
Knockdown of AKT3 Activates HER2 and DDR Kinases in Bone-Seeking Breast Cancer Cells, Promotes Metastasis In Vivo and Attenuates the TGFβ/CTGF Axis

 , , ,
, , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture
2.3. Lentiviral Knockdown
2.4. Inhibitor Testing and Cell Viability Assay
2.5. Proliferation Assay
2.6. Migration and Invasion Assay
2.7. Chemotaxis Assay
2.8. Western Blot
2.9. AKT Isoform-Specific In Vitro Kinase Assay
2.10. Functional Kinome Profiling
2.11. Intracardiac Mouse Model and Luciferase In Vivo Analysis
2.12. Statistical Analysis
3. Results
3.1. The Bone-Seeking Subline MDA-MB-231 BO Exhibits Constitutively Elevated pAKT Levels and Is Responsive for panAKT and mTOR Inhibition
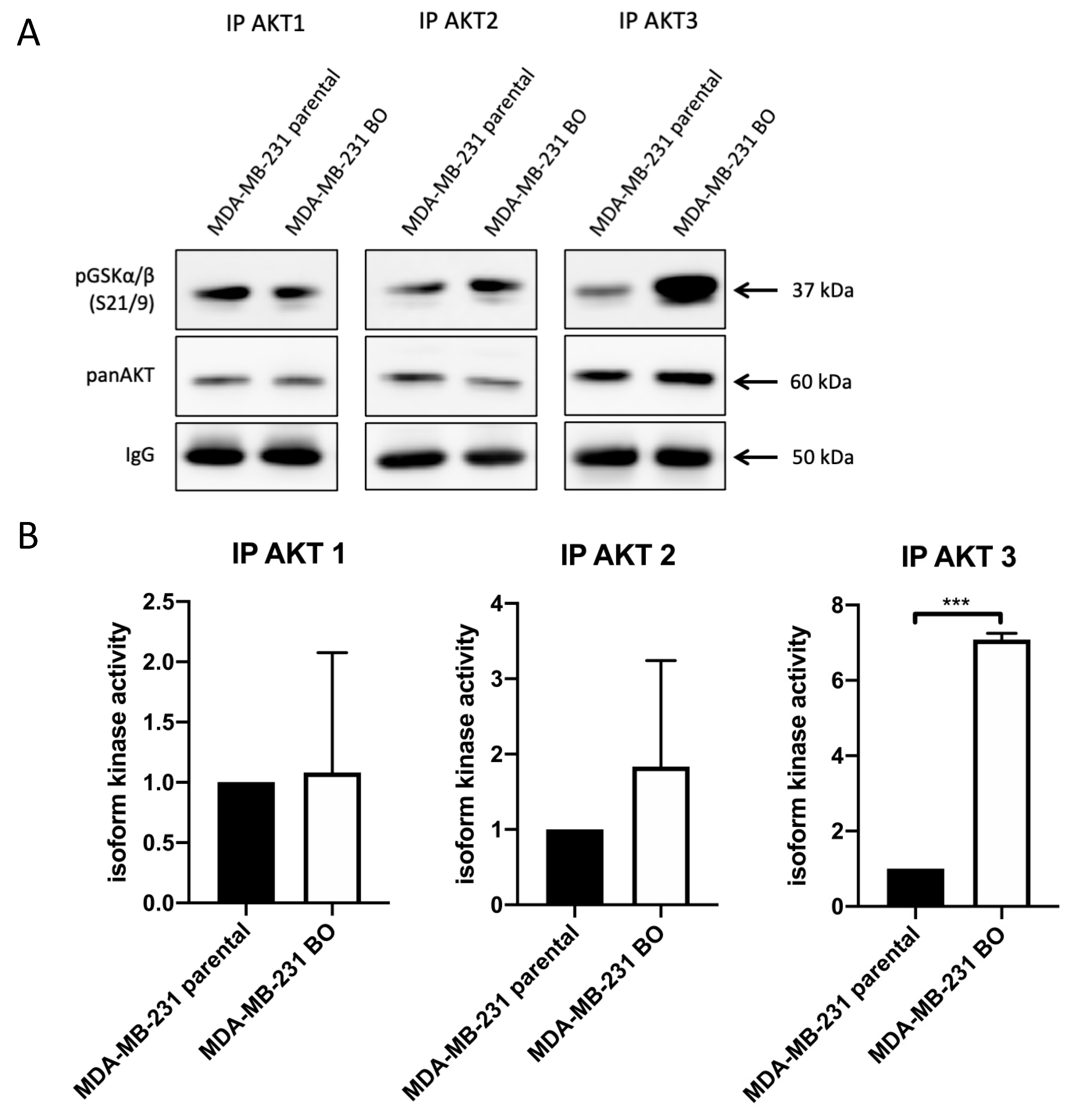
3.2. Bone-Seeking 231-BO Cells Show an Increased AKT3 Activity in an In Vitro Kinase Assay
3.3. Knockdown of AKT3 in Bone-Seeking 231-BO Cells Increases Migration, Invasion, and Chemotaxis towards EGF but Had No Effect on Proliferation
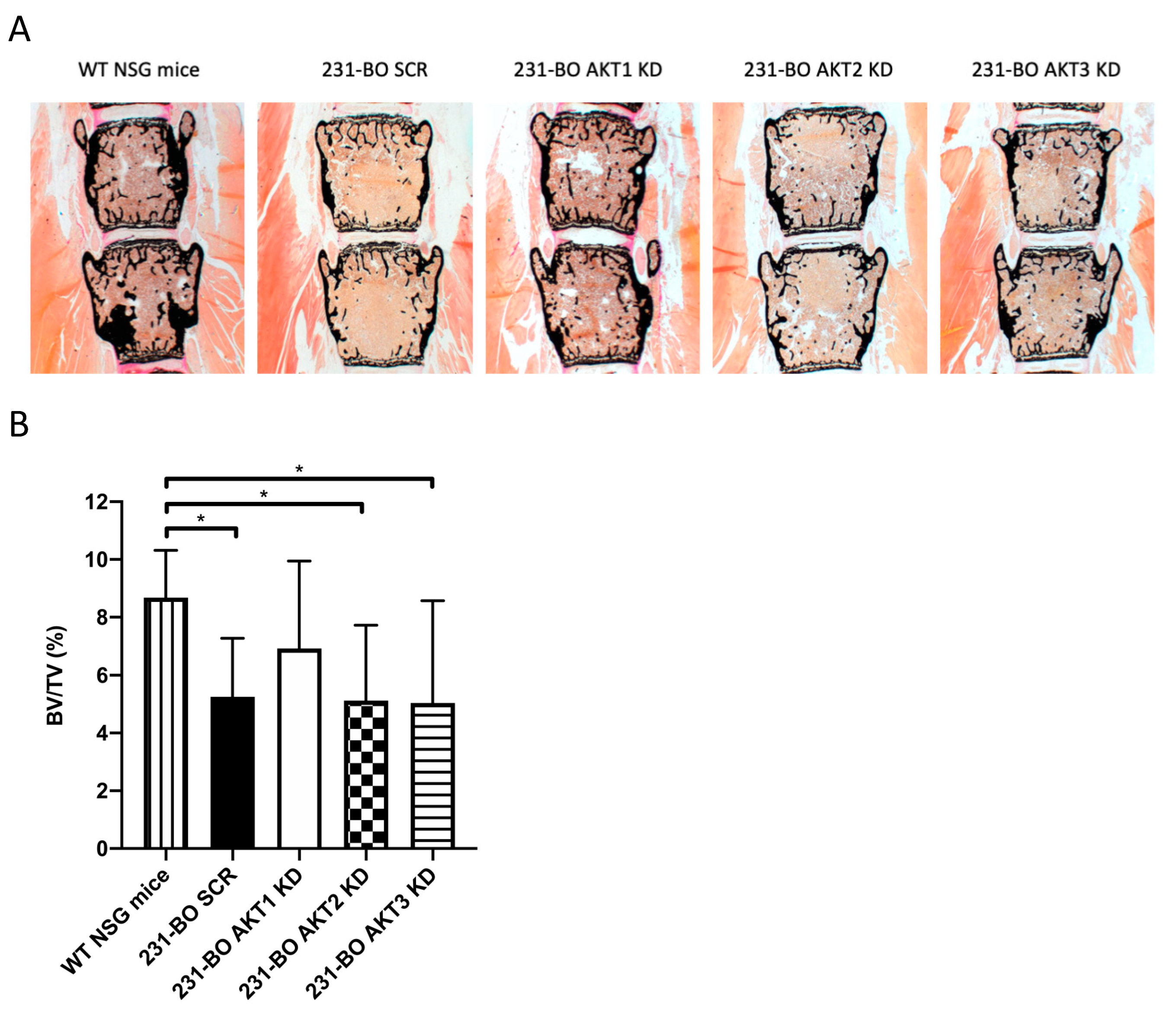
3.4. Knockdown of AKT3 in 231-BO Cells Promotes Metastasis to Bone after Intracardiac Inoculation but Does Not Increase Osteolysis
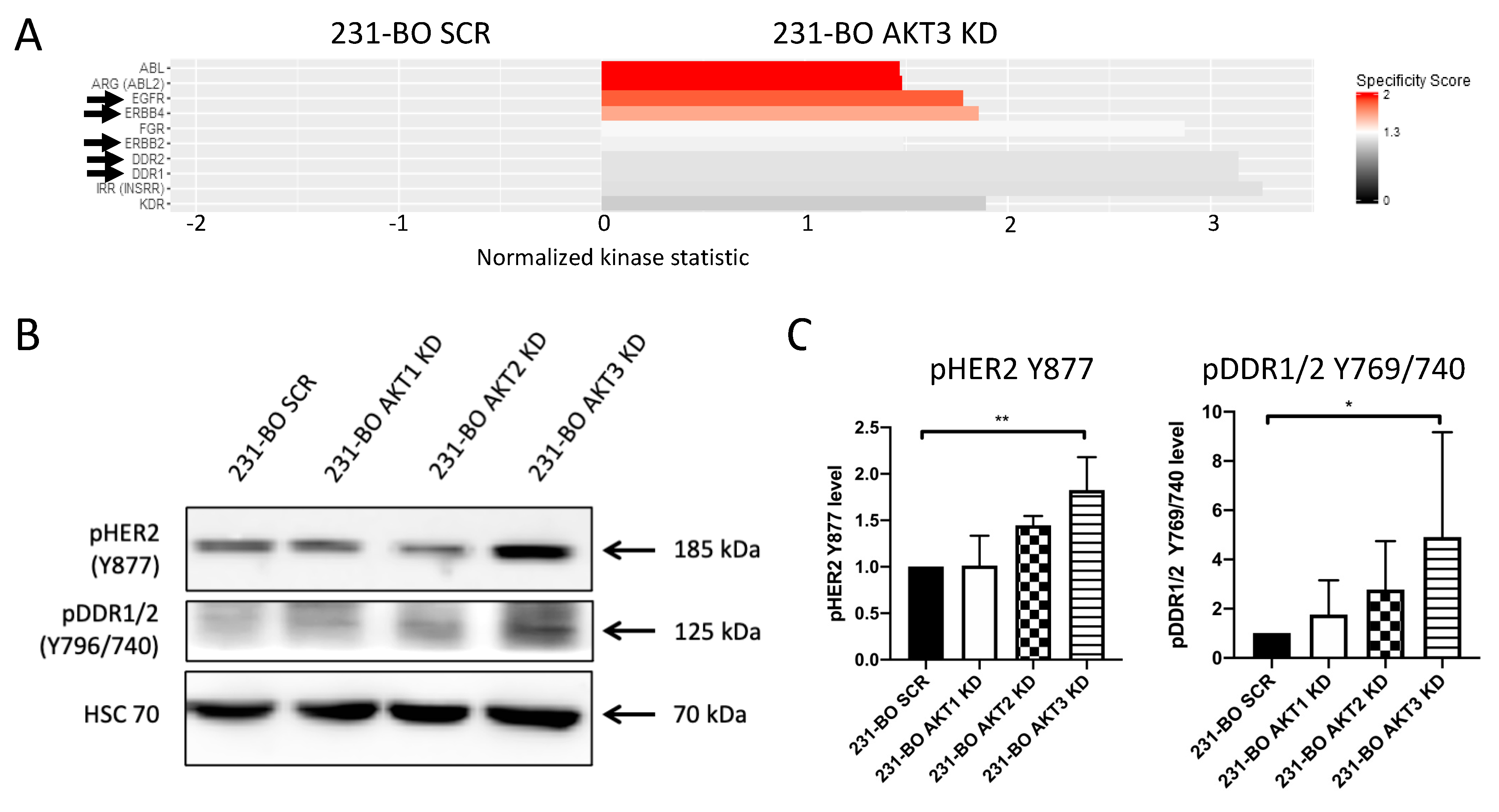
3.5. 231-BO Cells with an AKT3 Knockdown Exhibit Elevated Activity and Phosphorylation of Metastasis-Associated HER2 and DDR1/2
3.6. Knockdown of AKT3 in the 231-BO Cell Line Results in a Diminished Increase in the Vicious Cycle-Associated CTGF after TGFβ-Stimulation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Berman, A.T.; Thukral, A.D.; Hwang, W.T.; Solin, L.J.; Vapiwala, N. Incidence and patterns of distant metastases for patients with early-stage breast cancer after breast conservation treatment. Clin. Breast Cancer 2013, 13, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Mundy, G.R. Metastasis to bone: Causes, consequences and therapeutic opportunities. Nat. Rev. Cancer 2002, 2, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Hiraga, T.; Williams, P.J.; Mundy, G.R.; Yoneda, T. The bisphosphonate ibandronate promotes apoptosis in MDA-MB-231 human breast cancer cells in bone metastases. Cancer Res. 2001, 61, 4418–4424. [Google Scholar] [PubMed]
- Goldhirsch, A.; Wood, W.C.; Coates, A.S.; Gelber, R.D.; Thurlimann, B.; Senn, H.J.; Panel, m. Strategies for subtypes--dealing with the diversity of breast cancer: Highlights of the St. Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Ann. Oncol. 2011, 22, 1736–1747. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.; Deng, G.; Huang, X.; Li, X.; Xie, X.; Wang, J.; Shuang, Z.; Wang, X. Bone metastasis pattern in initial metastatic breast cancer: A population-based study. Cancer Manag. Res. 2018, 10, 287–295. [Google Scholar] [CrossRef] [Green Version]
- Pareek, A.; Singh, O.P.; Yogi, V.; Ghori, H.U.; Tiwari, V.; Redhu, P. Bone metastases incidence and its correlation with hormonal and human epidermal growth factor receptor 2 neu receptors in breast cancer. J. Cancer. Res. Ther. 2019, 15, 971–975. [Google Scholar] [CrossRef]
- Schmid-Alliana, A.; Schmid-Antomarchi, H.; Al-Sahlanee, R.; Lagadec, P.; Scimeca, J.C.; Verron, E. Understanding the Progression of Bone Metastases to Identify Novel Therapeutic Targets. Int. J. Mol. Sci. 2018, 19, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneda, T.; Williams, P.J.; Hiraga, T.; Niewolna, M.; Nishimura, R. A bone-seeking clone exhibits different biological properties from the MDA-MB-231 parental human breast cancer cells and a brain-seeking clone in vivo and in vitro. J. Bone Miner. Res. 2001, 16, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.P.; Massague, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigelt, B.; Peterse, J.L.; van ‘t Veer, L.J. Breast cancer metastasis: Markers and models. Nat. Rev. Cancer 2005, 5, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Eccles, S.A. The role of c-erbB-2/HER2/neu in breast cancer progression and metastasis. J. Mammary Gland Biol. Neoplasia 2001, 6, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Guy, C.T.; Webster, M.A.; Schaller, M.; Parsons, T.J.; Cardiff, R.D.; Muller, W.J. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc. Natl. Acad. Sci. USA 1992, 89, 10578–10582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadiya, M.; Chakraborty, G. Signaling by discoidin domain receptor 1 in cancer metastasis. Cell Adh Migr 2018, 12, 315–323. [Google Scholar] [CrossRef]
- Leitinger, B. Discoidin domain receptor functions in physiological and pathological conditions. Int. Rev. Cell Mol. Biol. 2014, 310, 39–87. [Google Scholar] [CrossRef] [Green Version]
- Paget, S. The distribution of secondary growths in cancer of the breast. Cancer Metastasis Rev. 1889, 8, 98–101. [Google Scholar] [CrossRef] [Green Version]
- Yoneda, T.; Sasaki, A.; Mundy, G.R. Osteolytic bone metastasis in breast cancer. Breast Cancer Res. Treat. 1994, 32, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Kang, Y. Organotropism of breast cancer metastasis. J. Mammary Gland Biol. Neoplasia 2007, 12, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordón-Cardo, C.; Guise, T.A.; Massagué, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Weilbaecher, K.N.; Guise, T.A.; McCauley, L.K. Cancer to bone: A fatal attraction. Nat. Rev. Cancer 2011, 11, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Chirgwin, J.M.; Mohammad, K.S.; Guise, T.A. Tumor-bone cellular interactions in skeletal metastases. J Musculoskelet. Neuronal. Interact. 2004, 4, 308–318. [Google Scholar] [PubMed]
- Kan, C.; Vargas, G.; Pape, F.L.; Clezardin, P. Cancer Cell Colonisation in the Bone Microenvironment. Int. J. Mol. Sci. 2016, 17, 1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roodman, G.D. Mechanisms of bone metastasis. N. Engl. J. Med. 2004, 350, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Kakonen, S.M.; Selander, K.S.; Chirgwin, J.M.; Yin, J.J.; Burns, S.; Rankin, W.A.; Grubbs, B.G.; Dallas, M.; Cui, Y.; Guise, T.A. Transforming growth factor-beta stimulates parathyroid hormone-related protein and osteolytic metastases via Smad and mitogen-activated protein kinase signaling pathways. J. Biol. Chem. 2002, 277, 24571–24578. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.; He, W.; Tulley, S.; Gupta, G.P.; Serganova, I.; Chen, C.R.; Manova-Todorova, K.; Blasberg, R.; Gerald, W.L.; Massague, J. Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc. Natl. Acad. Sci. USA 2005, 102, 13909–13914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takigawa, M. CCN2: A master regulator of the genesis of bone and cartilage. J. Cell Commun. Signal. 2013, 7, 191–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khotskaya, Y.B.; Goverdhan, A.; Shen, J.; Ponz-Sarvise, M.; Chang, S.S.; Hsu, M.C.; Wei, Y.; Xia, W.; Yu, D.; Hung, M.C. S6K1 promotes invasiveness of breast cancer cells in a model of metastasis of triple-negative breast cancer. Am. J. Transl. Res. 2014, 6, 361–376. [Google Scholar] [PubMed]
- Chen, X.; Pei, Z.; Peng, H.; Zheng, Z. Exploring the molecular mechanism associated with breast cancer bone metastasis using bioinformatic analysis and microarray genetic interaction network. Medicine 2018, 97, e12032. [Google Scholar] [CrossRef] [PubMed]
- Pascual, J.; Turner, N.C. Targeting the PI3-kinase pathway in triple-negative breast cancer. Ann. Oncol. 2019, 30, 1051–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ossovskaya, V.; Wang, Y.; Budoff, A.; Xu, Q.; Lituev, A.; Potapova, O.; Vansant, G.; Monforte, J.; Daraselia, N. Exploring molecular pathways of triple-negative breast cancer. Genes Cancer 2011, 2, 870–879. [Google Scholar] [CrossRef]
- Altomare, D.A.; Testa, J.R. Perturbations of the AKT signaling pathway in human cancer. Oncogene 2005, 24, 7455–7464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castaneda, C.A.; Cortes-Funes, H.; Gomez, H.L.; Ciruelos, E.M. The phosphatidyl inositol 3-kinase/AKT signaling pathway in breast cancer. Cancer Metastasis Rev. 2010, 29, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, K.M.; Anderson, N.G. The protein kinase B/Akt signalling pathway in human malignancy. Cell. Signal. 2002, 14, 381–395. [Google Scholar] [CrossRef]
- Dillon, R.L.; White, D.E.; Muller, W.J. The phosphatidyl inositol 3-kinase signaling network: Implications for human breast cancer. Oncogene 2007, 26, 1338–1345. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [Green Version]
- Hiraga, T.; Myoui, A.; Hashimoto, N.; Sasaki, A.; Hata, K.; Morita, Y.; Yoshikawa, H.; Rosen, C.J.; Mundy, G.R.; Yoneda, T. Bone-derived IGF mediates crosstalk between bone and breast cancer cells in bony metastases. Cancer Res. 2012, 72, 4238–4249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinz, N.; Jücker, M. Distinct functions of AKT isoforms in breast cancer: A comprehensive review. Cell Commun. Signal. 2019, 17, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Hurley, G.; Daly, E.; O’Grady, A.; Cummins, R.; Quinn, C.; Flanagan, L.; Pierce, A.; Fan, Y.; Lynn, M.A.; Rafferty, M.; et al. Investigation of molecular alterations of AKT-3 in triple-negative breast cancer. Histopathology 2014, 64, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.S.; Xu, P.Z.; Gottlob, K.; Chen, M.L.; Sokol, K.; Shiyanova, T.; Roninson, I.; Weng, W.; Suzuki, R.; Tobe, K.; et al. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev. 2001, 15, 2203–2208. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Mu, J.; Kim, J.K.; Thorvaldsen, J.L.; Chu, Q.; Crenshaw, E.B.; Kaestner, K.H.; Bartolomei, M.S.; Shulman, G.I.; Birnbaum, M.J. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science 2001, 292, 1728–1731. [Google Scholar] [CrossRef] [PubMed]
- Tschopp, O.; Yang, Z.-Z.; Brodbeck, D.; Dummler, B.A.; Hemmings-Mieszczak, M.; Watanabe, T.; Michaelis, T.; Frahm, J.; Hemmings, B.A. Essential role of protein kinase B gamma (PKB gamma/Akt3) in postnatal brain development but not in glucose homeostasis. Development 2005, 132, 2943–2954. [Google Scholar] [CrossRef] [Green Version]
- Hutchinson, J.N.; Jin, J.; Cardiff, R.D.; Woodgett, J.R.; Muller, W.J. Activation of Akt-1 (PKB-alpha) can accelerate ErbB-2-mediated mammary tumorigenesis but suppresses tumor invasion. Cancer Res. 2004, 64, 3171–3178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maroulakou, I.G.; Oemler, W.; Naber, S.P.; Tsichlis, P.N. Akt1 ablation inhibits, whereas Akt2 ablation accelerates, the development of mammary adenocarcinomas in mouse mammary tumor virus (MMTV)-ErbB2/neu and MMTV-polyoma middle T transgenic mice. Cancer Res. 2007, 67, 167–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riggio, M.; Perrone, M.C.; Polo, M.L.; Rodriguez, M.J.; May, M.; Abba, M.; Lanari, C.; Novaro, V. AKT1 and AKT2 isoforms play distinct roles during breast cancer progression through the regulation of specific downstream proteins. Sci. Rep. 2017, 7, 44244. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Ariss, M.M.; Ramakrishnan, G.; Nogueira, V.; Blaha, C.; Putzbach, W.; Islam, A.; Frolov, M.V.; Hay, N. Cell-Autonomous versus Systemic Akt Isoform Deletions Uncovered New Roles for Akt1 and Akt2 in Breast Cancer. Mol. Cell 2020, 80, 87–101 e105. [Google Scholar] [CrossRef]
- Liu, H.; Radisky, D.C.; Nelson, C.M.; Zhang, H.; Fata, J.E.; Roth, R.A.; Bissell, M.J. Mechanism of Akt1 inhibition of breast cancer cell invasion reveals a protumorigenic role for TSC2. Proc. Natl. Acad. Sci. USA 2006, 103, 4134–4139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, Y.R.; Toker, A. The actin-bundling protein palladin is an Akt1-specific substrate that regulates breast cancer cell migration. Mol. Cell 2010, 38, 333–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irie, H.Y.; Pearline, R.V.; Grueneberg, D.; Hsia, M.; Ravichandran, P.; Kothari, N.; Natesan, S.; Brugge, J.S. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. J. Cell Biol. 2005, 171, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Arboleda, M.J.; Lyons, J.F.; Kabbinavar, F.F.; Bray, M.R.; Snow, B.E.; Ayala, R.; Danino, M.; Karlan, B.Y.; Slamon, D.J. Overexpression of AKT2/protein kinase Bbeta leads to up-regulation of beta1 integrins, increased invasion, and metastasis of human breast and ovarian cancer cells. Cancer Res. 2003, 63, 196–206. [Google Scholar] [PubMed]
- Chin, Y.R.; Yoshida, T.; Marusyk, A.; Beck, A.H.; Polyak, K.; Toker, A. Targeting Akt3 signaling in triple-negative breast cancer. Cancer Res. 2014, 74, 964–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grottke, A.; Ewald, F.; Lange, T.; Nörz, D.; Herzberger, C.; Bach, J.; Grabinski, N.; Gräser, L.; Höppner, F.; Nashan, B.; et al. Downregulation of AKT3 Increases Migration and Metastasis in Triple Negative Breast Cancer Cells by Upregulating S100A4. PLoS ONE 2016, 11, e0146370. [Google Scholar] [CrossRef] [Green Version]
- Fritsche, H.; Heilmann, T.; Tower, R.J.; Hauser, C.; von Au, A.; El-Sheikh, D.; Campbell, G.M.; Alp, G.; Schewe, D.; Hubner, S.; et al. TRAIL-R2 promotes skeletal metastasis in a breast cancer xenograft mouse model. Oncotarget 2015, 6, 9502–9516. [Google Scholar] [CrossRef]
- Albers, J.; Keller, J.; Baranowsky, A.; Beil, F.T.; Catala-Lehnen, P.; Schulze, J.; Amling, M.; Schinke, T. Canonical Wnt signaling inhibits osteoclastogenesis independent of osteoprotegerin. J. Cell Biol. 2013, 200, 537–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- Bijnsdorp, I.V.; Giovannetti, E.; Peters, G.J. Analysis of drug interactions. Methods Mol. Biol. 2011, 731, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Puchalapalli, M.; Zeng, X.; Mu, L.; Anderson, A.; Hix Glickman, L.; Zhang, M.; Sayyad, M.R.; Mosticone Wangensteen, S.; Clevenger, C.V.; Koblinski, J.E. NSG Mice Provide a Better Spontaneous Model of Breast Cancer Metastasis than Athymic (Nude) Mice. PLoS ONE 2016, 11, e0163521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kretschmann, K.L.; Welm, A.L. Mouse models of breast cancer metastasis to bone. Cancer Metastasis Rev. 2012, 31, 579–583. [Google Scholar] [CrossRef]
- Xue, C.; Wyckoff, J.; Liang, F.; Sidani, M.; Violini, S.; Tsai, K.L.; Zhang, Z.Y.; Sahai, E.; Condeelis, J.; Segall, J.E. Epidermal growth factor receptor overexpression results in increased tumor cell motility in vivo coordinately with enhanced intravasation and metastasis. Cancer Res. 2006, 66, 192–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mata, R.; Palladino, C.; Nicolosi, M.L.; Lo Presti, A.R.; Malaguarnera, R.; Ragusa, M.; Sciortino, D.; Morrione, A.; Maggiolini, M.; Vella, V.; et al. IGF-I induces upregulation of DDR1 collagen receptor in breast cancer cells by suppressing MIR-199a-5p through the PI3K/AKT pathway. Oncotarget 2016, 7, 7683–7700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.; Kim, H.; Jung, S.; Moon, A.; Noh, D.Y.; Lee, Z.H.; Kim, H.J.; Kim, H.H. A CTGF-RUNX2-RANKL Axis in Breast and Prostate Cancer Cells Promotes Tumor Progression in Bone. J. Bone Miner. Res. 2020, 35, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.H.; Nakashima, T.; Sanchez, O.H.; Kozieradzki, I.; Komarova, S.V.; Sarosi, I.; Morony, S.; Rubin, E.; Sarao, R.; Hojilla, C.V.; et al. Regulation of cancer cell migration and bone metastasis by RANKL. Nature 2006, 440, 692–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewald, F.; Grabinski, N.; Grottke, A.; Windhorst, S.; Norz, D.; Carstensen, L.; Staufer, K.; Hofmann, B.T.; Diehl, F.; David, K.; et al. Combined targeting of AKT and mTOR using MK-2206 and RAD001 is synergistic in the treatment of cholangiocarcinoma. Int. J. Cancer 2013, 133, 2065–2076. [Google Scholar] [CrossRef] [PubMed]
- Holler, M.; Grottke, A.; Mueck, K.; Manes, J.; Jucker, M.; Rodemann, H.P.; Toulany, M. Dual Targeting of Akt and mTORC1 Impairs Repair of DNA Double-Strand Breaks and Increases Radiation Sensitivity of Human Tumor Cells. PLoS ONE 2016, 11, e0154745. [Google Scholar] [CrossRef]
- Smit, D.J.; Cayrefourcq, L.; Haider, M.T.; Hinz, N.; Pantel, K.; Alix-Panabieres, C.; Jucker, M. High Sensitivity of Circulating Tumor Cells Derived from a Colorectal Cancer Patient for Dual Inhibition with AKT and mTOR Inhibitors. Cells 2020, 9, 2129. [Google Scholar] [CrossRef]
- Suyama, K.; Yao, J.; Liang, H.; Benard, O.; Loudig, O.D.; Amgalan, D.; McKimpson, W.M.; Phillips, G.R.; Segall, J.; Wang, Y.; et al. An Akt3 Splice Variant Lacking the Serine 472 Phosphorylation Site Promotes Apoptosis and Suppresses Mammary Tumorigenesis. Cancer Res. 2018, 78, 103–114. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.; Yao, J.; Suyama, K.; Bajaj, S.; Qian, X.; Loudig, O.D.; Eugenin, E.A.; Phillips, G.R.; Hazan, R.B. N-cadherin regulates mammary tumor cell migration through Akt3 suppression. Oncogene 2013, 32, 422–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phung, T.L.; Du, W.; Xue, Q.; Ayyaswamy, S.; Gerald, D.; Antonello, Z.; Nhek, S.; Perruzzi, C.A.; Acevedo, I.; Ramanna-Valmiki, R.; et al. Akt1 and akt3 exert opposing roles in the regulation of vascular tumor growth. Cancer Res. 2015, 75, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nutter, F.; Holen, I.; Brown, H.K.; Cross, S.S.; Evans, C.A.; Walker, M.; Coleman, R.E.; Westbrook, J.A.; Selby, P.J.; Brown, J.E.; et al. Different molecular profiles are associated with breast cancer cell homing compared with colonisation of bone: Evidence using a novel bone-seeking cell line. Endocr. Relat. Cancer 2014, 21, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Bellahcene, A.; Bachelier, R.; Detry, C.; Lidereau, R.; Clezardin, P.; Castronovo, V. Transcriptome analysis reveals an osteoblast-like phenotype for human osteotropic breast cancer cells. Breast Cancer Res. Treat. 2007, 101, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Santi, S.A.; Lee, H. Ablation of Akt2 induces autophagy through cell cycle arrest, the downregulation of p70S6K, and the deregulation of mitochondria in MDA-MB231 cells. PLoS ONE 2011, 6, e14614. [Google Scholar] [CrossRef]
- Kim, D.; Rhee, S. Matrix metalloproteinase2 regulates MDAMB231 breast cancer cell invasion induced by active mammalian diaphanous-related formin 1. Mol. Med. Rep. 2016, 14, 277–282. [Google Scholar] [CrossRef] [Green Version]
- Turner, K.M.; Sun, Y.; Ji, P.; Granberg, K.J.; Bernard, B.; Hu, L.; Cogdell, D.E.; Zhou, X.; Yli-Harja, O.; Nykter, M.; et al. Genomically amplified Akt3 activates DNA repair pathway and promotes glioma progression. Proc. Natl. Acad. Sci. USA 2015, 112, 3421–3426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Luca, A.; Carotenuto, A.; Rachiglio, A.; Gallo, M.; Maiello, M.R.; Aldinucci, D.; Pinto, A.; Normanno, N. The role of the EGFR signaling in tumor microenvironment. J. Cell. Physiol. 2008, 214, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Nickerson, N.K.; Mohammad, K.S.; Gilmore, J.L.; Crismore, E.; Bruzzaniti, A.; Guise, T.A.; Foley, J. Decreased autocrine EGFR signaling in metastatic breast cancer cells inhibits tumor growth in bone and mammary fat pad. PLoS ONE 2012, 7, e30255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, F.; Yang, J.; Wen, S.; An, Y.L.; Ding, J.; Ju, S.H.; Zhao, Z.; Chen, H.J.; Peng, X.G.; Wong, S.T.; et al. Involvement of epidermal growth factor receptor overexpression in the promotion of breast cancer brain metastasis. Cancer 2012, 118, 5198–5209. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; He, B.; Weber, G.F. Growth factor signaling induces metastasis genes in transformed cells: Molecular connection between Akt kinase and osteopontin in breast cancer. Mol. Cell Biol. 2003, 23, 6507–6519. [Google Scholar] [CrossRef] [Green Version]
- Foley, J.; Nickerson, N.K.; Nam, S.; Allen, K.T.; Gilmore, J.L.; Nephew, K.P.; Riese, D.J., 2nd. EGFR signaling in breast cancer: Bad to the bone. Semin. Cell Dev. Biol. 2010, 21, 951–960. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.; Yao, J.; Yu, D. Overexpression of the c-erbB-2 gene enhanced intrinsic metastasis potential in human breast cancer cells without increasing their transformation abilities. Cancer Res. 1997, 57, 1199–1205. [Google Scholar]
- Klos, K.S.; Wyszomierski, S.L.; Sun, M.; Tan, M.; Zhou, X.; Li, P.; Yang, W.; Yin, G.; Hittelman, W.N.; Yu, D. ErbB2 increases vascular endothelial growth factor protein synthesis via activation of mammalian target of rapamycin/p70S6K leading to increased angiogenesis and spontaneous metastasis of human breast cancer cells. Cancer Res. 2006, 66, 2028–2037. [Google Scholar] [CrossRef] [Green Version]
- Zeng, P.; Sun, S.; Li, R.; Xiao, Z.X.; Chen, H. HER2 Upregulates ATF4 to Promote Cell Migration via Activation of ZEB1 and Downregulation of E-Cadherin. Int. J. Mol. Sci. 2019, 20, 2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Li, J.; Zhu, S.; Wu, J.; Chen, C.; Liu, Q.; Wei, W.; Zhang, Y.; Sun, S. Breast cancer subtypes predict the preferential site of distant metastases: A SEER based study. Oncotarget 2017, 8, 27990–27996. [Google Scholar] [CrossRef] [Green Version]
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic behavior of breast cancer subtypes. J. Clin. Oncol. 2010, 28, 3271–3277. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Hoffmann, A.D.; Liu, H.; Liu, X. Organotropism: New insights into molecular mechanisms of breast cancer metastasis. NPJ Precis. Oncol. 2018, 2, 4. [Google Scholar] [CrossRef]
- Zhang, K.; Corsa, C.A.; Ponik, S.M.; Prior, J.L.; Piwnica-Worms, D.; Eliceiri, K.W.; Keely, P.J.; Longmore, G.D. The collagen receptor discoidin domain receptor 2 stabilizes SNAIL1 to facilitate breast cancer metastasis. Nat. Cell Biol. 2013, 15, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Chakraborty, G.; Zhang, Z.; Akalay, I.; Gadiya, M.; Gao, Y.; Sinha, S.; Hu, J.; Jiang, C.; Akram, M.; et al. Multi-organ Site Metastatic Reactivation Mediated by Non-canonical Discoidin Domain Receptor 1 Signaling. Cell 2016, 166, 47–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, J.J.; Castel, P.; Radosevic-Robin, N.; Elkabets, M.; Auricchio, N.; Aceto, N.; Weitsman, G.; Barber, P.; Vojnovic, B.; Ellis, H.; et al. Antagonism of EGFR and HER3 enhances the response to inhibitors of the PI3K-Akt pathway in triple-negative breast cancer. Sci. Signal. 2014, 7, ra29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandarlapaty, S.; Sawai, A.; Scaltriti, M.; Rodrik-Outmezguine, V.; Grbovic-Huezo, O.; Serra, V.; Majumder, P.K.; Baselga, J.; Rosen, N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 2011, 19, 58–71. [Google Scholar] [CrossRef] [Green Version]
- Er, E.E.; Mendoza, M.C.; Mackey, A.M.; Rameh, L.E.; Blenis, J. AKT facilitates EGFR trafficking and degradation by phosphorylating and activating PIKfyve. Sci. Signal. 2013, 6, ra45. [Google Scholar] [CrossRef] [Green Version]
- Massague, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, P.M.; Massague, J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat. Rev. Cancer 2003, 3, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Siegel, P.M.; Shu, W.; Cardiff, R.D.; Muller, W.J.; Massague, J. Transforming growth factor beta signaling impairs Neu-induced mammary tumorigenesis while promoting pulmonary metastasis. Proc. Natl. Acad. Sci. USA 2003, 100, 8430–8435. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-beta Family. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.Y.; Chen, Y.J.; Hsiao, Y.C.; Huang, Y.C.; Lai, T.H.; Tang, C.H. Osteoblasts-derived TGF-beta1 enhance motility and integrin upregulation through Akt, ERK, and NF-kappaB-dependent pathway in human breast cancer cells. Mol. Carcinog. 2008, 47, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.H.; Su, Y.H.; Chuang, R.L.; Chang, T.Y. Suppression of transforming growth factor-beta-induced apoptosis through a phosphatidylinositol 3-kinase/Akt-dependent pathway. Oncogene 1998, 17, 1959–1968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remy, I.; Montmarquette, A.; Michnick, S.W. PKB/Akt modulates TGF-beta signalling through a direct interaction with Smad3. Nat. Cell Biol. 2004, 6, 358–365. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, F.; ten Dijke, P. Signaling interplay between transforming growth factor-beta receptor and PI3K/AKT pathways in cancer. Trends Biochem. Sci. 2013, 38, 612–620. [Google Scholar] [CrossRef] [PubMed]
- Blom, I.E.; Goldschmeding, R.; Leask, A. Gene regulation of connective tissue growth factor: New targets for antifibrotic therapy? Matrix Biol. 2002, 21, 473–482. [Google Scholar] [CrossRef]
- Shimo, T.; Kubota, S.; Yoshioka, N.; Ibaragi, S.; Isowa, S.; Eguchi, T.; Sasaki, A.; Takigawa, M. Pathogenic role of connective tissue growth factor (CTGF/CCN2) in osteolytic metastasis of breast cancer. J. Bone Miner. Res. 2006, 21, 1045–1059. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, E.; Kubota, S.; Khattab, H.M.; Nishida, T.; Takigawa, M. CCN2 enhances RANKL-induced osteoclast differentiation via direct binding to RANK and OPG. Bone 2015, 73, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Teng, Y.; Zhang, Y.; Liu, J.; Xu, L.; Qu, J.; Hou, K.; Yang, X.; Liu, Y.; Qu, X. C-Src-mediated RANKL-induced breast cancer cell migration by activation of the ERK and Akt pathway. Oncol. Lett. 2012, 3, 395–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baselga, J.; Campone, M.; Piccart, M.; Burris, H.A., 3rd; Rugo, H.S.; Sahmoud, T.; Noguchi, S.; Gnant, M.; Pritchard, K.I.; Lebrun, F.; et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N. Engl. J. Med. 2012, 366, 520–529. [Google Scholar] [CrossRef] [Green Version]
- Ju, J.; Zhu, A.J.; Yuan, P. Progress in targeted therapy for breast cancer. Chronic Dis. Transl. Med. 2018, 4, 164–175. [Google Scholar] [CrossRef] [PubMed]
- McKenna, M.; McGarrigle, S.; Pidgeon, G.P. The next generation of PI3K-Akt-mTOR pathway inhibitors in breast cancer cohorts. Biochim. Biophys. Acta – Rev. Cancer 2018, 1870, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, Y.; Morita, T.Y.; Ohashi, A.; Haeno, H.; Hakozaki, Y.; Fujii, M.; Kashima, Y.; Kobayashi, S.S.; Mukohara, T. Combination treatment with a PI3K/Akt/mTOR pathway inhibitor overcomes resistance to anti-HER2 therapy in PIK3CA-mutant HER2-positive breast cancer cells. Sci. Rep. 2020, 10, 21762. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Hou, J.-Z.; Niu, J.; Xi, Z.-Q.; Ma, C.; Sun, H.; Wang, C.-J.; Fang, D.; Li, Q.; Xie, S.-Q. Akt1 inhibition promotes breast cancer metastasis through EGFR-mediated β-catenin nuclear accumulation. Cell Commun. Signal. 2018, 16, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnett, S.F.; Defeo-Jones, D.; Fu, S.; Hancock, P.J.; Haskell, K.M.; Jones, R.E.; Kahana, J.A.; Kral, A.M.; Leander, K.; Lee, L.L.; et al. Identification and characterization of pleckstrin-homology-domain-dependent and isoenzyme-specific Akt inhibitors. Biochem. J. 2005, 385, 399–408. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hinz, N.; Baranowsky, A.; Horn, M.; Kriegs, M.; Sibbertsen, F.; Smit, D.J.; Clezardin, P.; Lange, T.; Schinke, T.; Jücker, M. Knockdown of AKT3 Activates HER2 and DDR Kinases in Bone-Seeking Breast Cancer Cells, Promotes Metastasis In Vivo and Attenuates the TGFβ/CTGF Axis. Cells 2021, 10, 430. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10020430
Hinz N, Baranowsky A, Horn M, Kriegs M, Sibbertsen F, Smit DJ, Clezardin P, Lange T, Schinke T, Jücker M. Knockdown of AKT3 Activates HER2 and DDR Kinases in Bone-Seeking Breast Cancer Cells, Promotes Metastasis In Vivo and Attenuates the TGFβ/CTGF Axis. Cells. 2021; 10(2):430. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10020430
Chicago/Turabian StyleHinz, Nico, Anke Baranowsky, Michael Horn, Malte Kriegs, Freya Sibbertsen, Daniel J. Smit, Philippe Clezardin, Tobias Lange, Thorsten Schinke, and Manfred Jücker. 2021. "Knockdown of AKT3 Activates HER2 and DDR Kinases in Bone-Seeking Breast Cancer Cells, Promotes Metastasis In Vivo and Attenuates the TGFβ/CTGF Axis" Cells 10, no. 2: 430. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10020430