Single-Molecule Long-Read Sequencing of Avocado Generates Microsatellite Markers for Analyzing the Genetic Diversity in Avocado Germplasm

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection, DNA Extraction, and RNA Extraction
2.2. PacBiocDNA Library Construction and Sequencing
2.3. IlluminacDNA Library Construction and Sequencing
2.4. Quality Filtering and Correction of PacBio Long-Reads
2.5. Functional Annotation
2.6. Mining of EST-SSR Markers
2.7. Analyses of Detected Coding Sequences, Transcription Factors, and Long Non-Coding RNA Features
2.8. Assignment of the Native Avocado Accessions with an Unknown Race
2.9. Identification of EST-SSR Markers
2.10. Data Analysis
3. Results
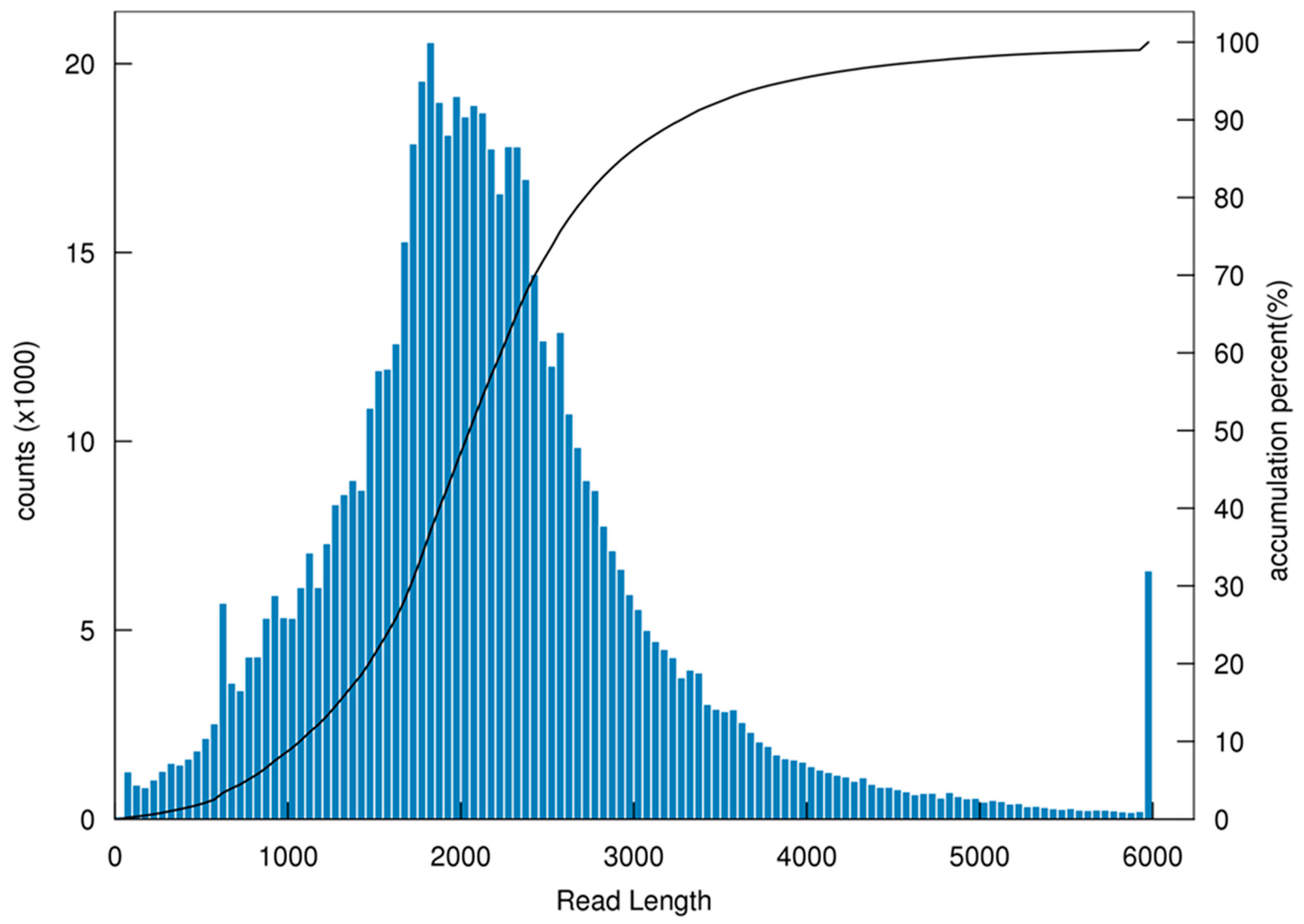
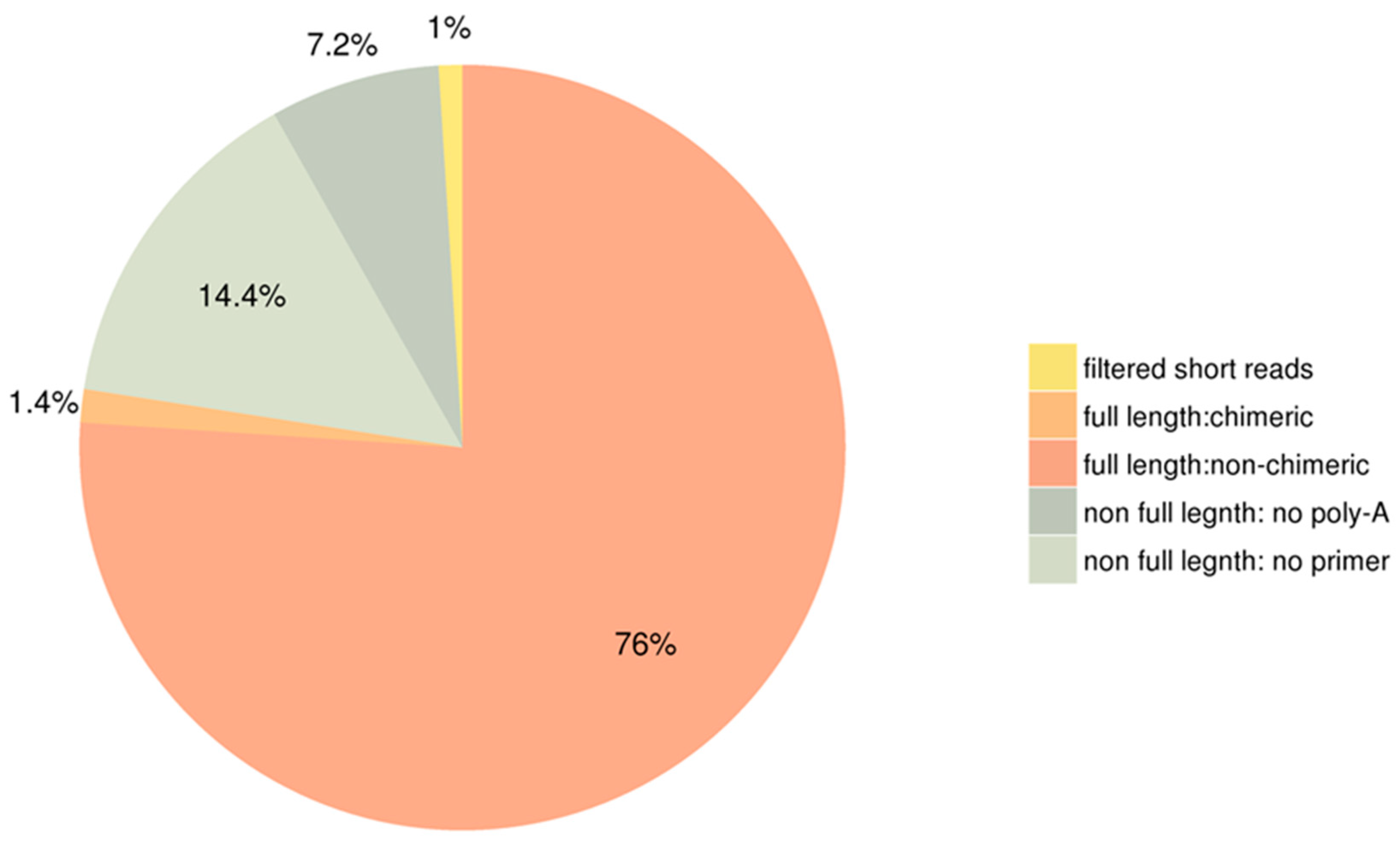
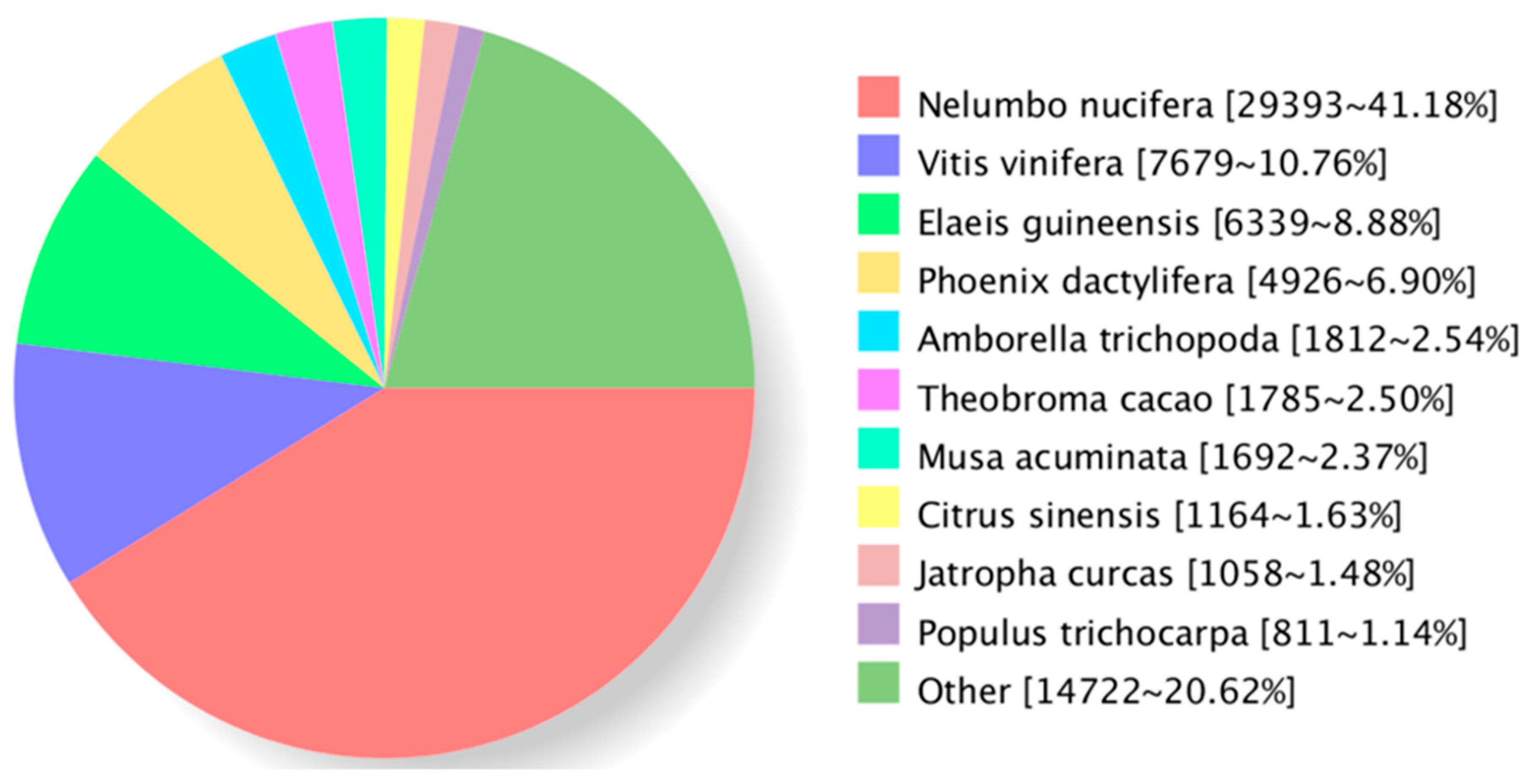
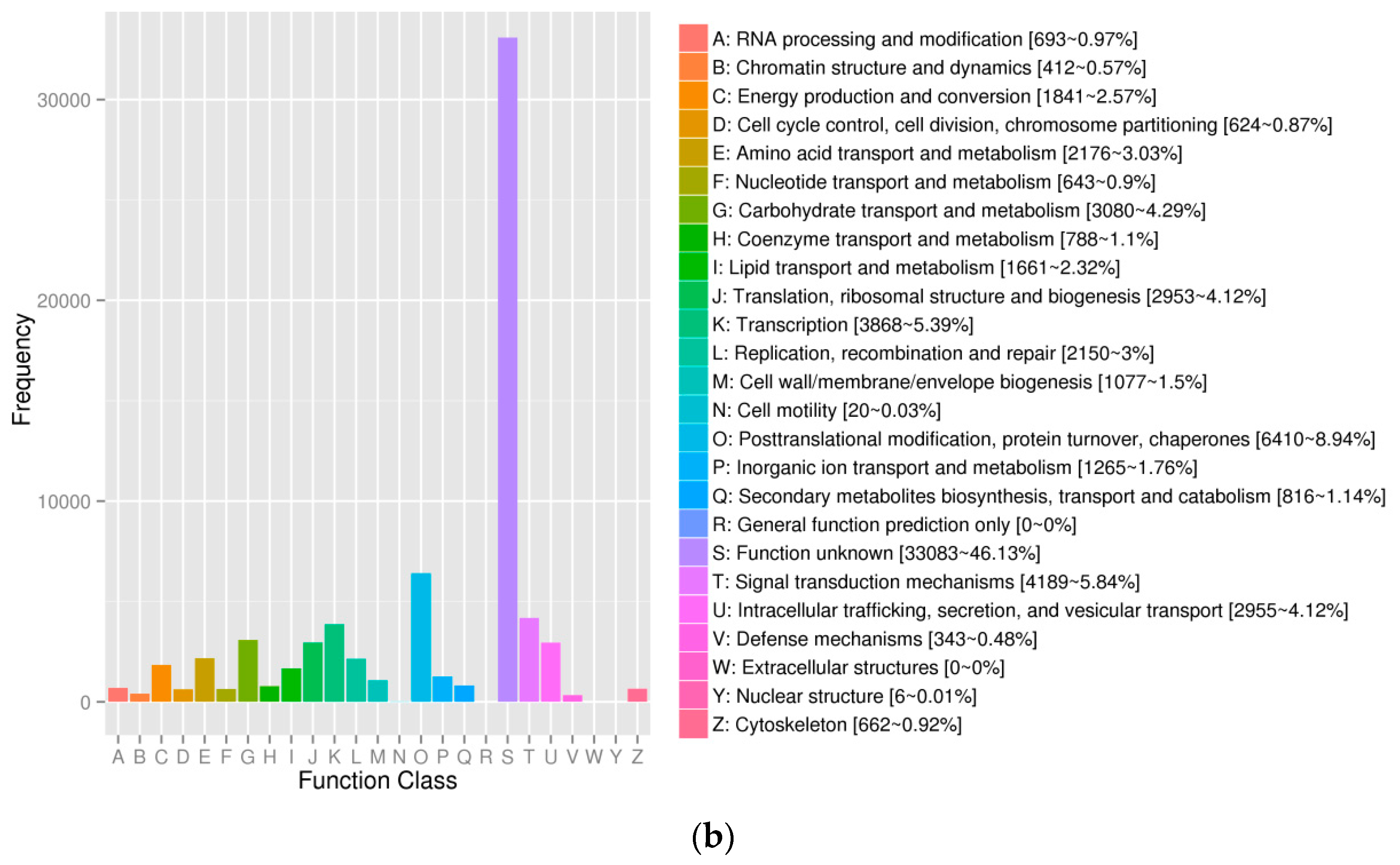
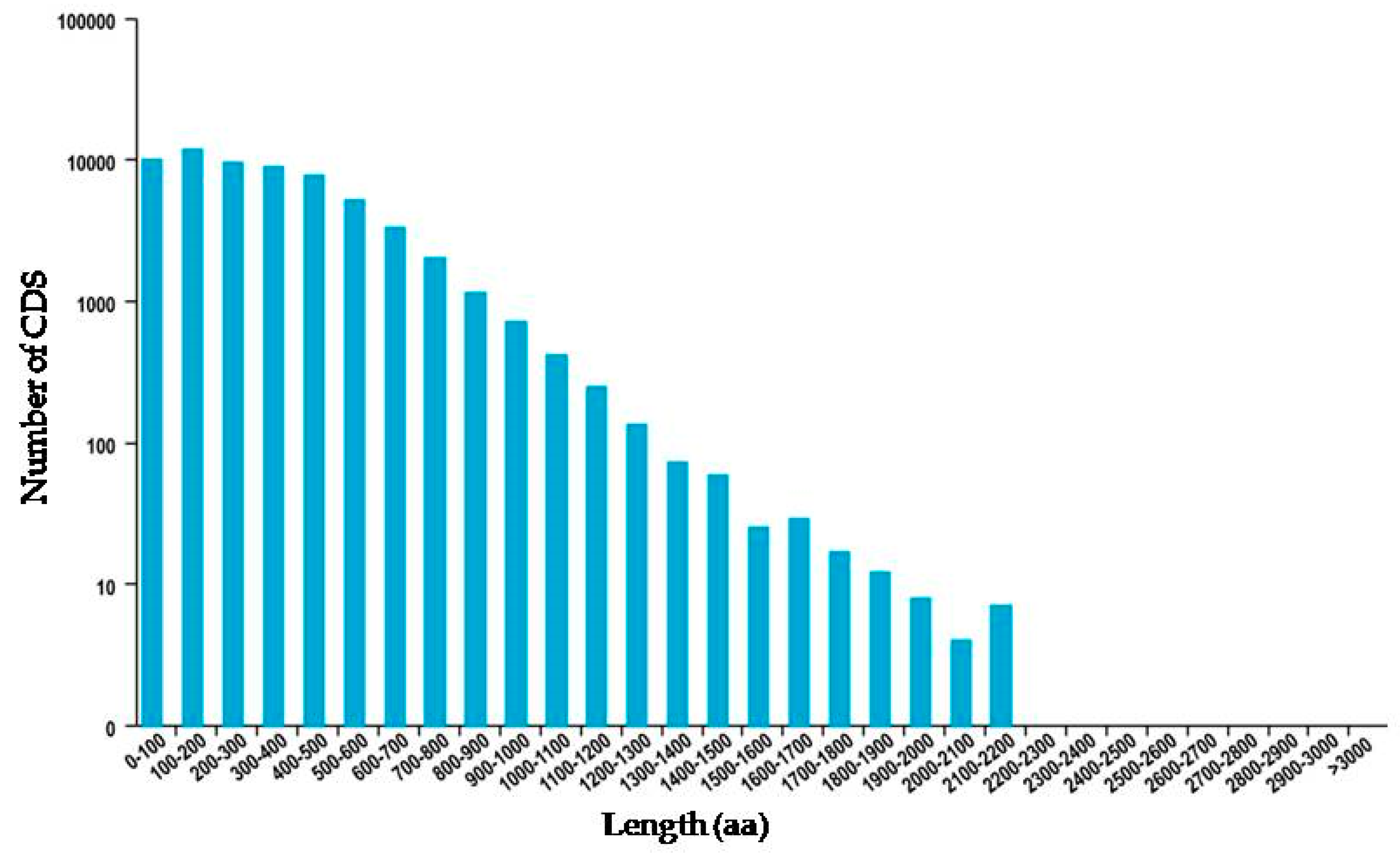
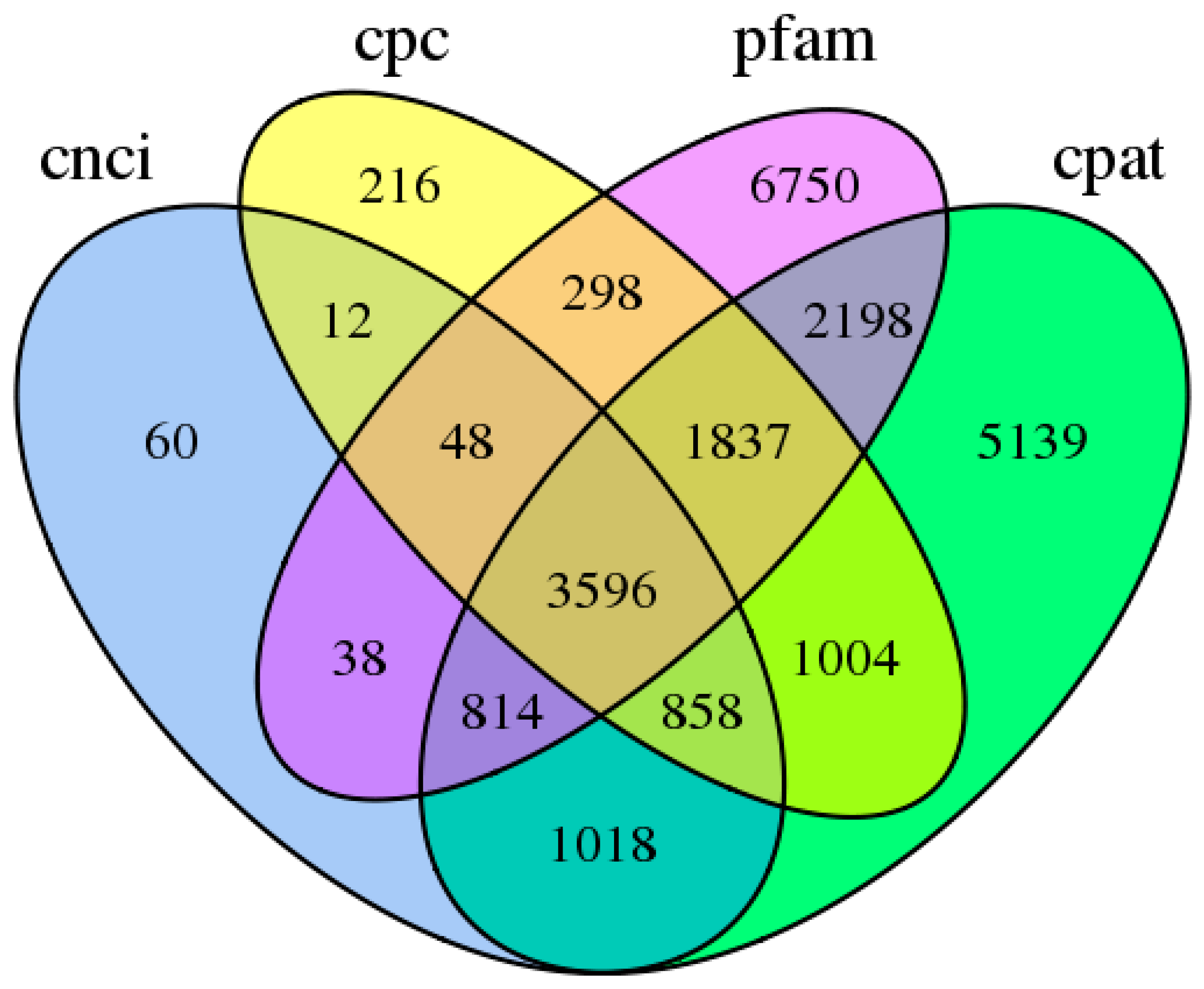
3.1. General Properties and Functional Annotations Based on Public Databases of Single-Molecule Long-Reads
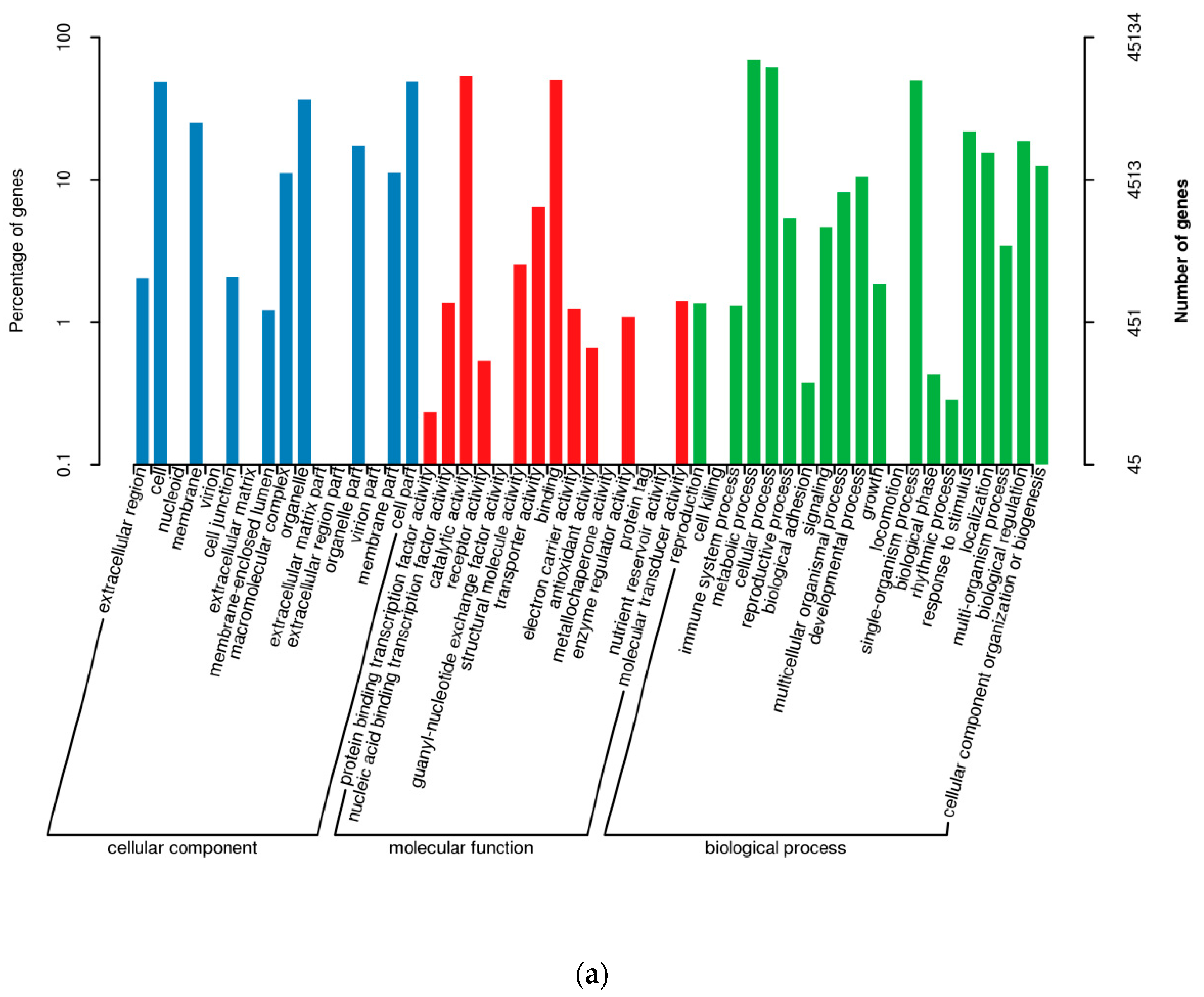
3.2. Predictions of ORFs, TFs, and lncRNAs
3.3. Frequency and Distribution of Various Types of EST-SSR Loci
3.4. Development of Polymorphic EST-SSR Markers, Analysis of Genetic Diversity, and KASP genotyping
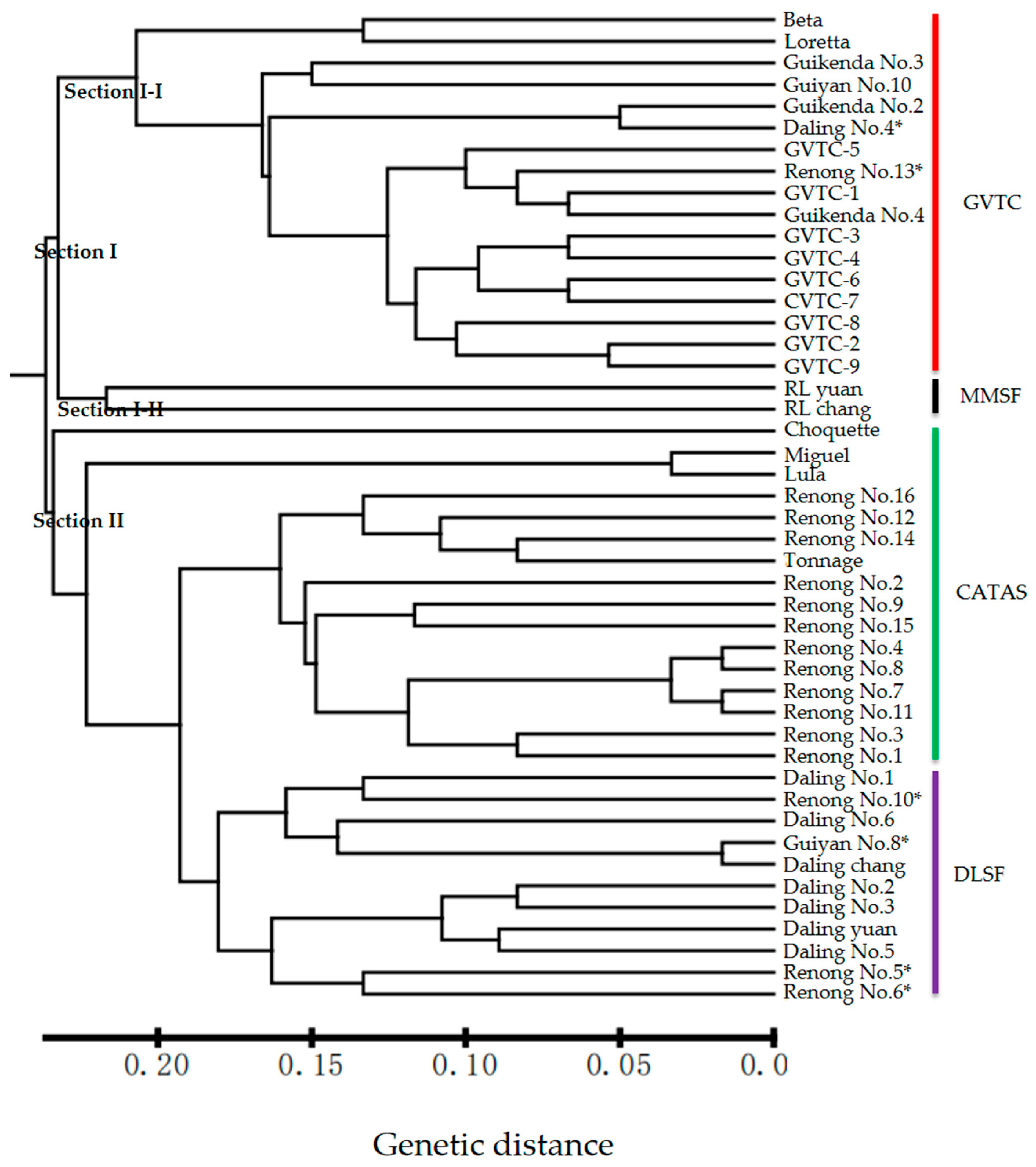
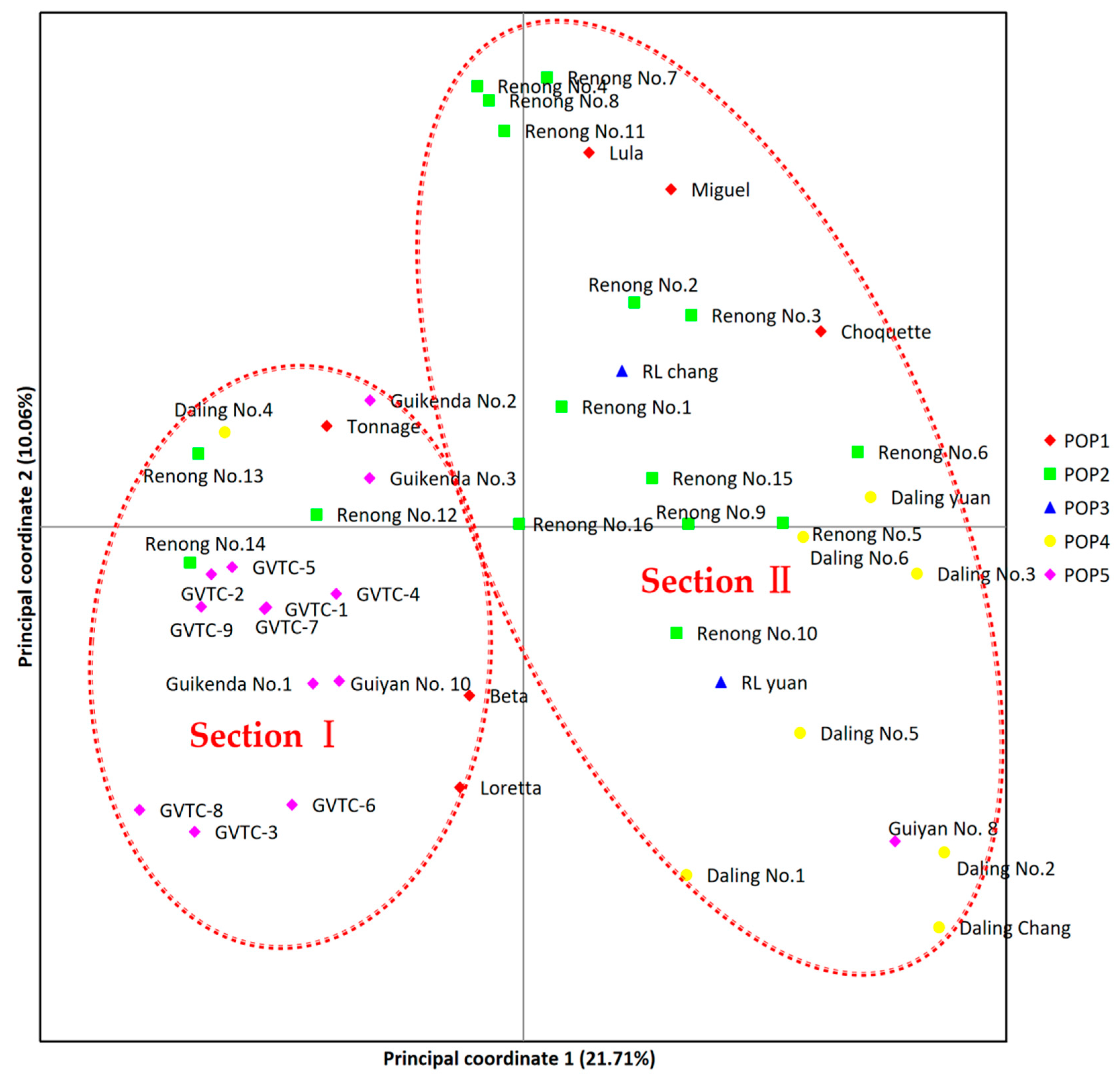
3.5. Analyses of Genetic Relationships Based on Polymorphic EST-SSRs from SMRT Sequencing Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schaffer, B.; Wolstenholme, B.N.; Whiley, A.W. The Avocado: Botany, Production and Uses, 2nd ed.; CPI Group (UK) Ltd.: Croydon, UK, 2012. [Google Scholar]
- Kopp, L.E. A taxonomic revision of the genus Persea in the western hemisphere (Persea-Lauraceae). Mem. N. Y. Bot. Gard. 1966, 14, 1–120. [Google Scholar]
- Williams, L.O. The avocado, a synopsis of the genus Persea, subg. Persea Econ. Bot. 1977, 31, 315–320. [Google Scholar] [CrossRef]
- Schaffer, B.; Wolstenholme, B.N. The Avocado: Botany, Production and Uses; CAB International: Wallingford, UK, 2002. [Google Scholar]
- Van der Werff, H. A synopsis of Persea (Lauraceae) in Central America. Novon 2002, 12, 575–586. [Google Scholar] [CrossRef]
- Dreher, M.L.; Davenport, A.J. Hass avocado composition and potential health effects. Crit. Rev. Food Sci. 2013, 53, 738–750. [Google Scholar] [CrossRef]
- Galvão, M.D.S.; Narain, N.; Nigam, N. Influence of different cultivars on oil quality and chemical characteristics of avocado fruit. Food Sci. Technol. 2014, 34, 539–546. [Google Scholar] [CrossRef] [Green Version]
- Ge, Y.; Si, X.Y.; Cao, J.Q.; Zhou, Z.X.; Wang, W.L.; Ma, W.H. Morphological characteristics, nutritional quality, and bioactive constituents in fruits of two avocado (Perseaamericana) varieties from hainan province, China. J. Agric. Sci. 2017, 9, 8–17. [Google Scholar] [CrossRef]
- Ge, Y.; Si, X.Y.; Lin, X.E.; Wang, J.S.; Zang, X.P.; Ma, W.H. Advances in avocado (Perseaamericana Mill.). South China Fruit 2017, 46, 63–70. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, D.S.; Liu, K.D. Environmental analysis and countermeasures for industrial development of Hainan avocado. Chin. J. Agric. Resour. Reg. Plan. 2015, 36, 78–84. [Google Scholar]
- Fiedler, J.; Bufler, G.; Bangerth, F. Genetic relationships of avocado (Perseaamericana Mill.) using RAPD markers. Euphytica 1998, 101, 249–255. [Google Scholar] [CrossRef]
- Mhameed, S.; Sharon, D.; Kaufman, D.; Lahav, E.; Hillel, J.; Degani, C.; Lavi, U. Genetic relationships within avocado (Perseaamericana Mill.) cultivars and between Persea species. Theor. Appl. Genet. 1997, 94, 279–286. [Google Scholar] [CrossRef]
- Furnier, G.R.; Cummings, M.P.; Clegg, M.T. Evolution of the avocados as revealed by DNA restriction site variation. J. Hered. 1990, 81, 183–188. [Google Scholar] [CrossRef]
- Davis, J.; Henderson, D.; Kobayashi, M. Genealogical relationships among cultivated avocado as revealed through RFLP analysis. J. Hered. 1998, 89, 319–323. [Google Scholar] [CrossRef]
- Ashworth, V.E.T.M.; Clegg, M.T. Microsatellite markers in avocado (Perseaamericana Mill.). genealogical relationships among cultivated avocado genotypes. J. Hered. 2003, 94, 407–415. [Google Scholar] [CrossRef]
- Schnell, R.J.; Brown, J.S.; Olano, C.T.; Power, E.J.; Krol, C.A.; Kuhn, D.N.; Motamayor, J.C. Evaluation of avocado germplasm using microsatellite markers. J. Am. Soc. Hortic. Sci. 2003, 128, 881–889. [Google Scholar] [CrossRef]
- Gross-German, E.; Viruel, M.A. Molecular characterization of avocado germplasm with a new set of SSR and EST-SSR markers: Genetic diversity, population structure, and identification of race-specific markers in a group of cultivated genotypes. Tree Genet. Genomes 2013, 9, 539–555. [Google Scholar] [CrossRef]
- Ge, Y.; Tan, L.; Wu, B.; Wang, T.; Zhang, T.; Chen, H.; Zou, M.; Ma, F.; Xu, Z.; Zhan, R. Transcriptome sequencing of different avocado ecotypes: De novo transcriptome assembly, annotation, identification and validation of EST-SSR markers. Forests 2019, 10, 411. [Google Scholar] [CrossRef]
- Chen, H.; Morrel, P.L.; Ashwoth, V.E.T.M.; De la Cruz, M.; Clegg, M.T. Nucleotide diversity and linkage disequilibrium in wild avocado (Perseaamericana Mill.). J. Hered. 2008, 99, 382–389. [Google Scholar] [CrossRef]
- Chen, H.; Morrel, P.L.; Ashwoth, V.E.T.M.; De la Cruz, M.; Clegg, M.T. Tracing the geographic origins of mayor avocado cultivars. J. Hered. 2009, 100, 56–65. [Google Scholar] [CrossRef]
- Hou, M.Y.; Mu, G.J.; Zhang, Y.J.; Cui, S.L.; Yang, X.L.; Liu, L.F. Evaluation of total flavonoid content and analysis of related EST-SSR in Chinese peanut germplasm. Crop Breed. Appl. Biotechnol. 2017, 17, 221–227. [Google Scholar] [CrossRef] [Green Version]
- Azevedo, A.O.N.; Azevedo, C.D.O.; Santos, P.H.A.D.; Ramos, H.C.C.; Boechat, M.S.B.; Arêdes, F.A.S.; Ramos, S.R.R.; Mirizola, L.A.; Perera, L.; Aragão, W.M.; et al. Selection of legitimate dwarf coconut hybrid seedlings using DNA fingerprinting. Crop Breed. Appl. Biotechnol. 2018, 18, 409–416. [Google Scholar] [CrossRef]
- Ahmad, A.; Wang, J.D.; Pan, Y.B.; Sharif, R.; Gao, S.J. Development and use of simple sequence repeats (SSRs) markers for sugarcane breeding and genetic studies. Agronomy 2018, 8, 260. [Google Scholar] [CrossRef]
- Ferreira, F.; Scapim, C.A.; Maldonado, C.; Mora, F. SSR-based genetic analysis of sweet corn inbred lines using artificial neural networks. Crop Breed. Appl. Biotechnol. 2018, 18, 309–313. [Google Scholar] [CrossRef]
- Ge, Y.; Hu, F.C.; Tan, L.; Wu, B.; Wang, T.; Zhang, T.; Ma, F.N.; Cao, J.Q.; Xu, Z.N.; Zhan, R.L. Molecular diversity in a germplasm collection of avocado accessions from the tropical and subtropical regions of China. Crop Breed. Appl. Biotechnol. 2019, 19, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Cai, C.F.; Cheng, F.Y.; Cui, H.L.; Zhou, H. Characterization and development of EST-SSR markers in tree peony using transcriptome sequences. Mol. Breed. 2014, 34, 1853–1866. [Google Scholar] [CrossRef]
- Biswas, M.K.; Nath, U.K.; Howlader, J.; Bagchi, M.; Natarajan, S.; Kayum, M.A.; Kim, H.T.; Park, J.I.; Kang, J.G.; Nou, I.S. Exploration and exploitation of novel SSR markers for candidate transcription factor genes in Lilium species. Genes 2018, 9, 97. [Google Scholar] [CrossRef]
- Ma, S.L.Y.; Dong, W.S.; Lyu, T.; Lyu, Y.M. An RNA sequencing transcriptome analysis and development of EST-SSR markers in Chinese hawthorn through Illumina sequencing. Forests 2019, 10, 82. [Google Scholar] [CrossRef]
- Li, X.; Li, M.; Hou, L.; Zhang, Z.Y.; Pang, X.M.; Li, Y.Y. De novo transcriptome assembly and population genetic analyses for an endangered Chinese endemic Acer miaotaiense (Aceraceae). Genes 2018, 9, 378. [Google Scholar] [CrossRef]
- Qi, W.C.; Chen, X.; Fang, P.H.; Shi, S.C.; Li, J.J.; Liu, X.T.; Cao, X.Q.; Zhao, N.; Hao, H.Y.; Li, Y.J.; et al. Genomic and transcriptomic sequencing of Rosa hybrida provides microsatellite markers for breeding, flower trait improvement and taxonomy studies. BMC Plant Biol. 2018, 18, 119. [Google Scholar] [CrossRef]
- Chen, J.; Tang, X.H.; Ren, C.X.; Wei, B.; Wu, Y.Y.; Wu, Q.H.; Pei, J. Full-length transcriptome sequences and the identification of putative genes for flavonoid biosynthesis in safflower. BMC Genom. 2018, 19, 548. [Google Scholar] [CrossRef]
- Tian, J.Y.; Feng, S.J.; Liu, Y.L.; Zhao, L.L.; Tian, L.; Hu, Y.; Yang, T.X.; Wei, A.Z. Single-molecule long-read sequencing of Zanthoxylumbungeanum maxim. transcriptome: Identification of aroma-related genes. Forests 2018, 9, 765. [Google Scholar] [CrossRef]
- Roberts, R.J.; Carneiro, M.O.; Schatz, M.C. The advantages of SMRT sequencing. Genome Biol. 2013, 14, 405–409. [Google Scholar] [CrossRef]
- Chao, Y.H.; Yuan, J.B.; Li, S.F.; Jia, S.Q.; Han, L.B.; Xu, L.X. Analysis of transcripts and splice isoforms in red clover (Trifoliumpratense L.) by single-molecule long-read sequencing. BMC Plant Biol. 2018, 18, 300. [Google Scholar] [CrossRef]
- Hoang, N.V.; Furtado, A.; Mason, P.J.; Marquardt, A.; Kasirajan, L.; Thirugnanasambandam, P.P.; Botha, F.C.; Henry, R.J. A survey of the complex transcriptome from the highly polyploid sugarcane genome using full-length isoform sequencing and de novo assembly from short read sequencing. BMC Genom. 2017, 18, 395. [Google Scholar] [CrossRef]
- Zuo, C.M.; Blow, M.; Sreedasyam, A.; Kuo, R.C.; Ramamoorthy, G.K.; Torres-Jerez, I.; Li, G.F.; Wang, M.; Dilworth, D.; Barry, K.; et al. Revealing the transcriptomic complexity of switchgrass by PacBio long-read sequencing. Biotechnol. Biofuels 2018, 11, 170. [Google Scholar] [CrossRef]
- Chao, Y.H.; Yuan, J.B.; Guo, T.; Xu, L.X.; Mu, Z.Y.; Han, L.B. Analysis of transcripts and splice isoforms in Medicago sativa L. by single-molecule long-read sequencing. Plant Mol. Biol. 2019, 99, 219–235. [Google Scholar] [CrossRef]
- Kim, J.A.; Roy, N.S.; Lee, I.H.; Choi, A.Y.; Choi, B.S.; Yu, Y.S.; Park, N.I.; Park, K.C.; Kim, S.; Yang, H.S.; et al. Genome-wide transcriptome profiling of the medicinal plant Zanthoxylumplanispinum using a single-molecule direct RNA sequencing approach. Genomics 2019, 111, 973–979. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, J.X.; Wang, X.S.; Wei, Z.W. Full-length RNA sequencing reveals unique transcriptome composition in bermudagrass. Plant Physiol. Biochem. 2018, 132, 95–103. [Google Scholar] [CrossRef]
- Xu, Q.S.; Zhu, J.Y.; Zhao, S.Q.; Hou, Y.; Li, F.D.; Tai, Y.L.; Wan, X.C.; Wei, C.L. Transcriptome profiling using single-molecule direct RNA sequencing approach for in-depth understanding of genes in secondary metabolism pathways of Camellia sinensis. Front. Plant Sci. 2017, 8, 1205. [Google Scholar] [CrossRef]
- Deng, Y.; Zheng, H.; Yan, Z.C.; Liao, D.Y.; Li, C.L.; Zhou, J.Y.; Liao, H. Full-length transcriptome survey and expression analysis of Cassia obtusifolia to discover putative genes related to aurantio-obtusin biosynthesis, seed formation and development, and stress response. Int. J. Mol. Sci. 2018, 19, 2476. [Google Scholar] [CrossRef]
- Ge, Y.; Cheng, Z.H.; Si, X.Y.; Ma, W.H.; Tan, L.; Zang, X.P.; Wu, B.; Xu, Z.N.; Wang, N.; Zhou, Z.X.; et al. Transcriptome profiling provides insight into the genes in carotenoid biosynthesis during the mesocarp and seed developmental stages of avocado (Persea Americana). Int. J. Mol. Sci. 2019, 20, 4117. [Google Scholar] [CrossRef]
- Ge, Y.; Ramchiary, N.; Wang, T.; Liang, C.; Wang, N.; Wang, Z.; Choi, S.R.; Lim, Y.P.; Piao, Z.Y. Development and linkage mapping of unigene-derived microsatellite markers in Brassica rapa L. Breed. Sci. 2011, 61, 160–167. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, 480–484. [Google Scholar] [CrossRef]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talon, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef]
- Perez-Rodriguez, P.; Riano-Pachon, D.M.; Correa, L.G.; Rensing, S.A.; Kersten, B.; Mueller-Roeber, B. PlnTFDB: Updated content and new features of the plant transcription factor database. Nucleic Acids Res. 2010, 38, 822–827. [Google Scholar] [CrossRef]
- Ge, Y.; Zhang, T.; Wu, B.; Tan, L.; Ma, F.N.; Zou, M.H.; Chen, H.H.; Pei, J.L.; Liu, Y.Z.; Chen, Z.H.; et al. Genome-wide assessment of avocado germplasm determined from specific length amplified fragment sequencing and transcriptomes: Population structure, genetic diversity, identification, and application of race-specific markers. Genes 2019, 10, 215. [Google Scholar] [CrossRef]
- Krawczak, M.; Nikolaus, S.; von Eberstein, H.; Croucher, P.J.; El Mokhtari, N.E.; Schreiber, S. PopGen: Population based recruitment of patients and controls for the analysis of complex genotype-phenotype relationships. Community Genet. 2006, 9, 55–61. [Google Scholar] [CrossRef]
- Liu, K.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Du, M.; Li, N.; Niu, B.; Liu, Y.; You, D.; Jiang, D.; Ruan, C.Q.; Qin, Z.Q.; Song, T.W.; Wang, W.T. De novo transcriptome analysis of Bagariusyarrelli (Siluriformes: Sisoridae) and the search for potential SSR markers using RNA-Seq. PLoS ONE 2018, 13, e0190343. [Google Scholar] [CrossRef]
- Liu, F.M.; Hong, Z.; Yang, Z.J.; Zhang, N.N.; Liu, X.J.; Xu, D.P. De Novo transcriptomeanalysis of Dalbergiaodorifera T. Chen (Fabaceae) and transferability of SSR markers developed from the transcriptome. Forests 2019, 10, 98. [Google Scholar] [CrossRef]
- Li, W.; Zhang, C.P.; Jiang, X.Q.; Liu, Q.C.; Liu, Q.H.; Wang, K.L. De Novotranscriptomicanalysis and development of EST–SSRs for Styrax japonicas. Forests 2018, 9, 748. [Google Scholar] [CrossRef]
- Kilaru, A.; Cao, X.; Dabbs, P.B.; Sung, H.J.; Rahman, M.M.; Thrower, N.; Zynda, G.; Podicheti, R.; Ibarra-Laclette, E.; Herrera-Estrella, L.; et al. Oil biosynthesis in a basal angiosperm: Transcriptome analysis of Persea Americana mesocarp. BMC Plant Biol. 2015, 15, 203. [Google Scholar] [CrossRef]
- Vergara-Pulgar, C.; Rothkegel, K.; González-Agüero, M.; Pedreschi, R.; Campos-Vargas, R.; Defilippi, B.G.; Meneses, C. De novo assembly of Perseaamericana cv. ‘Hass’ transcriptome during fruit development. BMC Genom. 2019, 20, 108. [Google Scholar] [CrossRef]
- Ibarra-Laclette, E.; Méndez-Bravo, A.; Pérez-Torres, C.A.; Albert, V.A.; Mockaitis, K.; Kilaru, A.; López-Gómez, R.; Cervantes-Luevano, J.I.; Herrera-Estrell, L. Deep sequencing of the Mexican avocado transcriptome, an ancient angiosperm with a high content of fatty acids. BMC Genom. 2015, 16, 599. [Google Scholar] [CrossRef]
- Yang, L.F.; Jin, Y.H.; Huang, W.; Sun, Q.; Liu, F.; Huang, X.Z. Full-length transcriptome sequences of ephemeral plant Arabidopsis pumila provides insight into gene expression dynamics during continuous salt stress. BMC Genom. 2018, 19, 717. [Google Scholar] [CrossRef]
- Liu, J.; Wang, H.; Chua, N.H. Long noncoding RNA transcriptome of plants. Plant Biotechnol. J. 2015, 13, 319–328. [Google Scholar] [CrossRef]
- Yandell, M.; Ence, D. A beginner’s guide to eukaryotic genome annotation. Nat. Rev. Genet. 2012, 13, 329–342. [Google Scholar] [CrossRef]
- Liu, J.; Jung, C.; Xu, J.; Wang, H.; Deng, S.; Bernad, L.; Arenas-Huertero, C.; Chua, N.H. Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in Arabidopsis. Plant Cell 2012, 24, 4333–4345. [Google Scholar] [CrossRef]
- Ochogavía, A.; Galla, G.; Seijo, J.G.; González, A.M.; Bellucci, M.; Pupilli, F.; Barcaccia, G.; Albertini, E.; Pessino, S. Structure, target-specifificity and expression of PN_LNC_N13, a long non-coding RNA differentially expressed in apomictic and sexual Paspalumnotatum. Plant Mol. Biol. 2018, 96, 53–67. [Google Scholar] [CrossRef]
- Alcaraz, M.L.; Hormaza, J.I. Molecular characterization and genetic diversity in an avocado collection of cultivars and local Spanish genotypes using SSRs. Heredity 2007, 144, 244–253. [Google Scholar] [CrossRef]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980, 32, 314–331. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source | Number |
|---|---|
| Total number of sequences examined | 75,956 |
| Total size of examined sequences (bp) | 170,959,769 |
| Total number of identified SSRs | 76,777 |
| Number of SSR containing sequences | 42,096 |
| Number of sequences containing more than 1 SSR | 19,825 |
| Number of SSRs present in compound formation | 12,675 |
| SSR Motif Length | Repeat Unit Number | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 5 | 6 | 7 | 8 | 9 | 10 | >10 | Total | % | |
| Mono- | - | - | - | - | - | 8951 | 35,849 | 44,800 | 58.35 |
| Di- | - | 4017 | 2773 | 2489 | 2013 | 1547 | 6064 | 18,903 | 24.62 |
| Tri- | 6129 | 2838 | 1237 | 720 | 403 | 193 | 204 | 11,724 | 15.27 |
| Tetra- | 541 | 175 | 41 | 21 | 8 | - | 2 | 788 | 0.01 |
| Penta- | 172 | 67 | 1 | 1 | - | - | - | 241 | 0.00 |
| Hexa- | 228 | 72 | 15 | 3 | 2 | - | 1 | 321 | 0.00 |
| Total | 7070 | 7169 | 4067 | 3234 | 2426 | 10,691 | 42,120 | 76,777 | |
| % | 9.21 | 9.34 | 5.30 | 4.21 | 3.16 | 13.92 | 45.14 | ||
| Marker Name | Transcript ID | Na 1 | Ne 2 | Ho 3 | He 4 | PIC 5 |
|---|---|---|---|---|---|---|
| Pa-eSSR-17 | F01_cb7709_c10/f1p0/2063 | 8 | 3.02 | 0.61 | 0.67 | 0.62 |
| Pa-eSSR-18 | F01_cb7876_c2/f1p0/2226 | 10 | 3.09 | 0.61 | 0.68 | 0.65 |
| Pa-eSSR-19 | F01_cb1803_c26/f1p0/2838 | 6 | 2.04 | 0.63 | 0.51 | 0.46 |
| Pa-eSSR-20 | F01_cb10663_c1/f1p0/2458 | 3 | 1.87 | 0.50 | 0.46 | 0.40 |
| Pa-eSSR-21 | F01_cb15691_c2/f1p0/2049 | 3 | 2.41 | 0.50 | 0.58 | 0.50 |
| Pa-eSSR-22 | F01_cb3034_c12/f2p0/2705 | 5 | 2.85 | 0.67 | 0.65 | 0.60 |
| Pa-eSSR-23 | F01_cb12182_c0/f6p2/1774 | 3 | 1.47 | 0.28 | 0.32 | 0.29 |
| Pa-eSSR-24 | F01_cb13109_c0/f3p0/1635 | 5 | 2.80 | 0.48 | 0.64 | 0.58 |
| Pa-eSSR-25 | F01_cb1901_c3/f1p1/2722 | 2 | 1.04 | 0.04 | 0.04 | 0.04 |
| Pa-eSSR-26 | F01_cb7204_c7/f10p1/2700 | 3 | 2.65 | 0.93 | 0.62 | 0.55 |
| Pa-eSSR-27 | F01_cb10594_c1/f1p0/4058 | 3 | 1.40 | 0.33 | 0.29 | 0.27 |
| Pa-eSSR-28 | F01_cb9432_c36/f1p2/1811 | 5 | 1.56 | 0.43 | 0.36 | 0.33 |
| Pa-eSSR-29 | F01_cb15387_c0/f3p0/1548 | 8 | 4.39 | 0.49 | 0.77 | 0.74 |
| Pa-eSSR-30 | F01_cb12814_c24/f1p0/3423 | 4 | 2.67 | 0.53 | 0.62 | 0.55 |
| Pa-eSSR-31 | F01_cb10835_c0/f4p0/2019 | 3 | 1.33 | 0.28 | 0.25 | 0.22 |
| Total | 71 | |||||
| Mean | 4.73 | 2.31 | 0.49 | 0.50 | 0.45 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ge, Y.; Zang, X.; Tan, L.; Wang, J.; Liu, Y.; Li, Y.; Wang, N.; Chen, D.; Zhan, R.; Ma, W. Single-Molecule Long-Read Sequencing of Avocado Generates Microsatellite Markers for Analyzing the Genetic Diversity in Avocado Germplasm. Agronomy 2019, 9, 512. https://0-doi-org.brum.beds.ac.uk/10.3390/agronomy9090512
Ge Y, Zang X, Tan L, Wang J, Liu Y, Li Y, Wang N, Chen D, Zhan R, Ma W. Single-Molecule Long-Read Sequencing of Avocado Generates Microsatellite Markers for Analyzing the Genetic Diversity in Avocado Germplasm. Agronomy. 2019; 9(9):512. https://0-doi-org.brum.beds.ac.uk/10.3390/agronomy9090512
Chicago/Turabian StyleGe, Yu, Xiaoping Zang, Lin Tan, Jiashui Wang, Yuanzheng Liu, Yanxia Li, Nan Wang, Di Chen, Rulin Zhan, and Weihong Ma. 2019. "Single-Molecule Long-Read Sequencing of Avocado Generates Microsatellite Markers for Analyzing the Genetic Diversity in Avocado Germplasm" Agronomy 9, no. 9: 512. https://0-doi-org.brum.beds.ac.uk/10.3390/agronomy9090512