
Fine-Mapping and Candidate Gene Analysis of qSERg-1b from O. glumaepatula to Improve Stigma Exsertion Rate in Rice


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Phenotyping
2.3. Genotyping
2.4. Fine-Mapping of qSERg-1b
2.5. Candidate Gene Prediction
2.6. Whole Genome Re-Sequencing
2.7. RNA Extraction and Quantitative Real-Time PCR (qRT-PCR)
2.8. Data Analysis and Illustration
3. Results
3.1. Validation of the Chromosomal Region and Effects of qSERg-1b in SG22
3.2. Fine-Mapping of qSERg-1b
3.3. Gene Annotation and Candidate Gene Prediction of qSERg-1b
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yuan, L.P.; Virmani, S.S. Status of hybrid rice research and development. In Hybrid Rice; IRRI: Manila, Philippines, 1988; pp. 7–24. [Google Scholar]
- Rashid, H.A.; Ali, M.; Gisselquist, D. Private-Sector Agricultural Research and Innovation in Bangladesh: Overview, Impact, and Policy Options; IFPRI: Washington, DC, USA, 2012; Available online: http://www.asti.cgiar.org/pdf/private-sector/Bangladesh-PS-Report.pdf (accessed on 17 August 2023).
- Abebrese, S.O.; Amoah, N.K.A.; Dartey, P.K.A.; Bimpong, I.K.; Akromah, R.; Gracen, V.E.; Offei, S.K.; Danquah, E.Y. Mapping chromosomal regions associated with anther indehiscence with exerted stigmas in CRI-48 and Jasmine 85 cross of rice (Oryza sativa L.). Heliyon 2021, 7, e06483. [Google Scholar] [CrossRef]
- Li, J.; Yuan, L. Hybrid rice: Genetics, breeding and seed production. In Plant Breeding Reviews; Janick, J., Ed.; John Wiley & Sons, Ltd.: Oxford, UK, 2000; Volume 17, pp. 15–158. [Google Scholar] [CrossRef]
- Anwar, M.; Zulfiqar, F.; Ferdous, Z.; Tsusaka, T.W.; Datta, A. Productivity, profitability, efficiency, and land utilization scenarios of rice cultivation: An assessment of hybrid rice in Bangladesh. Sustain. Prod. Consump. 2021, 26, 752–758. [Google Scholar] [CrossRef]
- Sarma, P.K.; Alam, M.J.; Begum, I.A. Farmers’ knowledge, attitudes, and practices towards the adoption of hybrid rice production in Bangladesh: An PLS-SEM approach. GM Crops Food 2022, 13, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.L.; Zheng, Y.C.; Liu, Y.; Guo, X.H.; Tan, Y.Y.; Qian, Q.P.; Shu, Q.Y.; Huang, J.Z. Identification of a major quantitative trait locus and its candidate underlying genetic variation for rice stigma exsertion rate. Crop J. 2019, 7, 350–359. [Google Scholar] [CrossRef]
- Takano-Kai, N.; Doi, K.; Yoshimura, A. GS3 participates in stigma exsertion as well as seed length in rice. Breed. Sci. 2011, 61, 244–250. [Google Scholar] [CrossRef]
- Marathi, B.; Jena, K.K. Floral traits to enhance outcrossing for higher hybrid seed production in rice: Present status and future prospects. Euphytica 2015, 201, 1–14. [Google Scholar] [CrossRef]
- Miyata, M.; Yamamoto, T.; Komori, T.; Nitta, N. Marker-assisted selection and evaluation of the QTL for stigma exsertion under japonica rice genetic background. Theor. Appl. Genet. 2007, 114, 539–548. [Google Scholar] [CrossRef]
- Yamamoto, T.; Takemori, N.; Sue, N.; Nitta, N. QTL analysis of stigma exsertion in rice. Rice Genet. Newsl. 2003, 20, 33–34. [Google Scholar]
- Rahman, M.H.; Zhang, Y.X.; Zhang, K.Q.; Rahman, M.S.; Barman, H.N.; Riaz, A.; Chen, Y.Y.; Wu, W.X.; Zhan, X.D.; Cao, L.Y.; et al. Genetic dissection of the major quantitative trait locus (qSE11), and its validation as the major influence on the rate of stigma exsertion in rice (Oryza sativa L.). Front. Plant Sci. 2017, 8, 1818–1827. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, A.N.; Wang, F.M.; Kong, D.Y.; Li, M.S.; Bi, J.G.; Zhang, F.Y.; Wang, J.H.; Luo, X.X.; Pan, Z.Q.; et al. Fine mapping a quantitative trait locus, qSER-7, that controls stigma exsertion rate in rice (Oryza sativa L.). Rice 2019, 12, 46–55. [Google Scholar] [CrossRef]
- Guo, N.H.; Wang, Y.K.; Chen, W.; Tang, S.J.; An, R.H.; Wei, X.J.; Hu, S.K.; Tang, S.Q.; Shao, G.N.; Jiao, G.A.; et al. Fine mapping and target gene identification of qSE4, a QTL for stigma exsertion rate in rice (Oryza sativa L.). Front. Plant Sci. 2022, 13, 959859. [Google Scholar] [CrossRef]
- Tan, Q.Y.; Zhu, H.T.; Liu, H.; Ni, Y.R.; Wu, S.Z.; Luan, X.; Liu, J.W.; Yang, W.F.; Yang, Z.F.; Zeng, R.Z.; et al. Fine mapping of QTLs for stigma exsertion rate from Oryza glaberrima by chromosome segment substitution. Rice Sci. 2022, 29, 55–66. [Google Scholar] [CrossRef]
- Oka, H.; Morishima, H. Wild and cultivated rice. In Science of the Rice Plant, 3rd ed.; Matuo, T., Hoshikawa, K., Eds.; Food and Agriculture Policy Research Center: Tokyo, Japan, 1997; pp. 88–111. [Google Scholar]
- Uga, Y.; Fukuta, Y.; Cai, H.W.; Iwata, H.; Ohsawa, R.; Morishima, H.; Fujimura, T. Mapping QTLs influencing rice floral morphology using recombinant inbred lines derived from a cross between Oryza sativa L. and Oryza ruftpogon Griff. Theor. Appl. Genet. 2003, 107, 218–226. [Google Scholar] [CrossRef]
- Xiong, L.Z.; Liu, K.D.; Dai, X.K.; Xu, C.G.; Zhang, Q. Identification of genetic factors controlling domestication-related traits of rice using an F2 population of a cross between Oryza sativa and O. rufipogon. Theor. Appl. Genet. 1999, 98, 243–251. [Google Scholar] [CrossRef]
- Uga, Y.; Fukuta, Y.; Ohsawa, R.; Fujimura, T. Variations of floral traits in Asian cultivated rice (Oryza sativa L.) and its wild relatives (O. rufipogon Griff.). Breed. Sci. 2003, 53, 345–352. [Google Scholar] [CrossRef]
- Bakti, C.; Tanaka, J. Detection of dominant QTLs for stigma exsertion ratio in rice derived from Oryza rufipogon accession ‘W0120’. Breed. Sci. 2019, 69, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Zou, T.; Zhao, H.W.; Li, X.H.; Zheng, M.M.; Zhang, S.D.; Sun, L.L.; He, N.; Pan, X.P.; Liu, Z.Q.; Fu, X.L. QTLs detection and pyramiding for stigma exsertion rate in wild rice species by using the single-segment substitution lines. Mol. Breed. 2020, 40, 74–87. [Google Scholar] [CrossRef]
- Li, H.B.; Gao, F.Y.; Zeng, L.H.; Li, Q.X.; Lu, X.J.; Li, Z.H.; Ren, J.S.; Su, X.W.; Ren, G.J. QTL Analysis of rice stigma morphology using an introgression line from Oryza longistaminata L. Mol. Plant Breed. 2010, 8, 1082–1089. [Google Scholar]
- Ali, M.L.; Sanchez, P.L.; Yu, S.; Lorieux, M.; Eizenga, G.C. Chromosome segment substitution lines: A powerful tool for the introgression of valuable genes from Oryza wild species into cultivated rice (O. sativa). Rice 2010, 3, 218–234. [Google Scholar] [CrossRef]
- Zhang, G.Q. Target chromosome-segment substitution: A way to breeding by design in rice. Crop J. 2021, 9, 658–668. [Google Scholar] [CrossRef]
- Tan, Q.Y.; Zou, T.; Zheng, M.M.; Ni, Y.R.; Luan, X.; Li, X.H.; Yang, W.F.; Yang, Z.F.; Zhu, H.T.; Zeng, R.Z.; et al. Substitution mapping of the major quantitative trait loci controlling stigma exsertion rate from Oryza Glumaepatula. Rice 2020, 13, 37–46. [Google Scholar] [CrossRef]
- He, H.G.; Ji, Y.Y.; Zhu, S.Y.; Li, B.; Zhao, R.H.; Jiang, Z.N.; Bie, T.D. Genetic, physical and comparative mapping of the powdery mildew resistance gene Pm21 originating from Dasypyrum villosum. Front. Plant Sci. 2017, 8, 1914–1922. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Hori, K.; Nonoue, Y.; Ono, N.; Shibaya, T.; Ebana, K.; Matsubara, K.; Ogiso-Tanaka, E.; Tanabata, T.; Sugimoto, K.; Taguchi-Shiobara, F.; et al. Genetic architecture of variation in heading date among Asian rice accessions. BMC Plant Biol. 2015, 15, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, N.; Kato, M.; Koyasaki, K.; Kawashima, T.; Nishimura, T.; Hirayama, Y.; Takamure, I.; Sato, T.; Kato, K. Identification of quantitative trait loci for rice grain quality and yield-related traits in two closely related Oryza sativa L. subsp japonica cultivars grown near the northernmost limit for rice paddy cultivation. Breed. Sci. 2017, 67, 191–206. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.N.; Wang, L.L.; Zhu, Y.J.; Fan, Y.Y.; Zhuang, J.Y.; Zhang, Z.H. Identification through fine mapping and verification using CRISPR/Cas9-targeted mutagenesis for a minor QTL controlling grain weight in rice. Theor. Appl. Genet. 2021, 134, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.W.; Zhang, K.M.; Gull, S.; Chen, C.Y.; Ran, J.H.; Zou, B.Y.; Wang, P.; Liang, G.H. Fine mapping of qTGW7b, a minor effect QTL for grain weight in rice (Oryza sativa L.). Int. J. Mol. Sci. 2022, 23, 8296. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.X.; Zheng, X.M.; Lu, G.W.; Zhong, Z.Z.; Gao, H.; Chen, L.P.; Wu, C.Y.; Wang, H.J.; Wang, Q.; Zhou, K.N.; et al. Association of functional nucleotide polymorphisms at DTH2 with the northward expansion of rice cultivation in Asia. Proc. Natl. Acad. Sci. USA 2013, 110, 2775–2780. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Jin, J.B.; Hasegawa, P.M. Sumoylation, a post-translational regulatory process in plants. Curr. Opin. Plant Biol. 2007, 10, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Lee, J.; Jin, J.B.; Yoo, C.Y.; Miura, T.; Hasegawa, P.M. Sumoylation of ABI5 by the Arabidopsis SUMO E3 ligase SIZ1 negatively regulates abscisic acid signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 5418–5423. [Google Scholar] [CrossRef]
- Kim, S.I.; Park, B.S.; Kim, D.Y.; Yeu, S.Y.; Song, S.I.; Song, J.T.; Seo, H.S. E3 SUMO ligase AtSIZ1 positively regulates SLY1-mediated GA signalling and plant development. Biochem. J. 2015, 469, 299–314. [Google Scholar] [CrossRef]
- Mutuku, J.M.; Nose, A. High activities and mRNA expression of pyrophosphate-fructose-6-phosphatephosphotransferase and 6-phosphofructokinase are induced as a response to Rhizoctonia solani infection in rice leaf sheaths. Physiol. Mol. Plant Pathol. 2012, 77, 41–51. [Google Scholar] [CrossRef]
- Li, J.; Li, X.Y.; Bai, Y.C.; Xie, Y.L.; Li, L.; Mu, S.H.; Gao, J. Transcriptome analysis of energy supply process during seed germination in Phyllostachys edulis. Plant Mol. Biol. Rep. 2023, 41, 489–511. [Google Scholar] [CrossRef]
- Song, W.M.; Hu, L.; Ma, Z.H.; Yang, L.; Li, J.M. Importance of tyrosine phosphorylation in hormone-regulated plant growth and development. Int. J. Mol. Sci. 2022, 23, 6603. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K.; Yamaguchi, M.; Kanamori, K.; Ariga, H.; Isono, K.; Kajino, T.; Tanaka, K.; Saijo, Y.; Yotsui, I.; Sakata, Y.; et al. MAP KINASE PHOSPHATASE1 promotes osmotolerance by suppressing PHYTOALEXIN DEFICIENT 4-independent immunity. Plant Physiol. 2022, 189, 1128–1138. [Google Scholar] [CrossRef] [PubMed]
- Blair, M.W.; Cortés, A.J.; This, D. Identification of an ERECTA gene and its drought adaptation associations with wild and cultivated common bean. Plant Sci. 2016, 242, 250–259. [Google Scholar] [CrossRef]
- Guo, T.; Lu, Z.Q.; Shan, J.X.; Ye, W.W.; Dong, N.Q.; Lin, H.X. ERECTA1 acts upstream of the OsMKKK10-OsMKK4-OsMPK6 cascade to control spikelet number by regulating cytokinin metabolism in rice. Plant Cell 2020, 32, 2763–2779. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Bai, G.; Rife, T.W.; Poland, J.; Lin, M.; Liu, S.; Chen, H.; Kumssa, T.; Fritz, A.; Trick, H.; et al. QTL mapping of pre-harvest sprouting resistance in a white wheat cultivar Danby. Theor. Appl. Genet. 2018, 131, 1683–1697. [Google Scholar] [CrossRef]
- Tan, Q.Y.; Wang, C.S.; Luan, X.; Zheng, L.J.; Ni, Y.R.; Yang, W.F.; Yang, Z.F.; Zhu, H.T.; Zeng, R.Z.; Liu, G.F.; et al. Dissection of closely linked QTLs controlling stigma exsertion rate in rice by substitution mapping. Theor. Appl. Genet. 2021, 134, 1253–1262. [Google Scholar] [CrossRef]
- Tan, Q.Y.; Bu, S.H.; Chen, G.D.; Yan, Z.G.; Chang, Z.Y.; Zhu, H.T.; Yang, W.F.; Zhan, P.L.; Lin, S.J.; Xiong, L.; et al. Reconstruction of the high stigma exsertion rate trait in rice by pyramiding multiple QTLs. Front. Plant Sci. 2022, 13, 921700. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
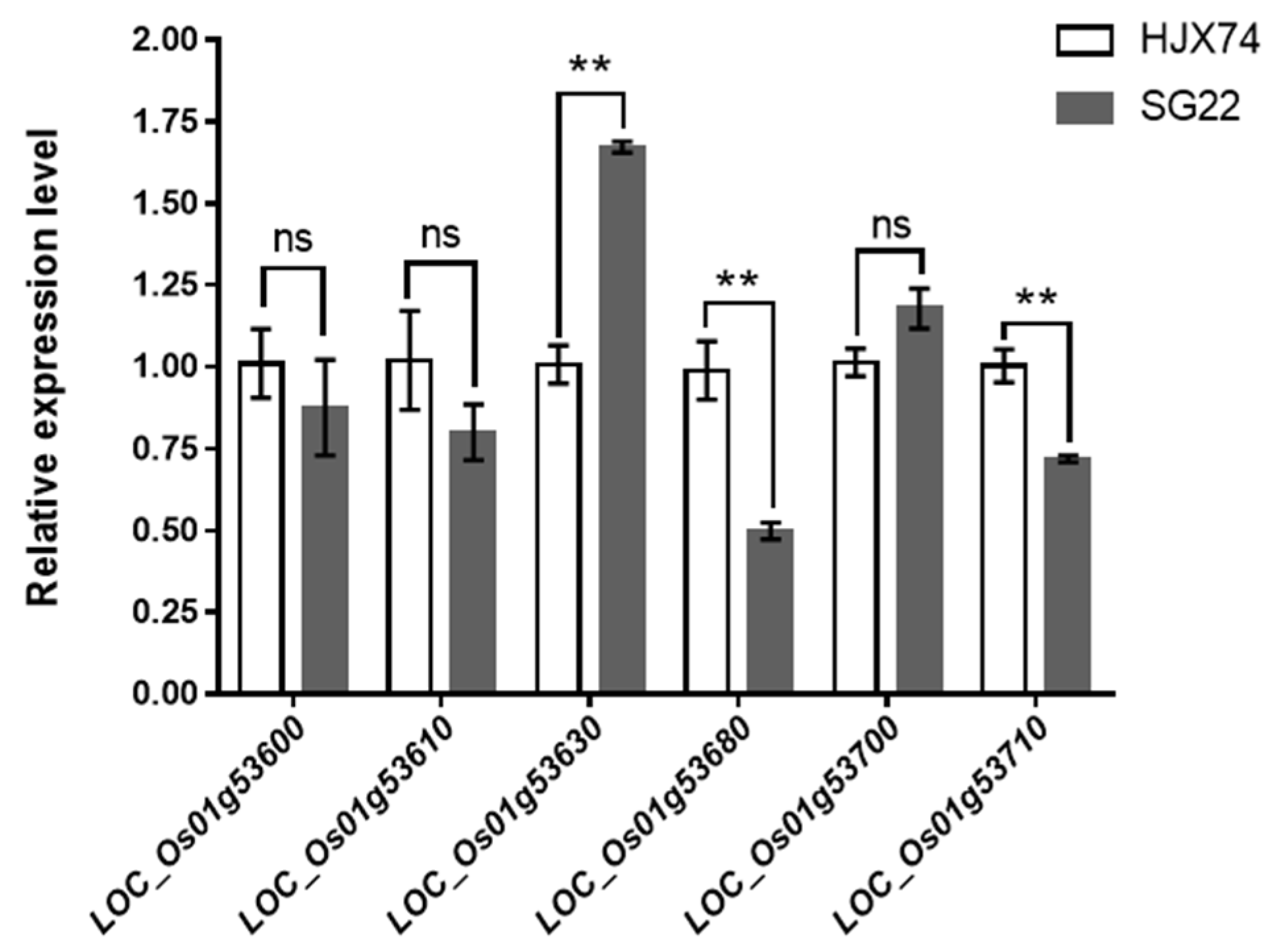{kind=link}
| MSU_Locus ID | Annotation | Nonsynonymous Mutations in Exon Regions |
|---|---|---|
| LOC_Os01g53630 | ulp1 protease family protein, putative, expressed | exon10: A1231G; I411V |
| exon8: C1068A; D356E | ||
| exon4: 724_741del; 242_247del | ||
| exon2: A376C; K126Q | ||
| exon1: T341C; L114P | ||
| exon1: T287G; L96W | ||
| LOC_Os01g53680 | 6-phosphofructokinase, putative, expressed | None |
| LOC_Os01g53710 | Dual-specificity protein phosphatase, putative, expressed | exon6: A977G; N326S |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, L.; Dan, J.; Li, X.; Tan, Q.; Zhang, S.; Song, R.; Fu, X. Fine-Mapping and Candidate Gene Analysis of qSERg-1b from O. glumaepatula to Improve Stigma Exsertion Rate in Rice. Agronomy 2024, 14, 323. https://0-doi-org.brum.beds.ac.uk/10.3390/agronomy14020323
Cao L, Dan J, Li X, Tan Q, Zhang S, Song R, Fu X. Fine-Mapping and Candidate Gene Analysis of qSERg-1b from O. glumaepatula to Improve Stigma Exsertion Rate in Rice. Agronomy. 2024; 14(2):323. https://0-doi-org.brum.beds.ac.uk/10.3390/agronomy14020323
Chicago/Turabian StyleCao, Lixia, Juncheng Dan, Xiaohui Li, Quanya Tan, Shaodi Zhang, Ruifeng Song, and Xuelin Fu. 2024. "Fine-Mapping and Candidate Gene Analysis of qSERg-1b from O. glumaepatula to Improve Stigma Exsertion Rate in Rice" Agronomy 14, no. 2: 323. https://0-doi-org.brum.beds.ac.uk/10.3390/agronomy14020323