) without centrifugation. Temperature increases from 25 to 215 °C from blue to orange lines.

Figure 6.
IR absorption spectra in the region 1060–950 cm−1 after ATR correction of Sigma-Aldrich (left) and Powhumus (right) without centrifugation. Temperature increases from 25 to 215 °C from blue to orange lines.
Figure 6.
IR absorption spectra in the region 1060–950 cm−1 after ATR correction of Sigma-Aldrich (left) and Powhumus (right) without centrifugation. Temperature increases from 25 to 215 °C from blue to orange lines.

Figure 7.
IR absorption spectra in the region 1150–1060 cm−1 after ATR correction of Sigma-Aldrich (left) and Powhumus (right) without centrifugation. Temperature increases from 25 to 215 °C from blue to orange lines.
Figure 7.
IR absorption spectra in the region 1150–1060 cm−1 after ATR correction of Sigma-Aldrich (left) and Powhumus (right) without centrifugation. Temperature increases from 25 to 215 °C from blue to orange lines.

Figure 8.
IR absorption spectra in the region 1500–1200 cm−1 after ATR correction of Sigma-Aldrich (left) and Powhumus (right) without centrifugation. Temperature increases from 25 to 215 °C from blue to orange lines.
Figure 8.
IR absorption spectra in the region 1500–1200 cm−1 after ATR correction of Sigma-Aldrich (left) and Powhumus (right) without centrifugation. Temperature increases from 25 to 215 °C from blue to orange lines.

Figure 9.
IR absorption spectra in the region 1750–1500 cm−1 after ATR correction of Sigma-Aldrich (left) and Powhumus (right) without centrifugation. Temperature increases from 25 to 215 °C from blue to orange lines.
Figure 9.
IR absorption spectra in the region 1750–1500 cm−1 after ATR correction of Sigma-Aldrich (left) and Powhumus (right) without centrifugation. Temperature increases from 25 to 215 °C from blue to orange lines.

Figure 10.
Blueshift of the smoothed band at 1573 cm−1 relative to 25 °C on temperature for the Sakhalin sample.
Figure 10.
Blueshift of the smoothed band at 1573 cm−1 relative to 25 °C on temperature for the Sakhalin sample.

Figure 11.
IR absorption spectra in the region 3050–2800 cm−1 after ATR correction of Sigma-Aldrich (left) and Powhumus (right) without centrifugation. Temperature increases from 25 to 215 °C from blue to orange lines.
Figure 11.
IR absorption spectra in the region 3050–2800 cm−1 after ATR correction of Sigma-Aldrich (left) and Powhumus (right) without centrifugation. Temperature increases from 25 to 215 °C from blue to orange lines.

Figure 12.
Redshift of the smoothed band at 3695 cm−1 and blueshift at 3618 cm−1 relative to 25 °C temperature for HS samples.
Figure 12.
Redshift of the smoothed band at 3695 cm−1 and blueshift at 3618 cm−1 relative to 25 °C temperature for HS samples.

Figure 13.
IR absorption spectra in the region 3720–3600 cm−1 after ATR correction of Sigma-Aldrich (left) and Powhumus (right) without centrifugation. Temperature increases from 25 to 215 °C from blue to orange lines.
Figure 13.
IR absorption spectra in the region 3720–3600 cm−1 after ATR correction of Sigma-Aldrich (left) and Powhumus (right) without centrifugation. Temperature increases from 25 to 215 °C from blue to orange lines.

Figure 14.
IR absorption spectra in the region 4000–400 cm−1 after ATR correction of Powhumus sample after centrifugation. Temperature increases from 25 to 215 °C from blue to orange lines.
Figure 14.
IR absorption spectra in the region 4000–400 cm−1 after ATR correction of Powhumus sample after centrifugation. Temperature increases from 25 to 215 °C from blue to orange lines.


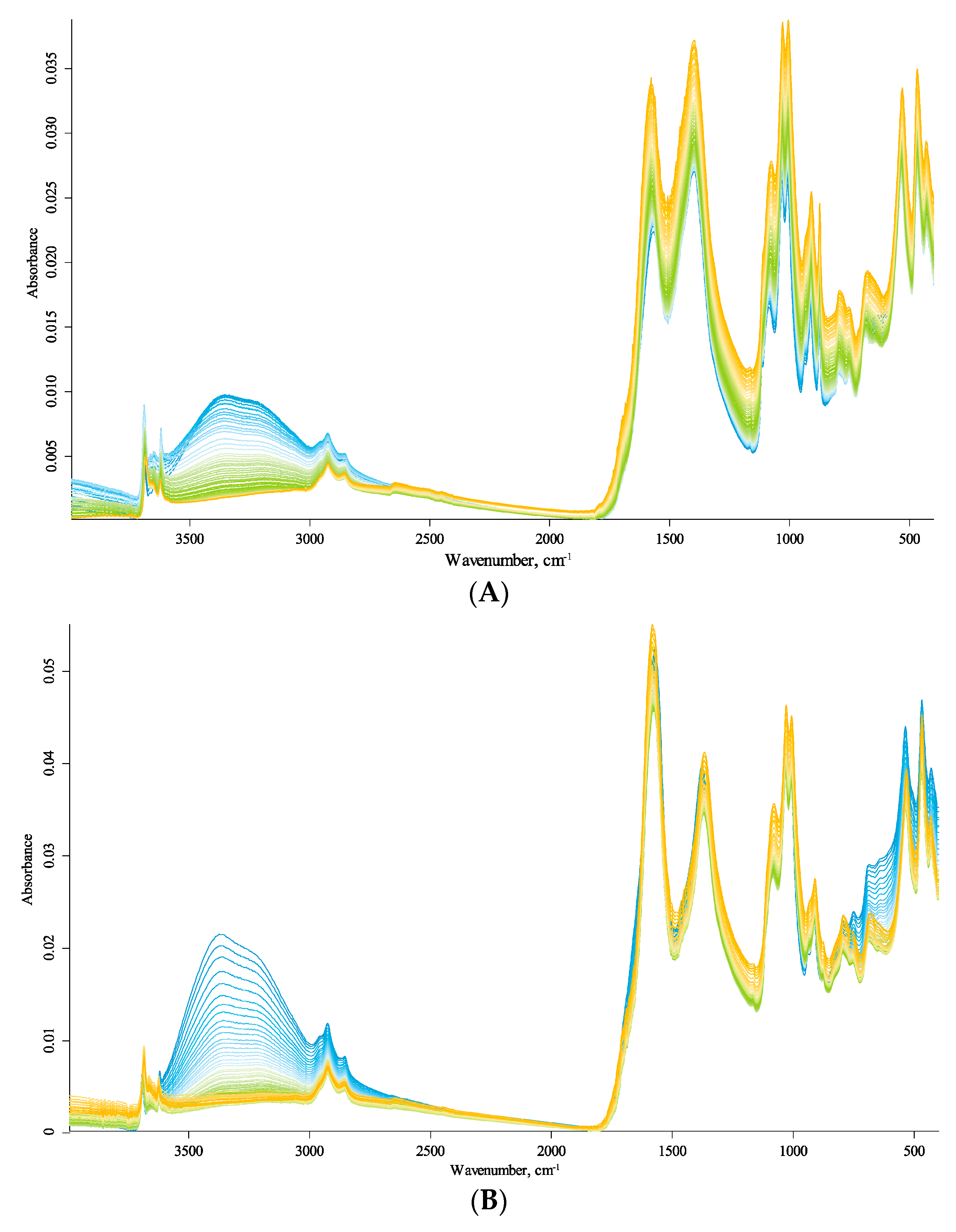{kind=link}
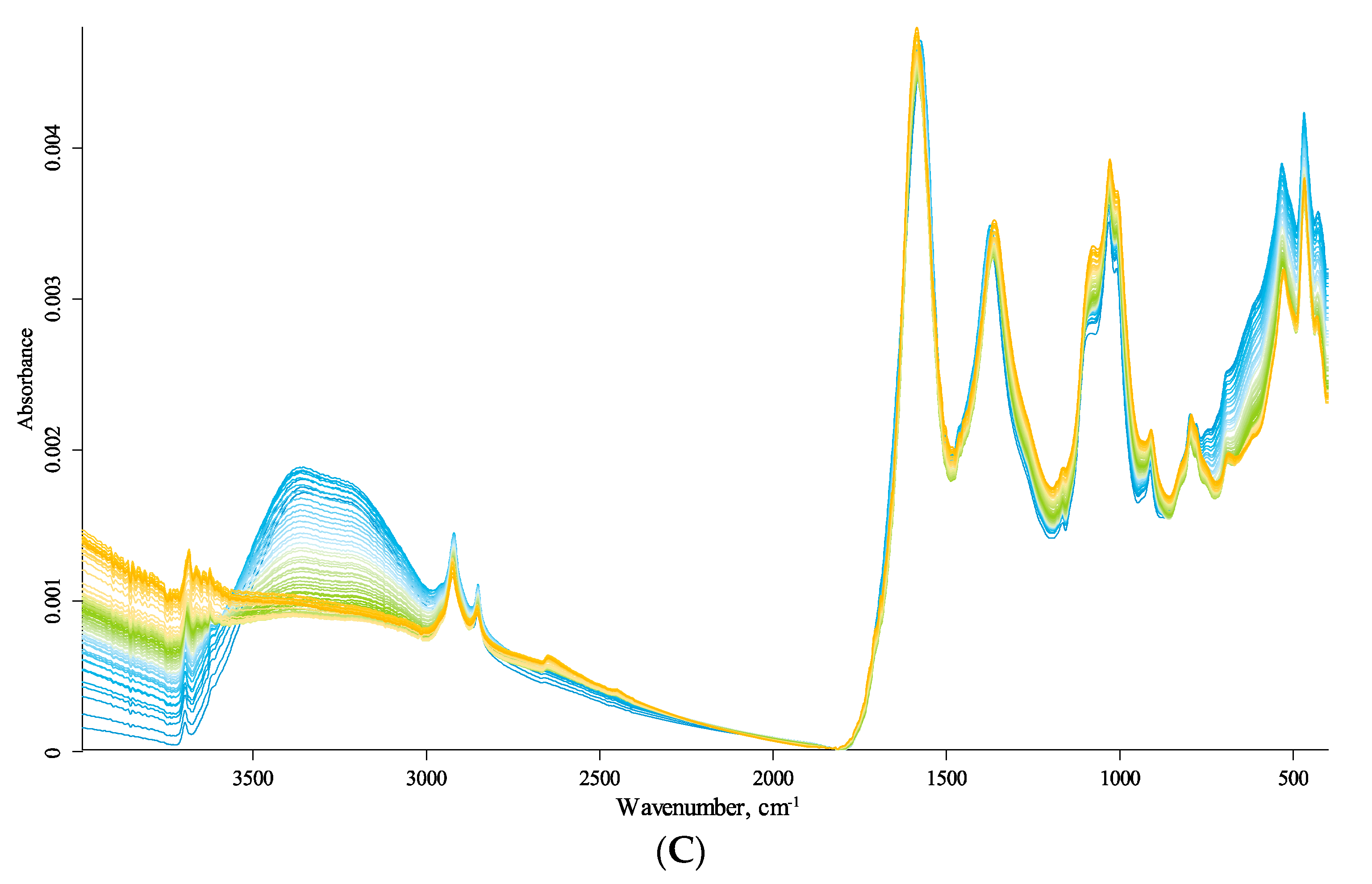{kind=link}
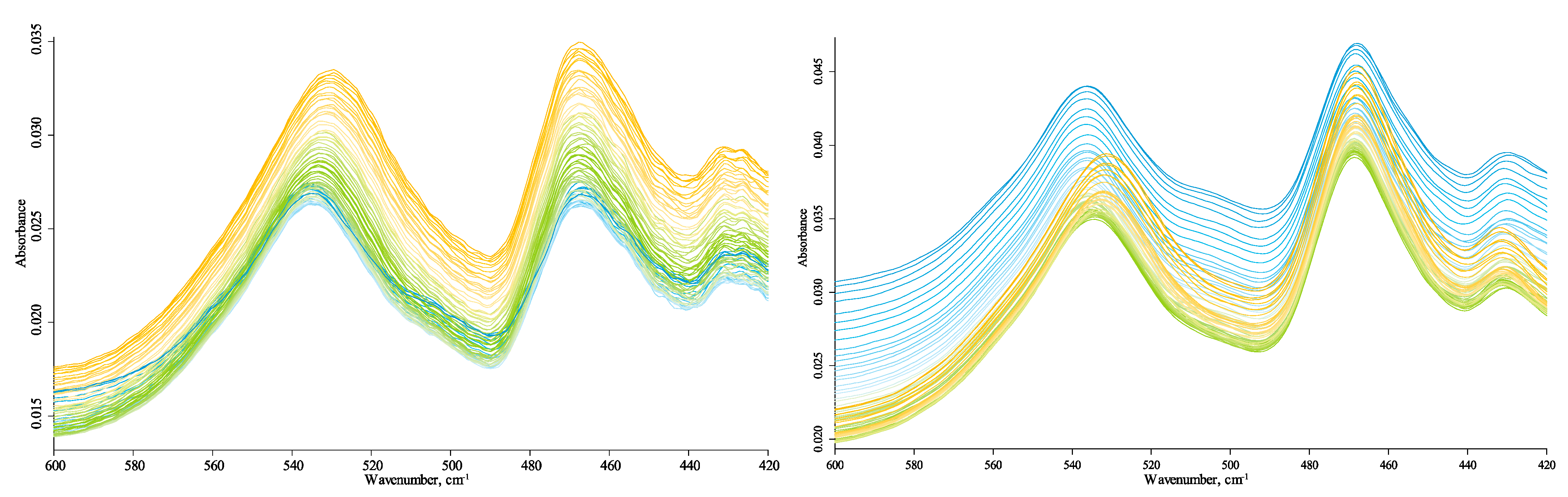{kind=link}
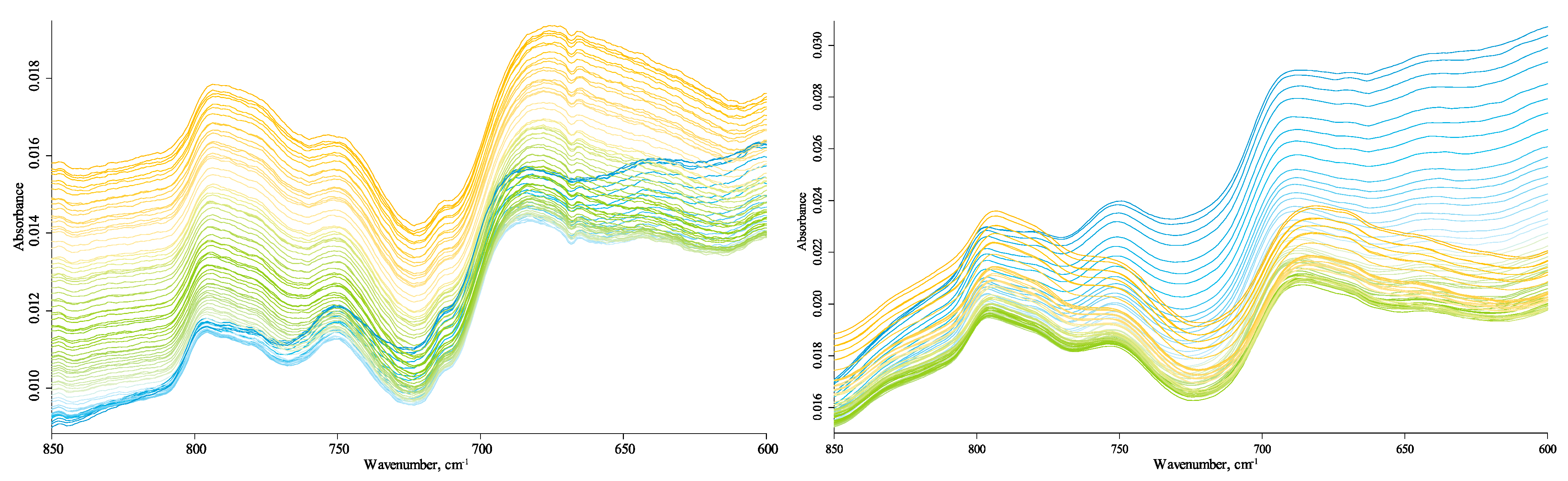{kind=link}
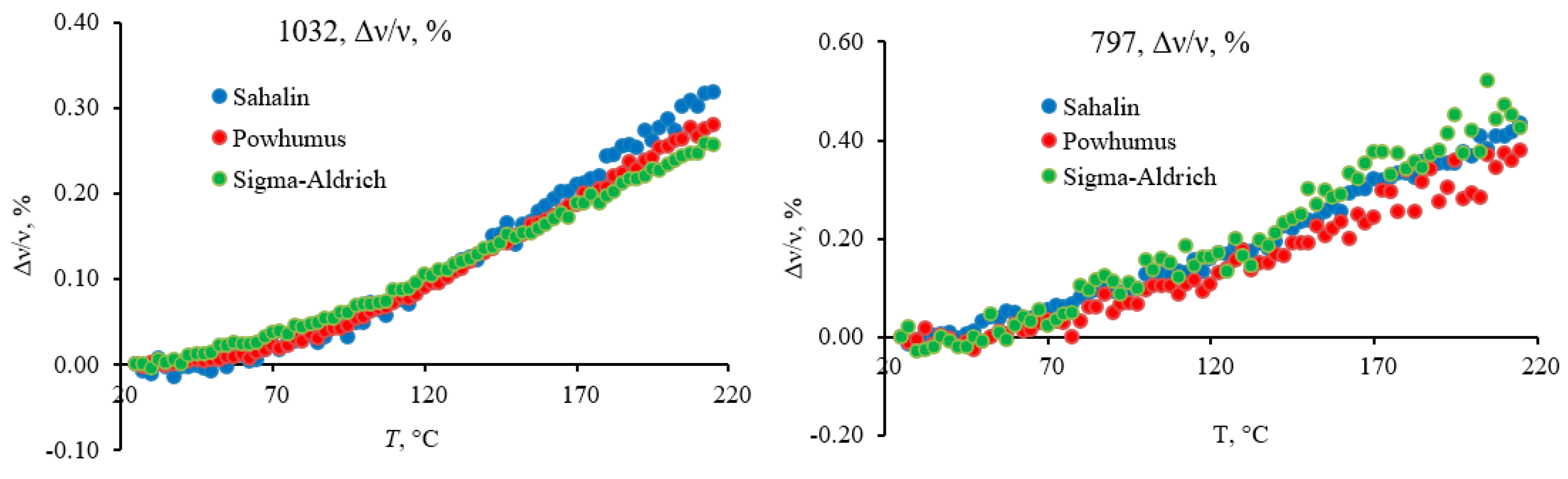{kind=link}
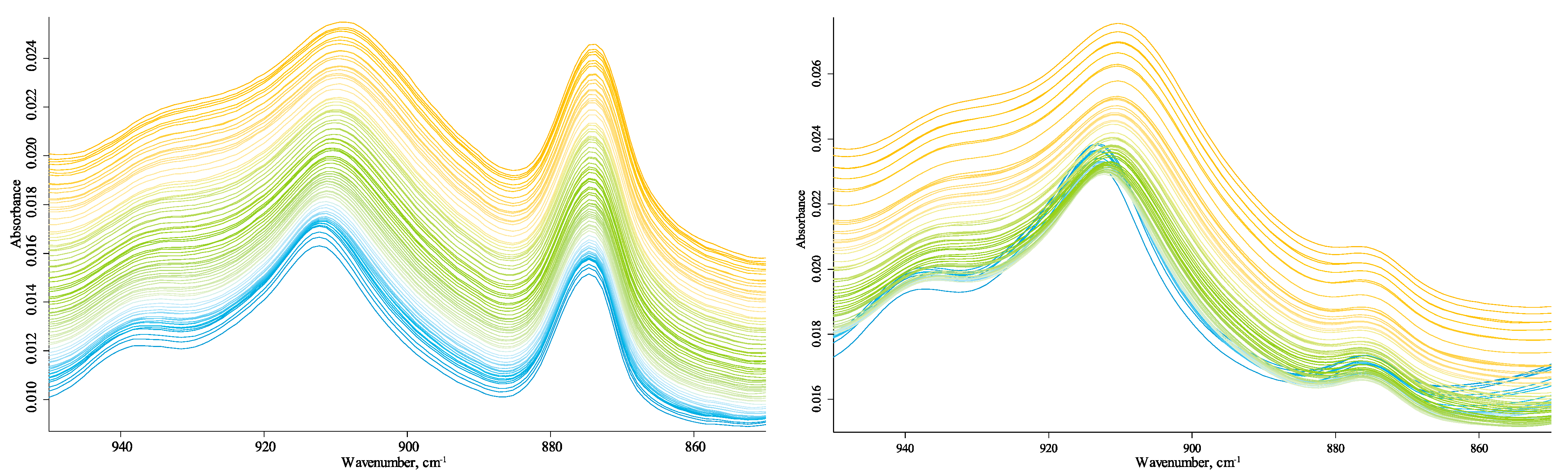{kind=link}
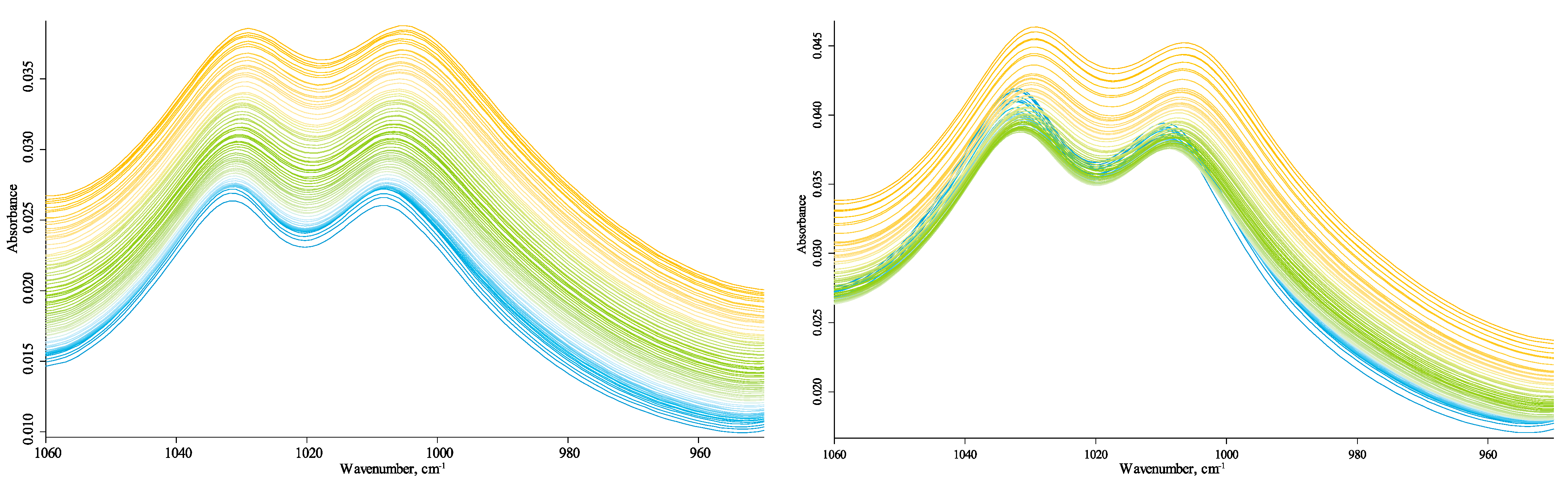{kind=link}
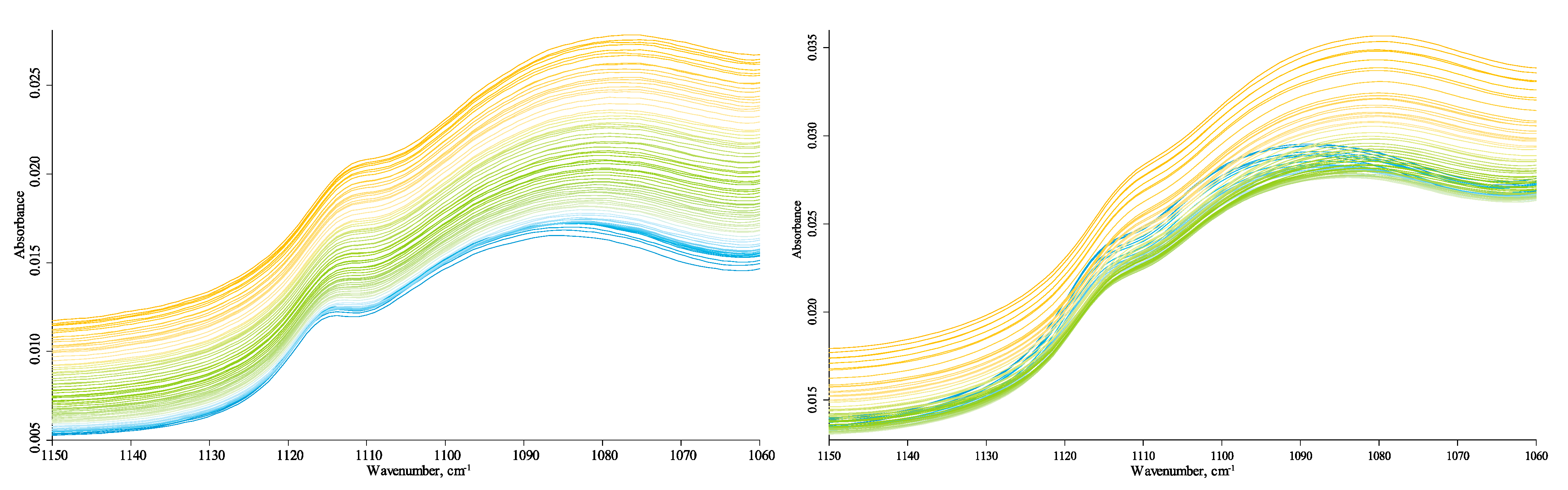{kind=link}
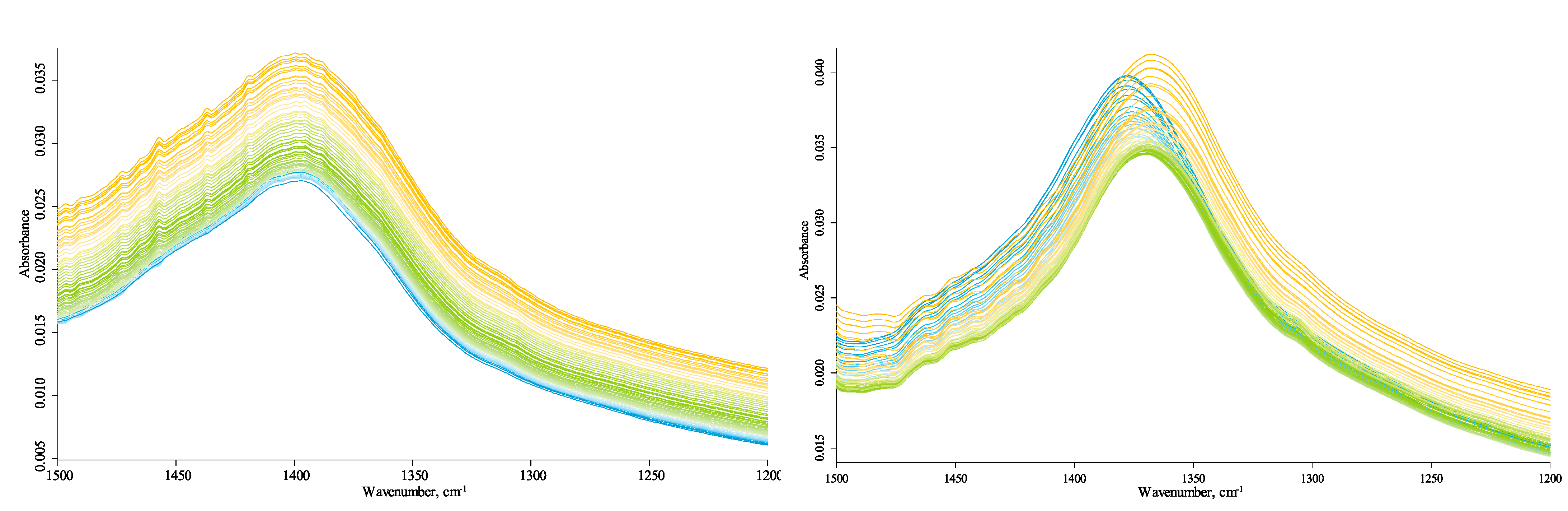{kind=link}
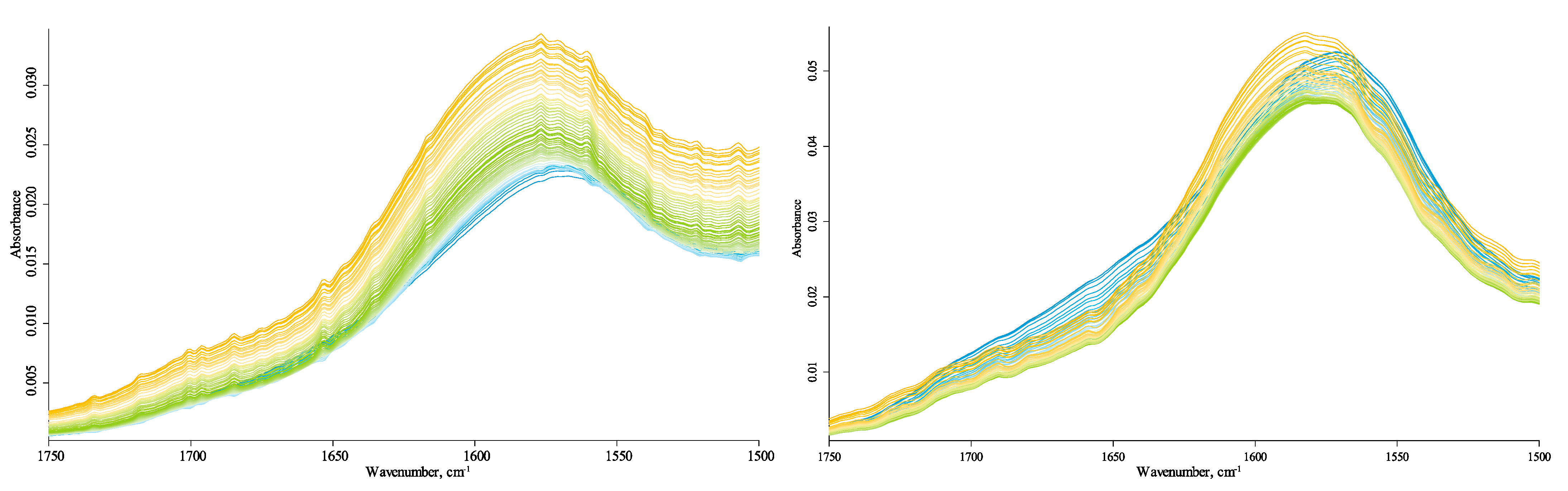{kind=link}
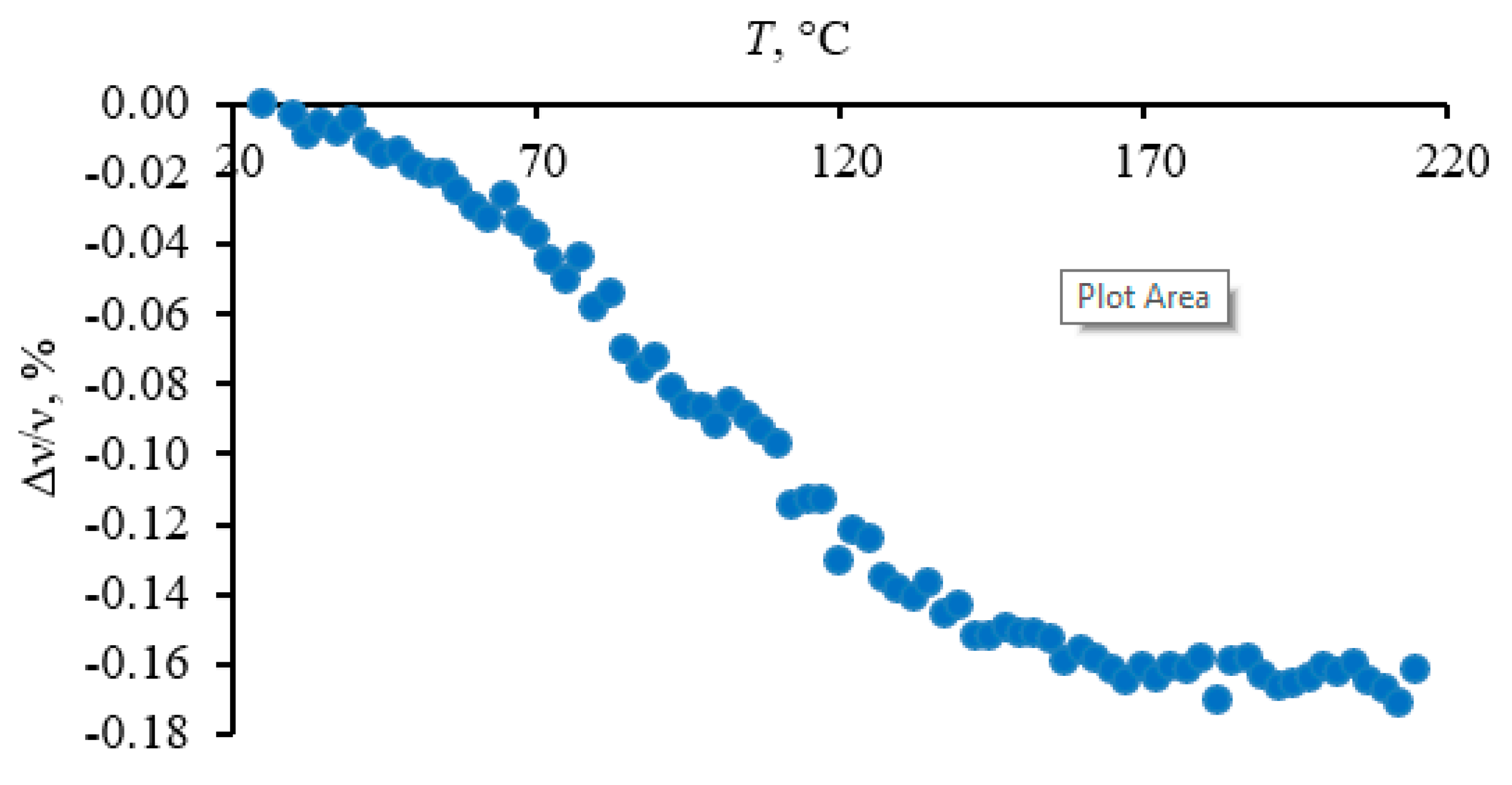{kind=link}
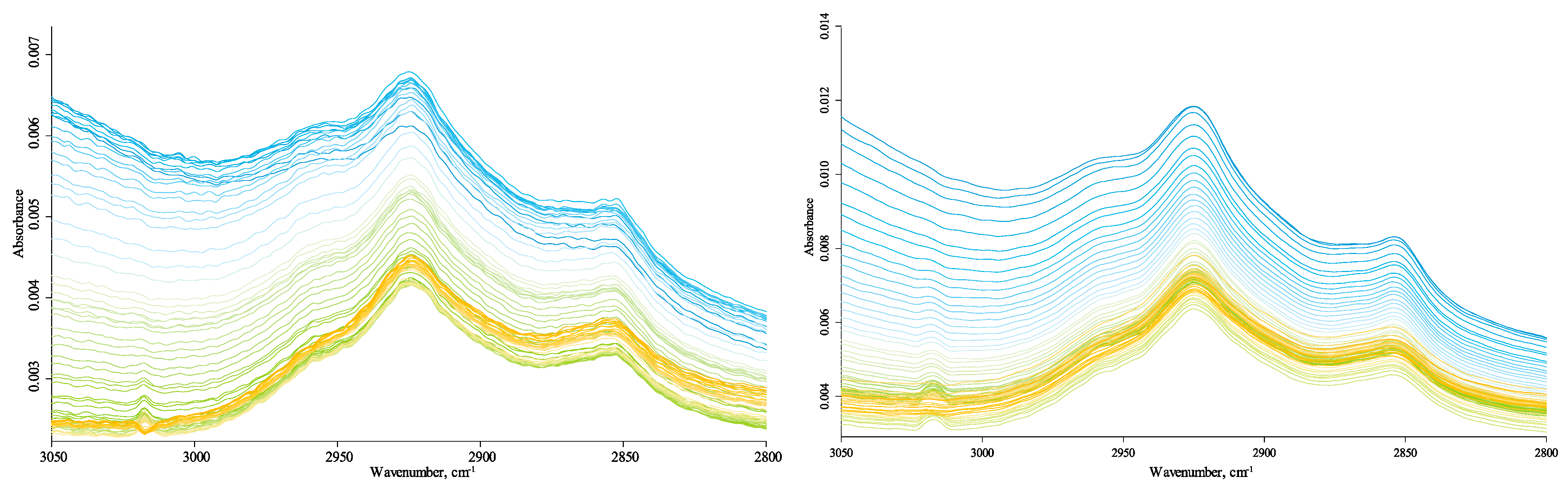{kind=link}
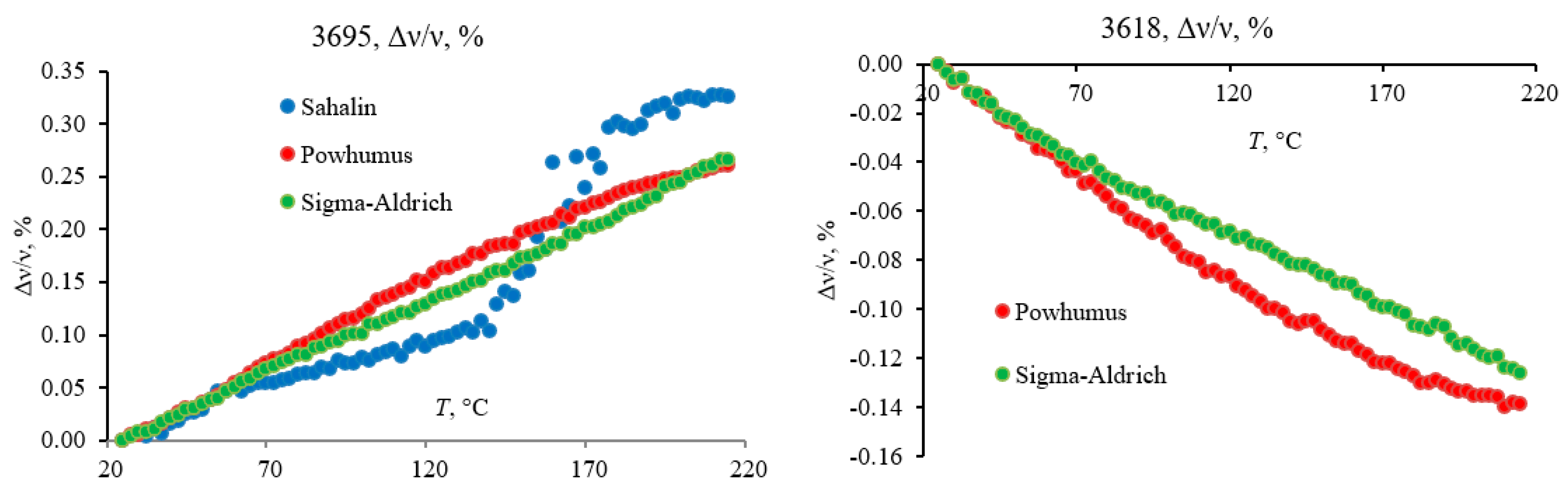{kind=link}
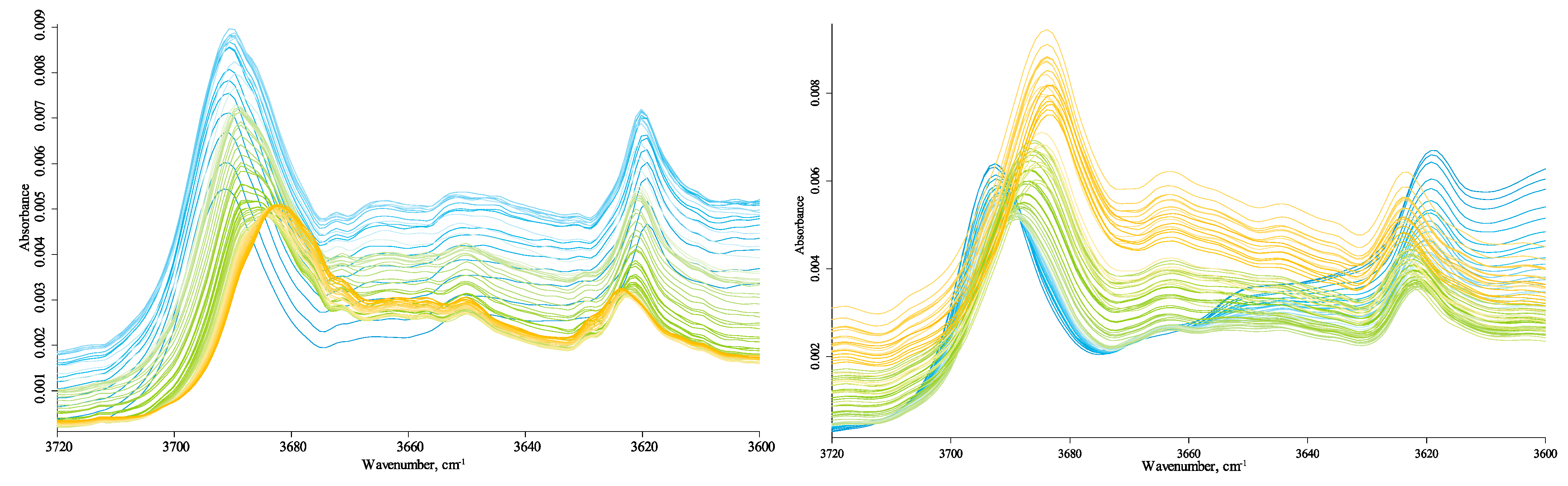{kind=link}
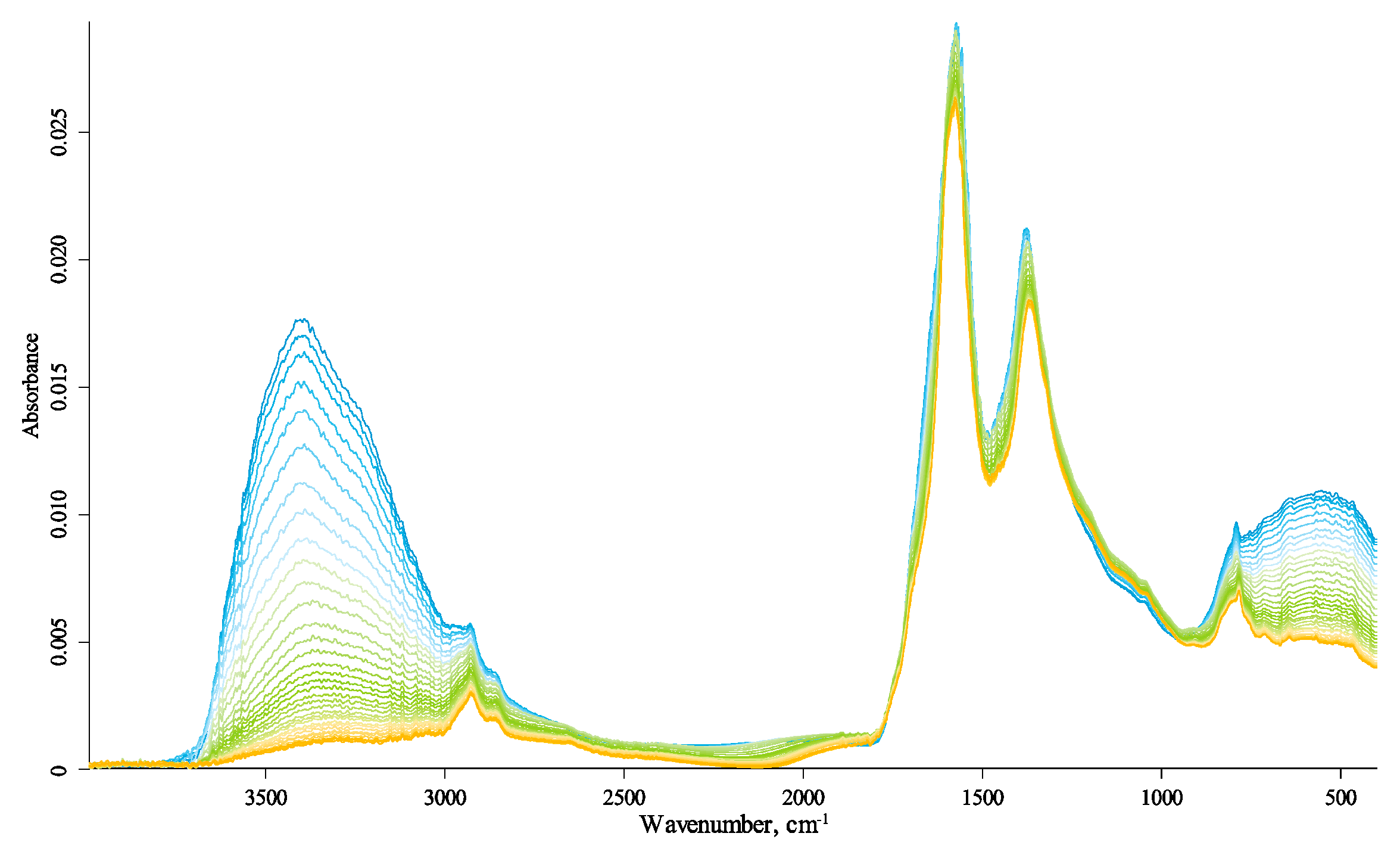{kind=link}
Table 1.
ATR–FTIR measurement conditions and parameters.
| Spectral range, cm−1 | 4000–400 |
| Resolution, cm−1 | 2 |
| Background scan | 128 |
| Sample scan | 128 |
| Acquisition mode | Double-sided, forward–backward |
| High-frequency limit | 15000 |
| Aperture setting | 8 mm |
| Phase resolution | 4 |
| Phase-correction mode | Mertz |
| Zero filling factor | 2 |
| Apodization function | Blackman–Harris 3-term |
| Sample and background pre-amplification gain | ‘Ref’ (without amplification) |
| Background signal gain | Auto |
| Sample signal gain | Auto |
| Scanner velocity | 10 kHz |
| Detector | Room temperature DLaTGS |
| Source | MIR, globar |
| Beam splitter | KBr |
| Background | Diamond crystal with a lowered pressure screw with a flat end |
Table 2.
Position, temperature-induced shifts, and assignment of the absorption bands in IR spectra of HS samples.
Table 2.
Position, temperature-induced shifts, and assignment of the absorption bands in IR spectra of HS samples.
| Sigma-Aldrich | Powhumus | Sakhalin | Assignment | Ref. | |||
|---|---|---|---|---|---|---|---|
| ν (25 °C) | ν (215 °C)–ν (25 °C) | ν (25 °C) | ν (215 °C)–ν (25 °C) | ν (25 °C) | ν (215 °C)–ν (25 °C) | ||
| 3691.4 | −9.84 | 3692.8 | −9.61 | 3695.9 | −12.04 | OH stretching of inner-surface hydroxyl groups, in-phase vibration with a transition moment nearly perpendicular to the (001) plane of kaolinite | [60,61] |
| 3666.7 | * | 3666.8 | * | - | - | OH stretching of inner-surface hydroxyl groups, anti-phase vibration with transition moment lying in the (001) plane of kaolinite | |
| 3650.0 | * | 3651.1 | * | - | - | ||
| 3618.7 | 4.57 | 3618.9 | 5.02 | 3613.3 | 10.63 | OH stretching of inner hydroxyl groups | |
| 2960 *** | * | 2960 *** | * | 2960 *** | * | Asymmetric and symmetric C–H stretching of aliphatic groups CHx | [18,62] |
| 2925.7 | −1.59 | 2925.2 | −0.13 | 2919.5 | 4.71 | ||
| 2854.9 | 0.47 | 2854.1 | 1.38 | 2850.7 | 2.60 | ||
| 1569.1 | 8.48 | 1571.2 | 11.15 | 1574.9 | 10.54 | Aromatic C=C stretching of the phenyl ring, CO carboxylate stretching; aromatic C=C skeletal stretching vibrations, C=O of quinone and/or H-bonded conjugated ketones; –COO– symmetric stretching | [20] |
| 1398.0 | 0.13 | - | - | - | - | 1450 –CH deformation of –CH3 and –CH bending of –CH; 1350 symmetric –COO~ stretch and/or –CH bending of aliphatics | [20] |
| - | - | 1378.1 | −10.39 | 1375.3 | −12.99 | A combination band, and is primarily attributed to CH absorption in aliphatics, as well as to CO–CH3 vibrations in lignin-derived phenols | [63] |
| 1164.5 | * | 1164.5 | * | 1164.5 | * | C–OH stretching of aliphatic O–H | [62] |
| 1114 ** | 1114 ** | - | - | - | Si–O stretching (longitudinal mode) of kaolinite | [61] | |
| 1085.2 | −8.42 | 1089.5 | −9.76 | ** | Alcoholic and polysaccharide CO stretch and OH deformation; Si–OH bend in silicate impurities | [62] | |
| 1031.5 | −2.64 | 1032.1 | −2.89 | 1032.2 | −3.27 | In-plane Si–O stretching (of kaolinite) | [61] |
| 1008.4 | −3.17 | 1009.5 | −3.06 | 1010.1 | −1.48 | In-plane Si–O stretching (of kaolinite) | [61] |
| 937.2 | * | 937.7 | * | 935 *** | * | OH deformation of the inner-surface hydroxyl group of kaolinite | [61] |
| 912.4 | −3.29 | 913.4 | −3.16 | 912.6 | −2.44 | OH deformation of inner hydroxyl groups of kaolinite | [61] |
| 875.0 | −0.80 | 875.9 | 1.03 | Si–O− or Si–O–Si bridge or carbonate, calcite, or polyaromatic bend vibrations | [60,64,65,66,67] | ||
| 795.7 | −3.38 | 796.4 | −3.02 | 797.5 | −3.46 | Si–O in kaolinite | [61] |
| 778 *** | * | 778 *** | * | 778.5 | * | Aromatic CH out of plane bending | [20] |
| 749.9 | 2.74 | 749.9 | * | 750 *** | * | Si–O, perpendicular | [61] |
| 713 | * | - | - | - | - | Al–O vibrations or C4+ alkanes | [68,69] |
| 690 ** | - | 690 ** | - | 690 ** | OH translation in kaolinite | [70] | |
| 640 *** | - | 640 *** | - | - | - | Si–O | [61] |
| 534.9 | −4.52 | 536.2 | −4.94 | 532.6 | −5.06 | Si–O–Al deformation in kaolinite | [61,70] |
| 466.6 | 0.84 | 468.3 | −0.44 | 468.9 | −0.95 | Si–O bending in kaolinite | [70] |
| 428.6 | 1.28 | 430.0 | 1.85 | Si–O deformation of kaolinite | [61] | ||
* The band intensity is low; when heated, becomes comparable to the noise, making it impossible to register the maxima accurately. ** A relatively intense band with a flat top, impossible to determine the exact position of the maximum. *** A weakly expressed band on the shoulder of another. - No band (indistinguishable from noise).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Volkov, D.S.; Rogova, O.B.; Proskurnin, M.A. Temperature Dependences of IR Spectra of Humic Substances of Brown Coal. Agronomy 2021, 11, 1822. https://0-doi-org.brum.beds.ac.uk/10.3390/agronomy11091822
AMA Style
Volkov DS, Rogova OB, Proskurnin MA. Temperature Dependences of IR Spectra of Humic Substances of Brown Coal. Agronomy. 2021; 11(9):1822. https://0-doi-org.brum.beds.ac.uk/10.3390/agronomy11091822
Chicago/Turabian StyleVolkov, Dmitry S., Olga B. Rogova, and Mikhail A. Proskurnin. 2021. "Temperature Dependences of IR Spectra of Humic Substances of Brown Coal" Agronomy 11, no. 9: 1822. https://0-doi-org.brum.beds.ac.uk/10.3390/agronomy11091822
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.