
Identification of the Genome-Wide Expression Patterns of Non-Coding RNAs Associated with Tanshinones Synthesis Pathway in Salvia miltiorrhiza
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Growth Conditions
2.2. HPLC Analysis of Tanshinone Content
2.3. Methods of RNA Extraction, Detection, and Profound Sequence of ncRNAs
2.4. Library Preparation for sRNA Sequencing, lncRNA-Seq and circRNA-Seq
2.5. Clustering, Sequencing, and Quality Control
2.6. Computational Identification of ncRNAs
2.7. Differential Expression Analysis
2.8. Validation by Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.9. Target Prediction and Functional Annotation
3. Results
3.1. Effective Constituent Contents in the White Root and Red Root
3.2. High-Throughput RNAs Sequencing and Different Expression RNAs
3.3. Functional Annotation of DE-RNAs
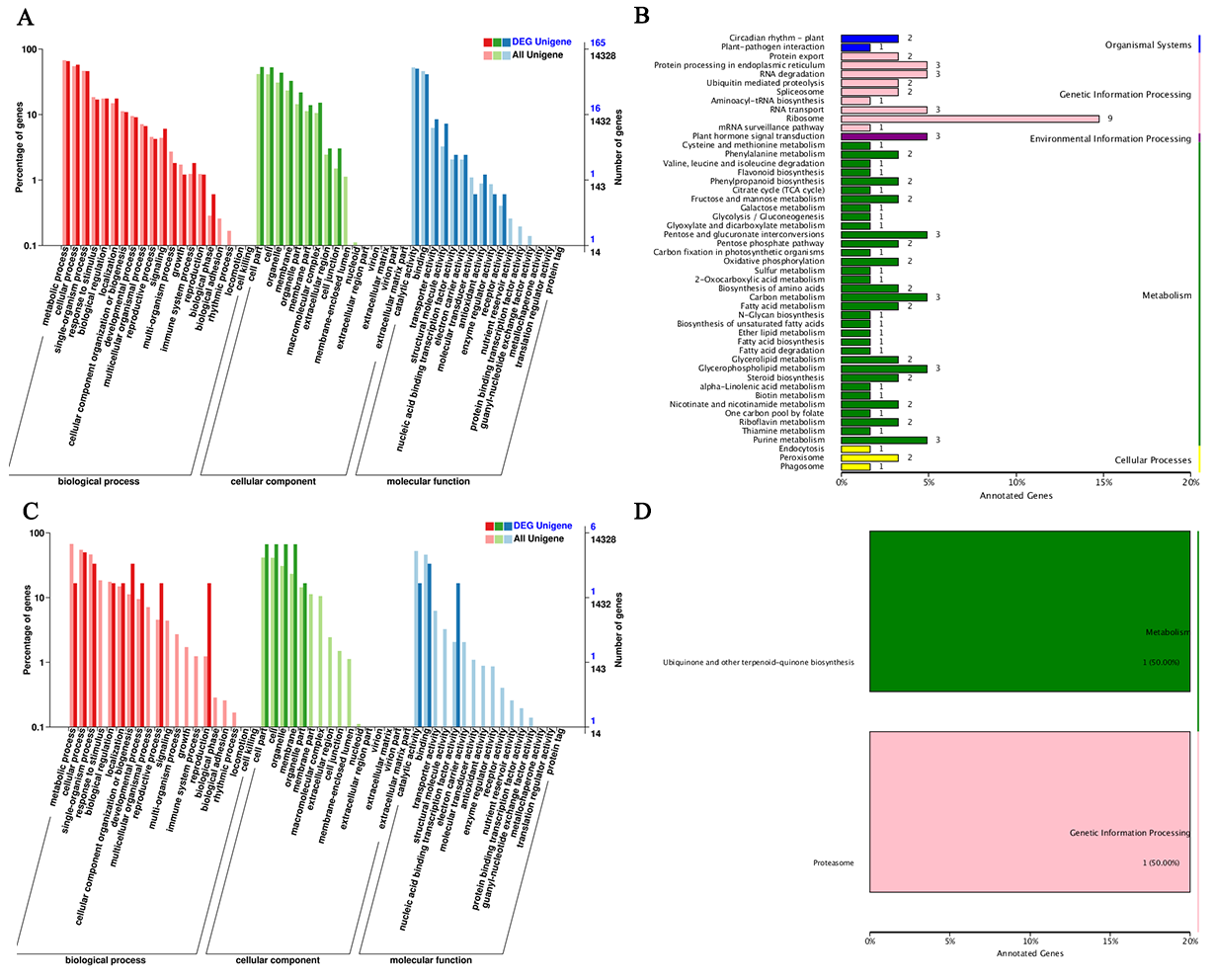
3.3.1. Functional Annotation of DEGs
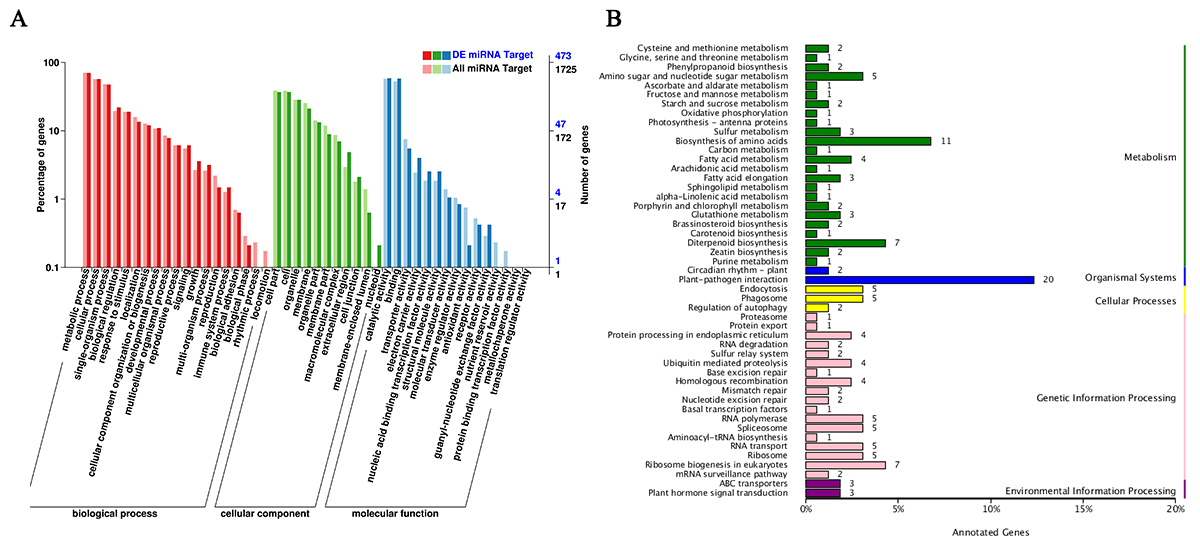
3.3.2. Functional Annotation of Target Genes of DE-miRNAs
3.3.3. Functional Annotation of Target Genes of DE-lncRNAs
3.3.4. Functional Annotation of Target Genes of DE-circRNAs
3.3.5. Functional Annotation of Target Genes of miRNAs Combined with DE-circRNAs
3.4. Tanshinone Biosynthesis Pathway and Its ncRNA Regulation
4. Discussion
4.1. A Large Number of ncRNAs Were Identified in Danshen
4.2. NcRNAs Expressed Differentially in Root Tissues and Their Target Genes
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Song, Z.Q.; Lin, C.C.; Xing, P.Y.; Feng, Y.Y.; Jin, H.; Zhou, C.H.; Gu, Y.Q.; Wang, J.H.; Li, X.F. A high-quality reference genome sequence of Salvia miltiorrhiza provides insights into tanshinone synthesis in its red rhizomes. Plant Genome 2020, 13, e20041. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.P.; Hao, X.L.; Shi, M.; Fu, R.; Wang, Y.; Zhang, Y.; Zhou, W.; Feng, Y.; Makunga, N.P.; Kai, G.Y. Tanshinone production could be increased by the expression of SmWRKY2 in Salvia miltiorrhiza hairy roots. Plant Sci. 2019, 284, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yang, D.F.; Xing, B.C.; Zhang, H.H.; Liang, Z.S. Salvia castanea hairy roots are more tolerant to phosphate deficiency than salvia miltiorrhiza hairy roots based on the secondary metabolism and antioxidant defenses. Molecules 2018, 3, 1132. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.Y.; Guo, L.L.; Jin, H.; Lin, C.C.; Zhou, C.H.; Fang, X.S.; Wang, J.H.; Song, Z.Q. Quantitative trait loci analysis of phenolic acids contents in Salvia miltiorrhiza based on genomic simple sequence repeat markers. Ind. Crop. Prod. 2019, 133, 365–372. [Google Scholar] [CrossRef]
- Zhang, J.H.; Lv, H.Z.; Liu, W.J.; Ji, A.J.; Zhang, X.; Song, J.Y.; Luo, H.M.; Chen, S.L. bHLH transcription factor SmbHLH92 negatively regulates biosynthesis of phenolic acids and tanshinones in Salvia miltiorrhiza. Chin. Herb. Med. 2020, 12, 237–246. [Google Scholar] [CrossRef]
- Wang, H.T.; Wang, Z.D.; Chen, H.T.; Yang, Y. Study on Tanshen red pigment (Ⅰ)—Extraction and chemical components. Food Sci. 2004, 25, 86–91. [Google Scholar]
- Wang, L.L.; Ma, R.F.; Liu, C.Y.; Liu, H.X.; Zhu, R.Y.; Guo, S.Z.; Tang, M.K.; Li, Y.; Niu, J.Z.; Fu, M.; et al. Salvia miltiorrhiza: A potential red light to the development of cardiovascular diseases. Curr. Pharm. Des. 2017, 23, 1077–1097. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.C.; Peters, R.J.; Weirather, J.; Luo, H.M.; Liao, B.S.; Zhang, X.; Zhu, Y.J.; Ji, A.j.; Zhang, B.; Hu, S.N.; et al. Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza and tanshinone biosynthesis. Plant J. 2015, 82, 951–961. [Google Scholar] [CrossRef]
- Wang, Q.R.; Li, X.Y.; Sun, Y.; Mu, D.X.; Yan, Z.Y.; Chen, X. Research on distribution of tanshinones in different parts of danshen’s root by raman spectroscopy. J. Light Scatt. 2018, 30, 351–356. [Google Scholar] [CrossRef]
- Li, S.L.; Zhu, N.L.; Tang, C.P.; Duan, H.N.; Wang, Y.W.; Zhao, G.R.; Liu, J.; Ye, Y. Differential distribution of characteristic constituents in root, stem and leaf tissues of Salvia miltiorrhiza using MALDI mass spectrometry imaging. Fitoterapia 2020, 146, 104679. [Google Scholar] [CrossRef]
- Guo, J.; Zhou, Y.J.; Hillwig, M.L.; Shen, Y.; Yang, L.; Wang, Y.J.; Zhang, X.N.; Liu, W.J.; Peters, R.J.; Chen, X.Y.; et al. CYP76AH1 catalyzes turnover of miltiradiene in tanshinones biosynthesis and enables heterologous production of ferruginol in yeasts. Proc. Natl. Acad. Sci. USA 2013, 110, 12108–12113. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Ma, X.; Cai, Y.; Ma, Y.; Zhan, Z.; Zhou, Y.J.; Liu, W.; Guan, M.; Yang, J.; Cui, G.; et al. Cytochrome P450 promiscuity leads to a bifurcating biosynthetic pathway for tanshinones. New Phytol. 2016, 210, 525–534. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Cui, G.; Chen, T.; Ma, X.; Wang, R.; Jin, B.; Yang, J.; Kang, L.; Tang, J.; Lai, C.; et al. Expansion within the CYP71D subfamily drives the heterocyclization of tanshinones synthesis in Salvia miltiorrhiza. Nat. Commun. 2021, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Fang, X.; Li, C.; Jiang, Y.; Li, J.; Wu, S.; Guo, J.; Liu, Y.; Fan, H.; Huang, Y.; et al. A 2-oxoglutarate-dependent dioxygenase converts dihydrofuran to furan in Salvia diterpenoids. Plant Physiol. 2022, 188, 1496–1506. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Liang, H.; Liu, X.; Lee, J.S.; Cho, J.Y.; Cheong, J.H.; Kim, H.; Li, M.; Downey, T.J.; Dyer, M.D.; et al. AMPKα modulation in cancer progression: Multilayer integrative analysis of the whole transcriptome in Asian gastric cancer. Cancer Res. 2012, 72, 2512–2521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guttman, M.; Rinn, J.L. Modular regulatory principles of large non-coding RNAs. Nature 2012, 482, 339–346. [Google Scholar] [CrossRef] [Green Version]
- Xue, Y.C.; Chen, R.S.; Qu, L.H.; Cao, X.F. Noncoding RNA: From dark matter to bright star. Sci. China Life Sci. 2020, 63, 463–468. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.P.; Tang, Z.H.; Ma, X.X.; Meng, Y.J. Investigating the regulatory roles of the microRNAs and the Argonaute 1-enriched small RNAs in plant metabolism. Gene 2017, 628, 180–189. [Google Scholar] [CrossRef]
- Pani, A.; Mahapatra, R.K.; Behera, N.; Naik, P.K. Computational identification of sweet wormwood (Artemisia annua) microRNA and their mRNA targets. Genom. Proteom. Bioinform. 2011, 9, 200–210. [Google Scholar] [CrossRef] [Green Version]
- Hao, D.C.; Yang, L.; Xiao, P.G.; Liu, M. Identification of Taxus microRNAs and their targets with high-throughput sequencing and degradome analysis. Physiol. Plant 2012, 146, 388–403. [Google Scholar] [CrossRef]
- Singh, N.; Srivastava, S.; Shasany, A.K.; Sharma, A. Identification of miRNAs and their targets involved in the secondary metabolic pathways of Mentha spp. Comput. Biol. Chem. 2016, 64, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.B.; Jiang, Q.H.; Ma, X.Y.; Ying, Q.C.; Shen, B.; Qian, Y.S.; Song, H.M.; Wang, H.Z. Deep sequencing identifies tissue-specific microRNAs and their target genes involving in the biosynthesis of tanshinones in Salvia miltiorrhiza. PLoS ONE 2014, 9, e111679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.H.; Jin, W.B.; Zhu, X.L.; Liu, L.; He, Z.G.; Yang, S.S.; Liang, Z.S.; Yan, X.J.; He, Y.F.; Liu, Y. Identification and characterization of Salvia miltiorrhizain miRNAs in response to replanting disease. PLoS ONE 2016, 11, e0159905. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, S.U.; Grote, P.; Herrmann, B.G. Mechanisms of long noncoding RNA function in development and disease. Cell Mol. Life Sci. 2016, 73, 2491–2509. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Du, Y. LncRNAs: From basic research to medical application. Int. J. Biol. Sci. 2017, 13, 295–307. [Google Scholar] [CrossRef]
- Delás, M.J.; Gregory, J.H. LncRNAs in development and disease: From functions to mechanisms. Open Biol. 2017, 7, 170121. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.P.; Wang, W.; Zhu, W.D.; Dong, J.; Cheng, Y.Y.; Yin, Z.J.; Shen, F.F. Mechanisms and functions of long non-coding RNAs at multiple regulatory levels. Int. J. Mol. Sci. 2019, 20, 5573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Zheng, H.X.; Sui, N. Regulation mechanism of long non-coding RNA in plant response to stress. Biochem. Biophys. Res. Commun. 2018, 503, 402–407. [Google Scholar] [CrossRef]
- Wang, H.; Niu, Q.W.; Wu, H.W.; Liu, J.; Ye, J.; Yu, N.; Chua, N.H. Analysis of non-coding transcriptome in rice and maize uncovers roles of conserved lncRNAs associated with agriculture traits. Plant J. 2015, 84, 404–416. [Google Scholar] [CrossRef]
- Zhu, Q.H.; Stephen, S.; Taylor, J.; Helliwell, C.A.; Wang, M.B. Long noncoding RNAs responsive to Fusarium oxysporum infection in Arabidopsis thaliana. New Phytol. 2014, 201, 574–584. [Google Scholar] [CrossRef]
- Huang, L.; Dong, H.; Zhou, D.; Li, M.; Liu, Y.; Zhang, F.; Feng, Y.Y.; Yu, D.L.; Lin, S.; Cao, J.S. Systematic identification of long non-coding RNAs during pollen development and fertilization in Brassica rapa. Plant J. 2018, 96, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Huanca-Mamani, W.; Arias-Carrasco, R.; Cárdenas-Ninasivincha, S.; Rojas-Herrera, M.; Sepúlveda-Hermosilla, G.; Caris-Maldonado, J.C.; Bastías, E.; Maracaja-Coutinho, V. Long non-coding RNAs responsive to salt and boron stress in the hyper-arid Lluteño maize from Atacama Desert. Genes 2018, 9, 170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, S.B.; Yang, X.S.; Li, X.L.; Wang, J.L.; Gao, Y.; Shang, R.Z.; Sun, W.; Dou, K.F.; Li, H.M. Circular RNA: A new star of noncoding RNAs. Cancer Lett. 2015, 365, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Panda, A.C.; Grammatikakis, I.; Munk, R.; Gorospe, M.; Abdelmohsen, K. Emerging roles and context of circular RNAs. Wiley Interdiscip. Rev. RNA 2017, 8, e1386. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.J.; Zhou, Z.J.; Niu, Y.J.; Sun, X.Y.; Deng, Z.P. Identification and functional characterization of tomato circRNAs derived from genes involved in fruit pigment accumulation. Sci. Rep. 2017, 7, 8594. [Google Scholar] [CrossRef] [Green Version]
- Tong, W.; Yu, J.; Hou, Y.; Li, F.D.; Zhou, Q.Y.; Wei, C.L.; Bennetzen, J.L. Circular RNA architecture and differentiation during leaf bud to young leaf development in tea (Camellia sinensis). Planta 2018, 248, 1417–1429. [Google Scholar] [CrossRef]
- Wang, J.X.; Lin, J.; Wang, H.; Li, X.G.; Yang, Q.S.; Li, H.; Chang, Y.H. Identification and characterization of circRNAs in Pyrus betulifolia Bunge under drought stress. PLoS ONE 2018, 13, e0200692. [Google Scholar] [CrossRef]
- Pan, T.; Sun, X.Q.; Liu, Y.X.; Li, H.; Deng, G.B.; Lin, H.H.; Wang, S.H. Heat stress alters genome-wide profiles of circular RNAs in Arabidopsis. Plant Mol. Biol. 2018, 96, 217–229. [Google Scholar] [CrossRef]
- Fang, X.S.; Wang, J.H.; Zhang, S.; Zhao, Q.Q.; Zheng, Z.K.; Song, Z.Q. Simultaneous extraction of hydrosoluble phenolic acids and liposoluble tanshinones from Salviae miltiorrhizae radix by an optimized microwave-assisted extraction method. Sep. Purif. Technol. 2012, 86, 149–156. [Google Scholar] [CrossRef]
- Li, H.Q.; Li, C.L.; Deng., Y.X.; Jiang., X.W.; Lu, S.F. The pentatricopeptide repeat gene family in Salvia miltiorrhiza: Genome-wide characterization and expression analysis. Molecules 2018, 23, 1364. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Guo, L.L.; Pan, Y.L.; Zhao, Q.; Wang, J.H.; Song, Z.Q. Construction of the first high-density genetic linkage map of Salvia miltiorrhiza using specific length amplified fragment (SLAF) sequencing. Sci. Rep. 2016, 6, 24070. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, W.; Xia, Y.S.; Li, R.H.; Bai, G.H.; Siddique, K.H.M.; Guo, P.G. Non-coding RNAs: Functional roles in the regulation of stress response in Brassica crops. Genomics 2020, 112, 1419–1424. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Jiang, Y.L.; Cui, W.T.; Jin, Q.J.; Zhang, Y.H.; Bu, D.; Fu, J.Y.; Wang, R.; Zhou, F.; Shen, W.B. Hydrogen enhances adaptation of rice seedlings to cold stress via the reestablishment of redox homeostasis mediated by miRNA expression. Plant Soil 2017, 414, 53–67. [Google Scholar] [CrossRef]
- Zhang, J.S.; Zhang, H.; Srivastava, A.K.; Pan, Y.J.; Bai, J.J.; Fang, J.J.; Shi, H.Z.; Zhu, J.K. Knockdown of rice microRNA166 confers drought resistance by causing leaf rolling and altering stem xylem development. Plant Physiol. 2018, 176, 2082–2094. [Google Scholar] [CrossRef] [Green Version]
- Zuo, J.H.; Wang, Y.X.; Zhu, B.Z.; Luo, Y.B.; Wang, Q.; Gao, L.P. Analysis of the coding and non-coding RNA transcriptomes in response to Bell Pepper chilling. Int. J. Mol. Sci. 2018, 19, 2001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, L.J.; Liu, Z.B.; Zhang, Z.Q.; Wei, G.; Zhang, Y.P.; Kang, L.Y.; Yang, Z.; Yang, S.; Lv, J.H.; Liu, Y.H.; et al. Noncoding and coding transcriptome analysis reveals the regulation roles of long noncoding RNAs in fruit development of hot pepper (Capsicum annuum L.). Plant Growth Regul. 2017, 83, 141–156. [Google Scholar] [CrossRef]
- Su, Y.; Zhang, J.; Xu, Z.; Li, J.; Wang, P.; Song, Z.; Tian, G.; Li, L.; Song, J.; Wang, J. Integrative analysis of metabolome and transcriptome reveals the mechanism of color formation in white root (Salvia miltiorrhiza). Ind. Crop. Prod. 2021, 110, 113784. [Google Scholar] [CrossRef]
- Xu, H.; Song, J.; Luo, H.; Zhang, Y.; Li, Q.; Zhu, Y.; Xu, J.; Li, Y.; Song, C.; Wang, B.; et al. Analysis of the Genome Sequence of the Medicinal Plant Salvia miltiorrhiza. Mol. Plant 2016, 9, 949–952. [Google Scholar] [CrossRef] [Green Version]
- Crouzet, J.; Roland, J.; Peeters, E.; Trombik, T.; Ducos, E.; Nader, J.; Boutry, M. NtPDR1, a plasma membrane ABC transporter from Nicotiana tabacum, is involved in diterpene transport. Plant Mol. Biol. 2013, 82, 181–192. [Google Scholar] [CrossRef]
- Yan, L.; Zhang, J.; Chen, H.; Luo, H. Genome-wide analysis of ATP-binding cassette transporter provides insight to genes related to bioactive metabolite transportation in Salvia miltiorrhiza. BMC Genom. 2021, 22, 315. [Google Scholar] [CrossRef]
- Li, D.Q.; Shao, F.J.; Lu, S.F. Identification and characterization of mRNA-like noncoding RNAs in Salvia miltiorrhiza. Planta 2015, 241, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, H.; Hou, Z.; Xu, L.; Liang, Z. Overexpression of ath-mir160b increased the biomass while reduced the content of tanshinones in Salvia miltiorrhiza hairy roots by targeting arfs genes. Plant Cell Tissue Organ Cult. 2020, 142, 327–338. [Google Scholar] [CrossRef]
- Zou, H.; Guo, X.; Yang, R.; Wang, S.; Li, L.; Niu, J.; Wang, D.; Cao, X. MiR408-SmLAC3 module participates in salvianolic acid B synthesis in Salvia miltiorrhiza. Int. J. Mol. Sci. 2021, 22, 7541. [Google Scholar] [CrossRef] [PubMed]
- Curtis, R.H.; Pankaj; Powers, S.J.; Napier, J.; Matthes, M.C. The Arabidopsis F-box/Kelch-repeat protein At2g44130 is upregulated in giant cells and promotes nematode susceptibility. Mol. Plant Microbe. Interact. 2013, 26, 36–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.B.; Zhai, X.Q.; Xu, E.K.; Zhao, Z.L.; Fan, G.Q. Genome-wide identification of candidate genes related to disease resistance and high biomass in tetraploid Paulownia. Acta Physiol. Plant 2020, 42, 171. [Google Scholar] [CrossRef]
- Ahuja, I.; Kissen, R.; Bones, A.M. Phytoalexins in defense against pathogens. Trends Plant Sci. 2012, 17, 73–90. [Google Scholar] [CrossRef]
- Zhan, C.S.; Lei, L.; Liu, Z.X.; Zhou, S.; Yang, C.K.; Zhu, X.T.; Guo, H.; Zhang, F.; Peng, M.; Zhang, M.; et al. Selection of a subspecies-specific diterpene gene cluster implicated in rice disease resistance. Nat. Plants 2020, 6, 1447–1454. [Google Scholar] [CrossRef]
- Lu, X.; Zhang, J.; Brown, B.; Li, R.Q.; Rodríguez-Romero, J.; Berasategui, A.; Liu, B.; Xu, M.M.; Luo, D.P.; Pan, Z.Q.; et al. Inferring roles in defense from metabolic allocation of rice diterpenoids. Plant Cell 2018, 30, 1119–1131. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Content (mg/g) | Phloem in Red Root | Xylem in Red Root | Phloem in White Root | Xylem in White Root | Wavelength Detected (nm) |
|---|---|---|---|---|---|
| Tanshinone IIA | 4.34 | 0.001 | 0 | 0 | 269 |
| Dihydrotanshinone | 0.5 | 0.001 | 0 | 0 | 269 |
| Cryptotanshinone | 1.51 | 0.002 | 0.001 | 269 | |
| Tanshinone I | 0.968 | 0.001 | 0 | 0 | 269 |
| Type | Annotated | COG | GO | KEGG | KOG | Swissprot | eggNOG | Nr |
|---|---|---|---|---|---|---|---|---|
| DE-mRNAs | 661 | 260 | 362 | 204 | 332 | 543 | 205 | 40 |
| Targets of DE-miRNAs | 1232 | 375 | 473 | 315 | 677 | 894 | 1116 | 143 |
| cis-targets of DE-LncRNAs | 304 | 125 | 165 | 111 | 184 | 210 | 0 | 0 |
| tran-targets of DE-LncRNAs | 11 | 2 | 6 | 3 | 9 | 5 | 0 | 0 |
| source genes of DE-circRNAs | 12 | 5 | 9 | 3 | 5 | 9 | 12 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, C.; Zhou, C.; Liu, Z.; Li, X.; Song, Z. Identification of the Genome-Wide Expression Patterns of Non-Coding RNAs Associated with Tanshinones Synthesis Pathway in Salvia miltiorrhiza. Agronomy 2023, 13, 321. https://0-doi-org.brum.beds.ac.uk/10.3390/agronomy13020321
Lin C, Zhou C, Liu Z, Li X, Song Z. Identification of the Genome-Wide Expression Patterns of Non-Coding RNAs Associated with Tanshinones Synthesis Pathway in Salvia miltiorrhiza. Agronomy. 2023; 13(2):321. https://0-doi-org.brum.beds.ac.uk/10.3390/agronomy13020321
Chicago/Turabian StyleLin, Caicai, Changhao Zhou, Zhongqian Liu, Xingfeng Li, and Zhenqiao Song. 2023. "Identification of the Genome-Wide Expression Patterns of Non-Coding RNAs Associated with Tanshinones Synthesis Pathway in Salvia miltiorrhiza" Agronomy 13, no. 2: 321. https://0-doi-org.brum.beds.ac.uk/10.3390/agronomy13020321