
Stem Cells as a Source of Pancreatic Cells for Production of 3D Bioprinted Bionic Pancreas in the Treatment of Type 1 Diabetes

 and
and
Abstract
:1. Introduction
2. Structure of the Pancreas and the Role of Individual Cells
3. Available Treatments for Patients with Type 1 Diabetes
4. Embryonic Stem Cells (ESC)
5. Adult Stem Cells
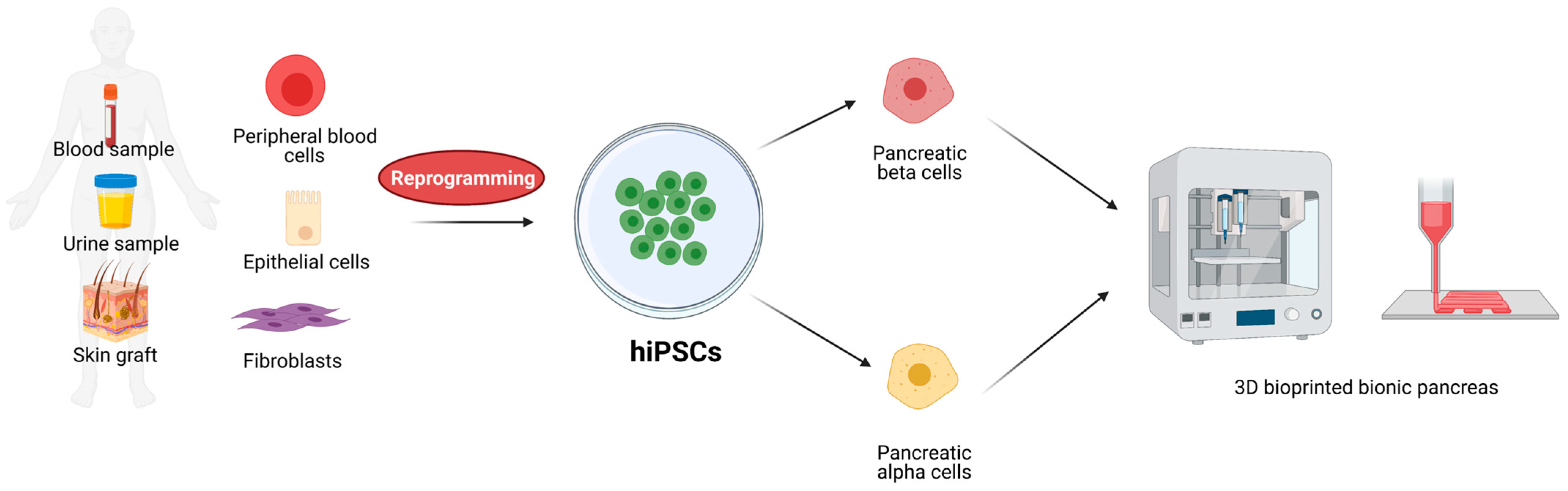
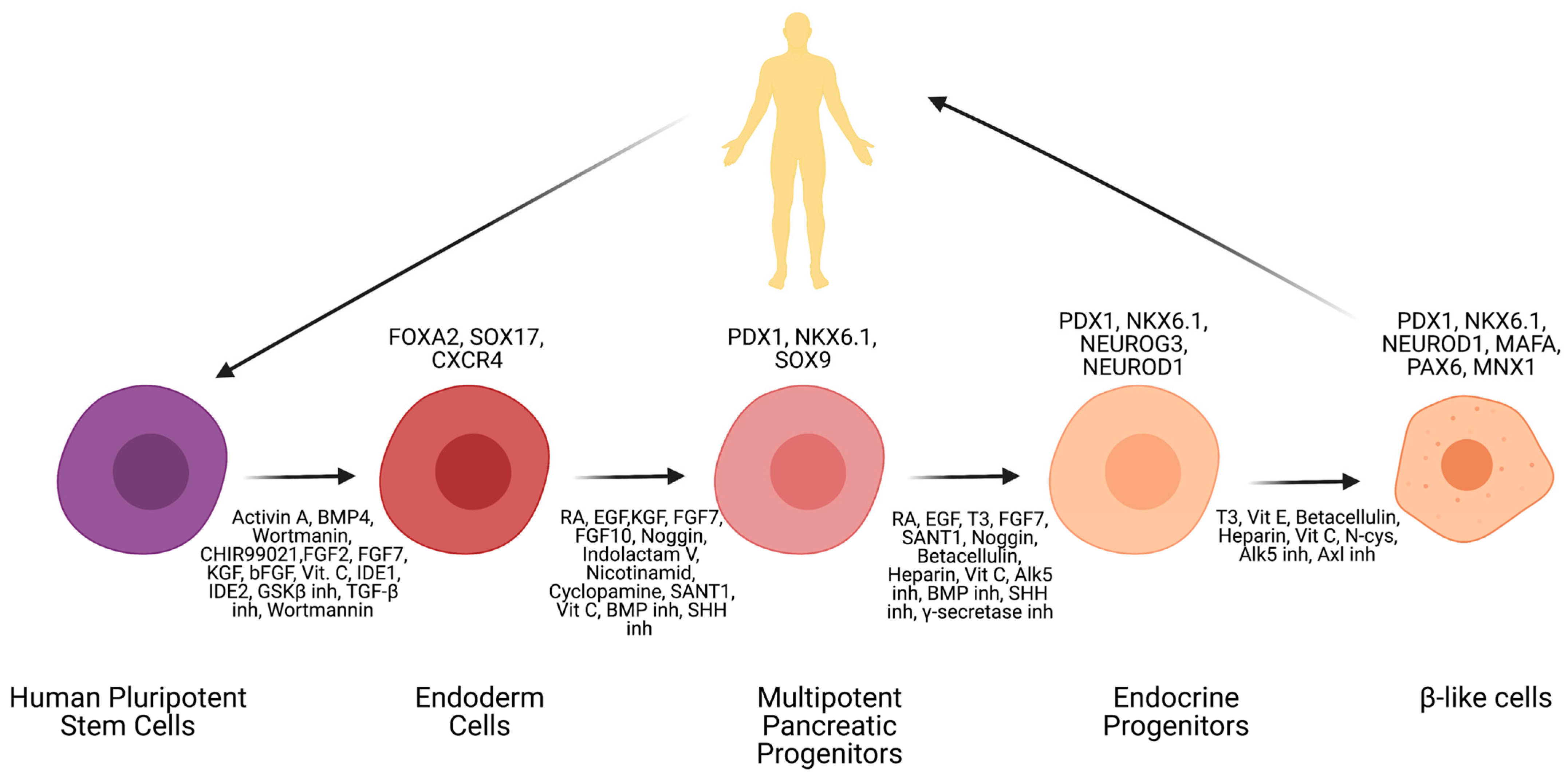
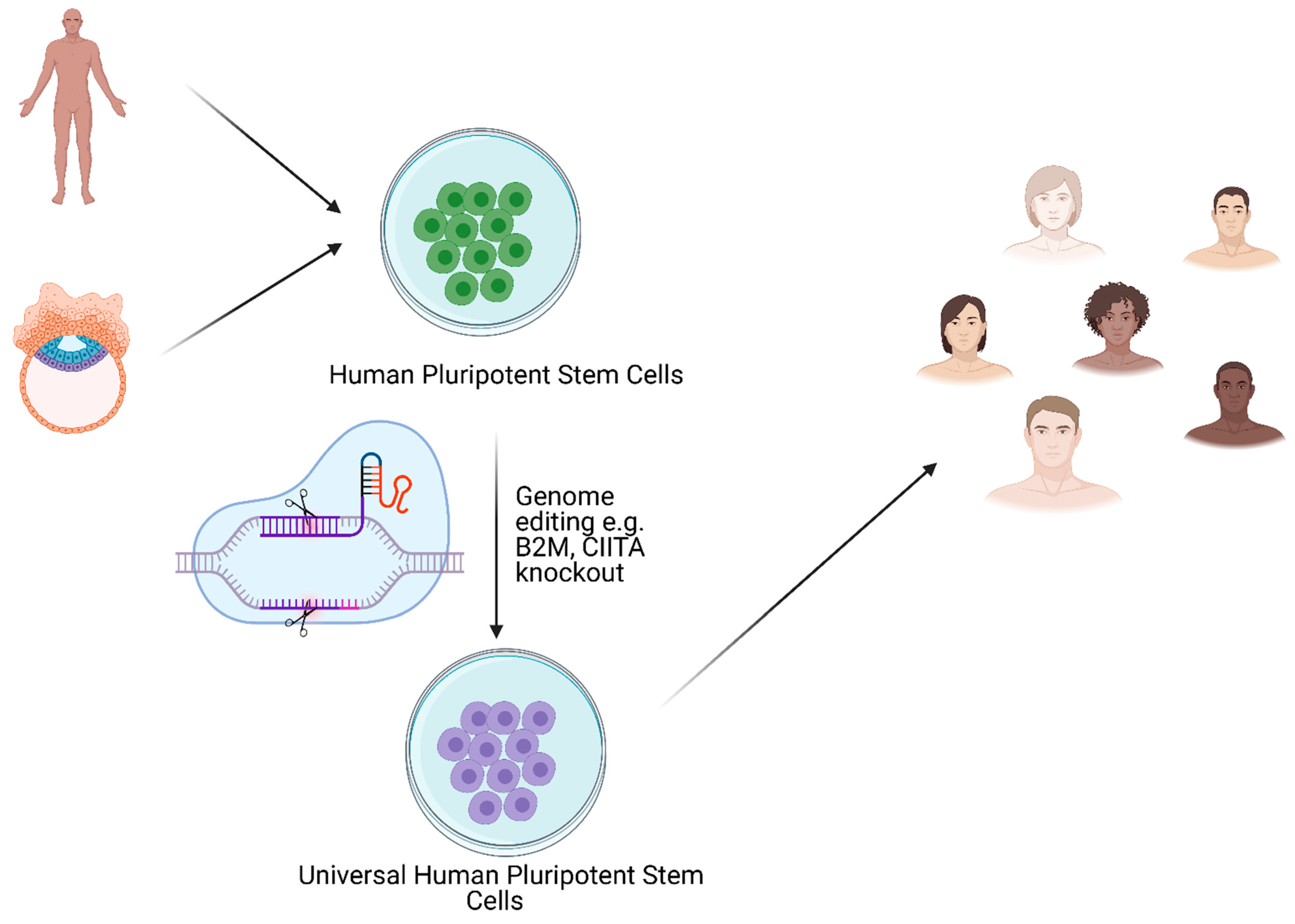
6. Induced Pluripotent Stem Cells (iPSC)
7. Available β-Cell Differentiation Protocols
8. Challenges in Obtaining Fully Maturated β-Cells
9. Available α-Cell Differentiation Protocols
10. Immune-Related Aspects of T1D and Possible Applications of Stem Cells in Therapy
11. Artificial Islets and 3D Bioprinted Pancreas for T1D Treatment
Author Contributions
Funding
Conflicts of Interest
References
- Martin, G.R. Isolation of a Pluripotent Cell Line from Early Mouse Embryos Cultured in Medium Conditioned by Teratocarcinoma Stem Cells. Proc. Natl. Acad. Sci. USA 1981, 78, 7634–7638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, M.J.; Kaufman, M.H. Establishment in Culture of Pluripotential Cells from Mouse Embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.A. Embryonic Stem Cell Lines Derived from Human Blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, V.K.; Kalsan, M.; Kumar, N.; Saini, A.; Chandra, R. Induced Pluripotent Stem Cells: Applications in Regenerative Medicine, Disease Modeling, and Drug Discovery. Front. Cell Dev. Biol. 2015, 3. [Google Scholar] [CrossRef] [Green Version]
- Potten, C.S.; Loeffler, M. Stem Cells: Attributes, Cycles, Spirals, Pitfalls and Uncertainties: Lessons for and from the Crypt. Development 1990, 110. [Google Scholar] [CrossRef]
- Yang, J.; Liu, H.; Sun, H.; Wang, Z.; Zhang, R.; Liu, Y.; Zhang, Q.; Zhang, S.; Zhang, J.; Shi, C.; et al. Construction of Induced Pluripotent Stem Cell Line (ZZUi0017-A) from the Fibroblast Cells of a Female Patient with CACNA1A Mutation by Unintegrated Reprogramming Approach. Stem Cell Res. 2020, 48. [Google Scholar] [CrossRef]
- Wobus, A.M.; Boheler, K.R. Embryonic Stem Cells: Prospects for Developmental Biology and Cell Therapy. Physiol. Rev. 2005, 85, 635–678. [Google Scholar] [CrossRef]
- Valetdinova, K.R.; Maretina, M.A.; Vyatkin, Y.V.; Perepelkina, M.P.; Egorova, A.A.; Baranov, V.S.; Kiselev, A.V.; Gershovich, P.M.; Zakian, S.M. Generation of Three Duchenne Muscular Dystrophy Patient-Derived Induced Pluripotent Stem Cell (IPSC) Lines ICGi002-A, ICGi002-B and ICGi002-C. Stem Cell Res. 2020, 48. [Google Scholar] [CrossRef]
- Wilmut, I.; Schnieke, A.E.; McWhir, J.; Kind, A.J.; Campbell, K.H.S. Viable Offspring Derived from Fetal and Adult Mammalian Cells. Nature 1997, 385, 810–813. [Google Scholar] [CrossRef]
- Murry, C.E.; Keller, G. Differentiation of Embryonic Stem Cells to Clinically Relevant Populations: Lessons from Embryonic Development. Cell 2008, 132, 661–680. [Google Scholar] [CrossRef] [Green Version]
- Vazin, T.; Freed, W.J. Human Embryonic Stem Cells: Derivation, Culture, and Differentiation: A Review. Restor. Neurol. Neurosci. 2010, 28, 589–603. [Google Scholar] [CrossRef]
- González, F.; Boué, S.; Belmonte, J.C.I. Methods for Making Induced Pluripotent Stem Cells: Reprogramming à La Carte. Nat. Rev. Genet. 2011, 12, 231–242. [Google Scholar] [CrossRef]
- Schlaeger, T.M.; Daheron, L.; Brickler, T.R.; Entwisle, S.; Chan, K.; Cianci, A.; DeVine, A.; Ettenger, A.; Fitzgerald, K.; Godfrey, M.; et al. A Comparison of Non-Integrating Reprogramming Methods. Nat. Biotechnol. 2015, 33, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Shafa, M.; Yang, F.; Fellner, T.; Rao, M.S.; Baghbaderani, B.A. Human-Induced Pluripotent Stem Cells Manufactured Using a Current Good Manufacturing Practice-Compliant Process Differentiate into Clinically Relevant Cells from Three Germ Layers. Front. Med. 2018, 5, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Cierpka-Kmiec, K.; Wronska, A.; Kmiec, Z. In Vitro Generation of Pancreatic β-Cells for Diabetes Treatment. I. β-like Cells Derived from Human Pluripotent Stem Cells. Folia Histochem. Cytobiol. 2019, 57, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, S.; Piemonti, L.; Sordi, V. Pluripotent Stem Cell Replacement Approaches to Treat Type 1 Diabetes. Curr. Opin. Pharmacol. 2018, 43, 20–26. [Google Scholar] [CrossRef]
- Millman, J.R.; Xie, C.; Van Dervort, A.; Gürtler, M.; Pagliuca, F.W.; Melton, D.A. Generation of Stem Cell-Derived β-Cells from Patients with Type 1 Diabetes. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Sneddon, J.B.; Tang, Q.; Stock, P.; Bluestone, J.A.; Roy, S.; Desai, T.; Hebrok, M. Stem Cell Therapies for Treating Diabetes: Progress and Remaining Challenges. Cell Stem Cell 2018, 22, 810–823. [Google Scholar] [CrossRef] [Green Version]
- Mesenchymal Stem Cell-Based Therapy for Type 1 Diabetes—Hao Wu—Discovery Medicine. Available online: https://www.discoverymedicine.com/Hao-Wu/2014/03/07/mesenchymal-stem-cell-based-therapy-for-type-1-diabetes/ (accessed on 20 August 2020).
- Liese, A.D. The Burden of Diabetes Mellitus among US Youth: Prevalence Estimates from the SEARCH for Diabetes in Youth Study: SEARCH for Diabetes in Youth Study Group. Pediatrics 2006, 118, 1510–1518. [Google Scholar] [CrossRef] [PubMed]
- Shih, H.P.; Wang, A.; Sander, M. Pancreas Organogenesis: From Lineage Determination to Morphogenesis. Annu. Rev. Cell Dev. Biol. 2013, 29, 81–105. [Google Scholar] [CrossRef] [Green Version]
- Guney, M.A.; Gannon, M. Pancreas Cell Fate. Birth Defects Res. Part C Embryo Today Rev. 2009, 87, 232–248. [Google Scholar] [CrossRef]
- American Diabetes Association. Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 2009, 32. [Google Scholar] [CrossRef] [Green Version]
- Maahs, D.M.; West, N.A.; Lawrence, J.M.; Mayer-Davis, E.J. Epidemiology of Type 1 Diabetes. Endocrinol. Metab. Clin. N. Am. 2010, 39, 481–497. [Google Scholar] [CrossRef] [Green Version]
- Mehers, K.L.; Gillespie, K.M. The Genetic Basis for Type 1 Diabetes. Br. Med. Bull. 2008, 88, 115–129. [Google Scholar] [CrossRef] [Green Version]
- Niedźwiedzka-Rystwej, P.; Wołącewicz, M.; Cywoniuk, P.; Klak, M.; Wszoła, M. Crosstalk Between Immunity System Cells and Pancreas. Transformation of Stem Cells Used in the 3D Bioprinting Process as a Personalized Treatment Method for Type 1 Diabetes. Arch. Immunol. Ther. Exp. 2020, 68, 1–9. [Google Scholar] [CrossRef]
- Diabetes Control and Complications Trial Research Group; Nathan, D.M.; Genuth, S.; Lachin, J.; Cleary, P.; Crofford, O.; Davis, M.; Rand, L.; Siebert, C. The Effect of Intensive Treatment of Diabetes on the Development and Progression of Long-Term Complications in Insulin-Dependent Diabetes Mellitus. N. Engl. J. Med. 1993, 329, 977–986. [Google Scholar] [CrossRef]
- Lachin, J.M.; Genuth, S.; Cleary, P.; Davis, M.D.; Nathan, D.M. Retinopathy and Nephropathy in Patients with Type I Diabetes Four Years after a Trial of Intensive Therapy. N. Engl. J. Med. 2000, 342, 381–389. [Google Scholar] [CrossRef] [Green Version]
- Diabetes Control and Complications Trial Research Group. The Effect of Intensive Treatment of Diabetes on the Development and Progression of Long-Term Complications in Insulin-Dependent Diabetes Mellitus. Endocrinologist 1994, 4, 154. [Google Scholar] [CrossRef]
- Nathan, D.M. Long-Term Complications of Diabetes Mellitus. N. Engl. J. Med. 1993, 328, 1676–1685. [Google Scholar] [CrossRef]
- Cryer, P.E. The Barrier of Hypoglycemia in Diabetes. Diabetes 2008, 57, 3169–3176. [Google Scholar] [CrossRef] [Green Version]
- Kawecki, D.; Kwiatkowski, A.; Michalak, G.; Sawicka-Grzelak, A.; Mlynarczyk, A.; Sokol-Leszczynska, B.; Kot, K.; Czerwinski, J.; Lisik, W.; Bieniasz, M.; et al. Etiologic Agents of Bacteremia in the Early Period After Simultaneous Pancreas-Kidney Transplantation. Transplant. Proc. 2009, 41, 3151–3153. [Google Scholar] [CrossRef]
- Michalak, G.; Kwiatkowski, A.; Bieniasz, M.; Meszaros, J.; Czerwinski, J.; Wszola, M.; Nosek, R.; Ostrowski, K.; Chmura, A.; Danielewicz, R.; et al. Infectious Complications after Simultaneous Pancreas-Kidney Transplantation. Transplant. Proc. 2005, 37, 3560–3563. [Google Scholar] [CrossRef]
- Michalak, G.; Kwiatkowski, A.; Czerwinski, J.; Chmura, A.; Wszola, M.; Nosek, R.; Ostrowski, K.; Danielewicz, R.; Lisik, W.; Adadynski, L.; et al. Surgical Complications of Simultaneous Pancreas-Kidney Transplantation: A 16-Year-Experience at One Center. Transplant. Proc. 2005, 37, 3555–3557. [Google Scholar] [CrossRef]
- Shapiro, A.M.J.; Lakey, J.R.T.; Ryan, E.A.; Korbutt, G.S.; Toth, E.; Warnock, G.L.; Kneteman, N.M.; Rajotte, R.V. Islet Transplantation in Seven Patients with Type 1 Diabetes Mellitus Using a Glucocorticoid-Free Immunosuppressive Regimen. N. Engl. J. Med. 2000, 343, 230–238. [Google Scholar] [CrossRef]
- Shapiro, A.M.J.; Ricordi, C.; Hering, B.J.; Auchincloss, H.; Lindblad, R.; Robertson, R.P.; Secchi, A.; Brendel, M.D.; Berney, T.; Brennan, D.C.; et al. International Trial of the Edmonton Protocol for Islet Transplantation. N. Engl. J. Med. 2006, 355, 1318–1330. [Google Scholar] [CrossRef] [Green Version]
- TransEndoscopic Gastric SubMucosa Islet Transplantation (EGSM-ITx) in Pigs with Streptozotocine Induced Diabetes—Technical Aspects of the Procedure—Preliminary Report—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/19487794/ (accessed on 18 August 2020).
- Wszola, M.; Berman, A.; Ostaszewska, A.; Gorski, L.; Serwanska-Swietek, M.; Gozdowska, J.; Bednarska, K.; Krajewska, M.; Lipinska, A.; Chmura, A.; et al. Islets Allotransplantation Into Gastric Submucosa in a Patient with Portal Hypertension: 4-Year Follow-Up. Transplant. Proc. 2018, 50, 1910–1913. [Google Scholar] [CrossRef] [PubMed]
- Wszola, M.; Berman, A.; Gorski, L.; Ostaszewska, A.; Serwanska-Swietek, M.; Krajewska, M.; Lipinska, A.; Chmura, A.; Kwiatkowski, A. Endoscopic Islet Autotransplantation into Gastric Submucosa—1000-Day Follow-up of Patients. Transplant. Proc. 2018, 50, 2119–2123. [Google Scholar] [CrossRef]
- Ye, Q.; Sung, T.C.; Yang, J.M.; Ling, Q.D.; He, Y.; Higuchi, A. Generation of Universal and Hypoimmunogenic Human Pluripotent Stem Cells. Cell Prolif. 2020, 53, 1–11. [Google Scholar] [CrossRef]
- Thomson, J.A.; Kalishman, J.; Golos, T.G.; Durning, M.; Harris, C.P.; Becker, R.A.; Hearn, J.P. Isolation of a Primate Embryonic Stem Cell Line. Proc. Natl. Acad. Sci. USA 1995, 92, 7844–7848. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, M.K.; Rosler, E.; Rao, M.S. Characterization and Differentiation of Human Embryonic Stem Cells. Cloning Stem Cells 2003, 5, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Klimanskaya, I.; Chung, Y.; Becker, S.; Lu, S.J.; Lanza, R. Human Embryonic Stem Cell Lines Derived from Single Blastomeres. Nature 2006, 444, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Baylis, F. Human Embryonic Stem Cell Lines: The Ethics of Derivation. J. Obstet. Gynaecol. Can. 2002, 24, 159–163. [Google Scholar] [CrossRef]
- Hovatta, O.; Stojkovic, M.; Nogueira, M.; Varela-Nieto, I. European Scientific, Ethical, and Legal Issues on Human Stem Cell Research and Regenerative Medicine. Stem Cells 2010, 28, 1005–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, Y.; Klimanskaya, I.; Becker, S.; Li, T.; Maserati, M.; Lu, S.J.; Zdravkovic, T.; Ilic, D.; Genbacev, O.; Fisher, S.; et al. Human Embryonic Stem Cell Lines Generated without Embryo Destruction. Cell Stem Cell 2008, 2, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Koivisto, H.; Hyvärinen, M.; Strömberg, A.M.; Inzunza, J.; Matilainen, E.; Mikkola, M.; Hovatta, O.; Teerijoki, H. Cultures of Human Embryonic Stem Cells: Serum Replacement Medium or Serum-Containing Media and the Effect of Basic Fibroblast Growth Factor. Reprod. Biomed. Online 2004, 9, 330–337. [Google Scholar] [CrossRef]
- Carpenter, M.K.; Rosler, E.S.; Fisk, G.J.; Brandenberger, R.; Ares, X.; Miura, T.; Lucero, M.; Rao, M.S. Properties of Four Human Embryonic Stem Cell Lines Maintained in a Feeder-Free Culture System. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2004, 229, 243–258. [Google Scholar] [CrossRef]
- Inzunza, J.; Gertow, K.; Strömberg, M.A.; Matilainen, E.; Blennow, E.; Skottman, H.; Wolbank, S.; Ährlund-Richter, L.; Hovatta, O. Derivation of Human Embryonic Stem Cell Lines in Serum Replacement Medium Using Postnatal Human Fibroblasts as Feeder Cells. Stem Cells 2005, 23, 544–549. [Google Scholar] [CrossRef] [Green Version]
- Rodin, S.; Antonsson, L.; Niaudet, C.; Simonson, O.E.; Salmela, E.; Hansson, E.M.; Domogatskaya, A.; Xiao, Z.; Damdimopoulou, P.; Sheikhi, M.; et al. Clonal Culturing of Human Embryonic Stem Cells on Laminin-521/E-Cadherin Matrix in Defined and Xeno-Free Environment. Nat. Commun. 2014, 5, 1–13. [Google Scholar] [CrossRef]
- Harrison, D.E.; Stone, M.; Astle, C.M. Effects of Transplantation on the Primitive Immunohematopoietic Stem Cell. J. Exp. Med. 1990, 172, 431–437. [Google Scholar] [CrossRef] [Green Version]
- Murrell, W.; Féron, F.; Wetzig, A.; Cameron, N.; Splatt, K.; Bellette, B.; Bianco, J.; Perry, C.; Lee, G.; Mackay-Sim, A. Multipotent Stem Cells from Adult Olfactory Mucosa. Dev. Dyn. 2005, 233, 496–515. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Dontu, G.; Wicha, M.S. Mammary Stem Cells, Self-Renewal Pathways, and Carcinogenesis. Breast Cancer Res. 2005, 7, 86–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezza, A.; Sennett, R.; Rendl, M. Adult Stem Cell Niches. Cellular and Molecular Components. Curr. Top. Dev. Biol. 2014, 107, 333–372. [Google Scholar] [PubMed]
- Friedenstein, A.J.; Chailakhyan, R.K.; Latsinik, N.V.; Panasyvk, A.F.; Keiliss-Borok, I.V. Stromal Cells Responsible for Transferring the Microenvironment of the Hemopoietic Tissues: Cloning In Vitro and Retransplantation In Vivo. Transplantation 1974, 17, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Sousa, B.R.; Parreira, R.C.; Fonseca, E.A.; Amaya, M.J.; Tonelli, F.M.P.; Lacerda, S.M.S.N.; Lalwani, P.; Santos, A.K.; Gomes, K.N.; Ulrich, H.; et al. Human Adult Stem Cells from Diverse Origins: An Overview from Multiparametric Immunophenotyping to Clinical Applications. Cytom. Part A 2014, 85, 43–77. [Google Scholar] [CrossRef] [Green Version]
- Zuk, P.A.; Zhu, M.; Mizuno, H.; Huang, J.; Futrell, J.W.; Katz, A.J.; Benhaim, P.; Lorenz, H.P.; Hedrick, M.H. Multilineage Cells from Human Adipose Tissue: Implications for Cell-Based Therapies. Tissue Eng. 2001, 7, 211–228. [Google Scholar] [CrossRef] [Green Version]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage Potential of Adult Human Mesenchymal Stem Cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef] [Green Version]
- Ha, D.H.; Kim, H.; Lee, J.; Kwon, H.H.; Park, G.-H.; Yang, S.H.; Jung, J.Y.; Choi, H.; Lee, J.H.; Sung, S.; et al. Mesenchymal Stem/Stromal Cell-Derived Exosomes for Immunomodulatory Therapeutics and Skin Regeneration. Cells 2020, 9, 1157. [Google Scholar] [CrossRef]
- Börger, V.; Bremer, M.; Ferrer-Tur, R.; Gockeln, L.; Stambouli, O.; Becic, A.; Giebel, B. Mesenchymal Stem/Stromal Cell-Derived Extracellular Vesicles and Their Potential as Novel Immunomodulatory Therapeutic Agents. Int. J. Mol. Sci. 2017, 18, 1450. [Google Scholar] [CrossRef] [Green Version]
- Maacha, S.; Sidahmed, H.; Jacob, S.; Gentilcore, G.; Calzone, R.; Grivel, J.C.; Cugno, C. Paracrine Mechanisms of Mesenchymal Stromal Cells in Angiogenesis. Stem Cells Int. 2020, 2020. [Google Scholar] [CrossRef]
- Poggi, A.; Zocchi, M.R. Immunomodulatory Properties of Mesenchymal Stromal Cells: Still Unresolved “Yin and Yang”. Curr. Stem Cell Res. Ther. 2018, 14, 344–350. [Google Scholar] [CrossRef]
- Goodarzi, P.; Larijani, B.; Alavi-Moghadam, S.; Tayanloo-Beik, A.; Mohamadi-Jahani, F.; Ranjbaran, N.; Payab, M.; Falahzadeh, K.; Mousavi, M.; Arjmand, B. Mesenchymal stem cells-derived exosomes for wound regeneration. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2018; Volume 1119, pp. 119–131. [Google Scholar]
- Fierabracci, A.; Del Fattore, A.; Luciano, R.; Muraca, M.; Teti, A.; Muraca, M. Recent Advances in Mesenchymal Stem Cell Immunomodulation: The Role of Microvesicles. Cell Transplant. 2015, 24, 133–149. [Google Scholar] [CrossRef]
- Prabakar, K.R.; Domínguez-Bendala, J.; Damaris Molano, R.; Pileggi, A.; Villate, S.; Ricordi, C.; Inverardi, L. Generation of Glucose-Responsive, Insulin-Producing Cells from Human Umbilical Cord Blood-Derived Mesenchymal Stem Cells. Cell Transplant. 2012, 21, 1321–1339. [Google Scholar] [CrossRef] [Green Version]
- Shivakumar, S.B.; Lee, H.J.; Son, Y.B.; Bharti, D.; Ock, S.A.; Lee, S.L.; Kang, Y.H.; Park, B.W.; Rho, G.J. In Vitro Differentiation of Single Donor Derived Human Dental Mesenchymal Stem Cells into Pancreatic β Cell-like Cells. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [Green Version]
- Kanafi, M.M.; Rajeshwari, Y.B.; Gupta, S.; Dadheech, N.; Nair, P.D.; Gupta, P.K.; Bhonde, R.R. Transplantation of Islet-like Cell Clusters Derived from Human Dental Pulp Stem Cells Restores Normoglycemia in Diabetic Mice. Cytotherapy 2013, 15, 1228–1236. [Google Scholar] [CrossRef]
- Guo, Q.S.; Zhu, M.Y.; Wang, L.; Fan, X.J.; Lu, Y.H.; Wang, Z.W.; Zhu, S.J.; Wang, Y.; Huang, Y. Combined Transfection of the Three Transcriptional Factors, PDX-1, NeuroD1, and MafA, Causes Differentiation of Bone Marrow Mesenchymal Stem Cells into Insulin-Producing Cells. Exp. Diabetes Res. 2012, 2012. [Google Scholar] [CrossRef] [Green Version]
- Govindasamy, V.; Ronald, V.S.; Abdullah, A.N.; Ganesan Nathan, K.R.; Aziz, Z.A.C.A.; Abdullah, M.; Musa, S.; Abu Kasim, N.H.; Bhonde, R.R. Differentiation of Dental Pulp Stem Cells into Islet-like Aggregates. J. Dent. Res. 2011, 90, 646–652. [Google Scholar] [CrossRef] [Green Version]
- Phadnis, S.M.; Joglekar, M.V.; Dalvi, M.P.; Muthyala, S.; Nair, P.D.; Ghaskadbi, S.M.; Bhonde, R.R.; Hardikar, A.A. Human Bone Marrow-Derived Mesenchymal Cells Differentiate and Mature into Endocrine Pancreatic Lineage In Vivo. Cytotherapy 2011, 13, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Timper, K.; Seboek, D.; Eberhardt, M.; Linscheid, P.; Christ-Crain, M.; Keller, U.; Müller, B.; Zulewski, H. Human Adipose Tissue-Derived Mesenchymal Stem Cells Differentiate into Insulin, Somatostatin, and Glucagon Expressing Cells. Biochem. Biophys. Res. Commun. 2006, 341, 1135–1140. [Google Scholar] [CrossRef] [PubMed]
- Lechner, A.; Yang, Y.-G.; Blacken, R.A.; Wang, L.; Nolan, A.L.; Habener, J.F. No Evidence for Significant Transdifferentiation of Bone Marrow into Pancreatic-Cells In Vivo. Diabetes 2004, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.B.; Uchino, H.; Azuma, K.; Iwashita, N.; Tanaka, Y.; Mochizuki, H.; Migita, M.; Shimada, T.; Kawamori, R.; Watada, H. Little Evidence of Transdifferentiation of Bone Marrow-Derived Cells into Pancreatic Beta Cells. Diabetologia 2003, 46, 1366–1374. [Google Scholar] [CrossRef] [Green Version]
- Ezquer, F.; Ezquer, M.; Contador, D.; Ricca, M.; Simon, V.; Conget, P. The Antidiabetic Effect of Mesenchymal Stem Cells Is Unrelated to Their Transdifferentiation Potential but to Their Capability to Restore Th1/Th2 Balance and to Modify the Pancreatic Microenvironment. Stem Cells 2012, 30, 1664–1674. [Google Scholar] [CrossRef]
- Dave, S.D.; Vanikar, A.V.; Trivedi, H.L.; Thakkar, U.G.; Gopal, S.C.; Chandra, T. Novel Therapy for Insulin-Dependent Diabetes Mellitus: Infusion of in Vitro-Generated Insulin-Secreting Cells. Clin. Exp. Med. 2015, 15, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, U.G.; Trivedi, H.L.; Vanikar, A.V.; Dave, S.D. Insulin-Secreting Adipose-Derived Mesenchymal Stromal Cells with Bone Marrow-Derived Hematopoietic Stem Cells from Autologous and Allogenic Sources for Type 1 Diabetes Mellitus. Cytotherapy 2015, 17, 940–947. [Google Scholar] [CrossRef]
- Dang, L.T.T.; Bui, A.N.T.; Le-Thanh Nguyen, C.; Truong, N.C.; van Bui, A.T.; Kim, N.P.; Truong, K.D.; van Pham, P. Intravenous infusion of human adipose tissue-derived mesenchymal stem cells to treat type 1 diabetic mellitus in mice: An evaluation of grafted cell doses. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2018; Volume 1083, pp. 145–156. [Google Scholar]
- Li, L.; Hui, H.; Jia, X.; Zhang, J.; Liu, Y.; Xu, Q.; Zhu, D. Infusion with Human Bone Marrow-Derived Mesenchymal Stem Cells Improves β-Cell Function in Patients and Non-Obese Mice with Severe Diabetes. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Yaochite, J.N.U.; Caliari-Oliveira, C.; de Souza, L.E.B.; Neto, L.S.; Palma, P.V.B.; Covas, D.T.; Malmegrim, K.C.R.; Donadi, E.A. Therapeutic Efficacy and Biodistribution of Allogeneic Mesenchymal Stem Cells Delivered by Intrasplenic and Intrapancreatic Routes in Streptozotocin-Induced Diabetic Mice. Stem Cell Res. Ther. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerby, A.; Jones, E.S.; Jones, P.M.; King, A.J. Co-Transplantation of Islets with Mesenchymal Stem Cells in Microcapsules Demonstrates Graft Outcome Can Be Improved in an Isolated-Graft Model of Islet Transplantation in Mice. Cytotherapy 2013, 15, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Madec, A.M.; Mallone, R.; Afonso, G.; Abou Mrad, E.; Mesnier, A.; Eljaafari, A.; Thivolet, C. Mesenchymal Stem Cells Protect NOD Mice from Diabetes by Inducing Regulatory T Cells. Diabetologia 2009, 52, 1391–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, R.H.; Seo, M.J.; Reger, R.L.; Spees, J.L.; Pulin, A.A.; Olson, S.D.; Prockop, D.J. Multipotent Stromal Cells from Human Marrow Home to and Promote Repair of Pancreatic Islets and Renal Glomeruli in Diabetic NODscid Mice. Proc. Natl. Acad. Sci. USA 2006, 83. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Chen, J.; Cheng, Y.; Fu, Y.; Zhao, H.; Tang, M.; Zhao, H.; Lin, N.; Shi, X.; Lei, Y.; et al. Mesenchymal Stem Cell-Derived Exosomes Protect Beta Cells against Hypoxia-Induced Apoptosis via MiR-21 by Alleviating ER Stress and Inhibiting P38 MAPK Phosphorylation. Stem Cell Res. Ther. 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Mesples, A.; Majeed, N.; Zhang, Y.; Xiang, H. Early Immunotherapy Using Autologous Adult Stem Cells Reversed the Effect of Anti-Pancreatic Islets in Recently Diagnosed Type 1 Diabetes Mellitus: Preliminary Results. Med. Sci. Monit. 2013, 19, 852–857. [Google Scholar] [CrossRef]
- Carlsson, P.O.; Schwarcz, E.; Korsgren, O.; le Blanc, K. Preserved β-Cell Function in Type 1 Diabetes by Mesenchymal Stromal Cells. Diabetes 2015, 64, 587–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collombat, P.; Xu, X.; Ravassard, P.; Sosa-Pineda, B.; Dussaud, S.; Billestrup, N.; Madsen, O.D.; Serup, P.; Heimberg, H.; Mansouri, A. The Ectopic Expression of Pax4 in the Mouse Pancreas Converts Progenitor Cells into α and Subsequently β Cells. Cell 2009, 138, 449–462. [Google Scholar] [CrossRef] [Green Version]
- Thorel, F.; Népote, V.; Avril, I.; Kohno, K.; Desgraz, R.; Chera, S.; Herrera, P.L. Conversion of Adult Pancreatic α-Cells to Β-Cells after Extreme Β-Cell Loss. Nature 2010, 464, 1149–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, E.J.; Leech, C.A.; Lin, J.C.; Zulewski, H.; Habener, J.F. Insulinotropic Hormone Glucagon-Like Peptide-1 Differentiation of Human Pancreatic Islet-Derived Progenitor Cells into Insulin-Producing Cells. Endocrinology 2002, 143. [Google Scholar] [CrossRef] [PubMed]
- Park, M.-K.; Han, C.; Lee, K.-H.; Hong, S.-H.; Kim, H.S.; Lee, Y.-J.; Jeong, I.K.; Noh, J.-H.; Yang, T.-Y.; Lee, M.-S.; et al. Effects of Activin A on Pancreatic Ductal Cells in Streptozotocin-Induced Diabetic Rats. Transplantation 2007, 83, 925–930. [Google Scholar] [CrossRef]
- Kim, H.S.; Hong, S.H.; Oh, S.H.; Kim, J.H.; Lee, M.S.; Lee, M.K. Activin a, Exendin-4, and Glucose Stimulate Differentiation of Human Pancreatic Ductal Cells. J. Endocrinol. 2013, 217, 241–252. [Google Scholar] [CrossRef] [Green Version]
- Baeyens, L.; de Breuck, S.; Lardon, J.; Mfopou, J.K.; Rooman, I.; Bouwens, L. In Vitro Generation of Insulin-Producing Beta Cells from Adult Exocrine Pancreatic Cells. Diabetologia 2005, 48, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Minami, K.; Okuno, M.; Miyawaki, K.; Okumachi, A.; Ishizaki, K.; Oyama, K.; Kawaguchi, M.; Ishizuka, N.; Iwanaga, T.; Seino, S. Lineage Tracing and Characterization of Insulin-Secreting Cells Generated from Adult Pancreatic Acinar Cells. Proc. Natl. Acad. Sci. USA 2005, 102. [Google Scholar] [CrossRef] [Green Version]
- Hao, E.; Tyrberg, B.; Itkin-Ansari, P.; Lakey, J.R.T.; Geron, I.; Monosov, E.Z.; Barcova, M.; Mercola, M.; Levine, F. Beta-Cell Differentiation from Nonendocrine Epithelial Cells of the Adult Human Pancreas. Nat. Med. 2006, 12, 310–316. [Google Scholar] [CrossRef]
- Zhou, Q.; Brown, J.; Kanarek, A.; Rajagopal, J.; Melton, D.A. In Vivo Reprogramming of Adult Pancreatic Exocrine Cells to β-Cells. Nature 2008, 455, 627–632. [Google Scholar] [CrossRef]
- Li, W.; Nakanishi, M.; Zumsteg, A.; Shear, M.; Wright, C.; Melton, D.A.; Zhou, Q. In Vivo Reprogramming of Pancreatic Acinar Cells to Three Islet Endocrine Subtypes. eLife 2014, 2014. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of Germline-Competent Induced Pluripotent Stem Cells. Nature 2007, 448, 313–317. [Google Scholar] [CrossRef]
- Ye, Z.; Cheng, L. Potential of Human Induced Pluripotent Stem Cells Derived from Blood and Other Postnatal Cell Types. Regen. Med. 2010, 5, 521–530. [Google Scholar] [CrossRef] [Green Version]
- Damdimopoulou, P.; Rodin, S.; Stenfelt, S.; Antonsson, L.; Tryggvason, K.; Hovatta, O. Human Embryonic Stem Cells. Best Pract. Res. Clin. Obstet. Gynaecol. 2016, 31, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; David, B.T.; Trawczynski, M.; Fessler, R.G. Advances in Pluripotent Stem Cells: History, Mechanisms, Technologies, and Applications. Stem Cell Rev. Rep. 2020, 16, 3–32. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Okita, K.; Yamakawa, T.; Matsumura, Y.; Sato, Y.; Amano, N.; Watanabe, A.; Goshima, N.; Yamanaka, S. An Efficient Nonviral Method to Generate Integration-Free Human-Induced Pluripotent Stem Cells from Cord Blood and Peripheral Blood Cells. Stem Cells 2013, 31, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Cai, X.; Wang, L.; Liao, B.; Zhang, H.; Shan, Y.; Chen, Q.; Zhou, T.; Li, X.; Hou, J.; et al. Generating a Non-Integrating Human Induced Pluripotent Stem Cell Bank from Urine-Derived Cells. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Okita, K.; Mochiduki, Y.; Takizawa, N.; Yamanaka, S. Generation of Induced Pluripotent Stem Cells without Myc from Mouse and Human Fibroblasts. Nat. Biotechnol. 2008, 26, 101–106. [Google Scholar] [CrossRef]
- Feng, B.; Ng, J.H.; Heng, J.C.D.; Ng, H.H. Molecules That Promote or Enhance Reprogramming of Somatic Cells to Induced Pluripotent Stem Cells. Cell Stem Cell 2009, 4, 301–312. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced Pluripotent Stem Cell Lines Derived from Human Somatic Cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Wang, J.; Rao, S.; Chu, J.; Shen, X.; Levasseur, D.N.; Theunissen, T.W.; Orkin, S.H. A Protein Interaction Network for Pluripotency of Embryonic Stem Cells. Nature 2006, 444, 364–368. [Google Scholar] [CrossRef]
- Tsai, S.Y.; Bouwman, B.A.; Ang, Y.S.; Kim, S.J.; Lee, D.F.; Lemischka, I.R.; Rendl, M. Single Transcription Factor Reprogramming of Hair Follicle Dermal Papilla Cells to Induced Pluripotent Stem Cells. Stem Cells 2011, 29, 964–971. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.B.; Sebastiano, V.; Wu, G.; Araúzo-Bravo, M.J.; Sasse, P.; Gentile, L.; Ko, K.; Ruau, D.; Ehrich, M.; van den Boom, D.; et al. Oct4-Induced Pluripotency in Adult Neural Stem Cells. Cell 2009, 136, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Greber, B.; Arazo-Bravo, M.J.; Meyer, J.; Park, K.I.; Zaehres, H.; Schöler, H.R. Direct Reprogramming of Human Neural Stem Cells by OCT4. Nature 2009, 461, 649–653. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Zaehres, H.; Wu, G.; Gentile, L.; Ko, K.; Sebastiano, V.; Araúzo-Bravo, M.J.; Ruau, D.; Han, D.W.; Zenke, M.; et al. Pluripotent Stem Cells Induced from Adult Neural Stem Cells by Reprogramming with Two Factors. Nature 2008, 454, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Wernig, M.; Meissner, A.; Cassady, J.P.; Jaenisch, R. C-Myc Is Dispensable for Direct Reprogramming of Mouse Fibroblasts. Cell Stem Cell 2008, 2, 10–12. [Google Scholar] [CrossRef] [Green Version]
- Woltjen, K.; Michael, I.P.; Mohseni, P.; Desai, R.; Mileikovsky, M.; Hämäläinen, R.; Cowling, R.; Wang, W.; Liu, P.; Gertsenstein, M.; et al. PiggyBac Transposition Reprograms Fibroblasts to Induced Pluripotent Stem Cells. Nature 2009, 458, 766–770. [Google Scholar] [CrossRef]
- Kaji, K.; Norrby, K.; Paca, A.; Mileikovsky, M.; Mohseni, P.; Woltjen, K. Virus-Free Induction of Pluripotency and Subsequent Excision of Reprogramming Factors. Nature 2009, 458, 771–775. [Google Scholar] [CrossRef] [Green Version]
- Yang, W. IPSC Reprogramming from Human Peripheral Blood Using Sendai Virus Mediated Gene Transfer. StemBook 2014. [Google Scholar] [CrossRef]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient Induction of Transgene-Free Human Pluripotent Stem Cells Using a Vector Based on Sendai Virus, an RNA Virus That Does Not Integrate into the Host Genome. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 348–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junying, Y.; Kejin, H.; Kim, S.O.; Shulan, T.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human Induced Pluripotent Stem Cells Free of Vector and Transgene Sequences. Science 2009, 324, 797–801. [Google Scholar] [CrossRef] [Green Version]
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.I.; et al. A More Efficient Method to Generate Integration-Free Human IPS Cells. Nat. Methods 2011, 8, 409–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly Efficient Reprogramming to Pluripotency and Directed Differentiation of Human Cells with Synthetic Modified MRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef] [Green Version]
- Mandal, P.K.; Rossi, D.J. Reprogramming Human Fibroblasts to Pluripotency Using Modified MRNA. Nat. Protoc. 2013, 8, 568–582. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, W.Z.; Liu, T.; Feng, X.; Yang, N.; Zhou, H.F. Signaling Pathway of MAPK/ERK in Cell Proliferation, Differentiation, Migration, Senescence and Apoptosis. J. Recept. Signal Transduct. 2015, 35, 600–604. [Google Scholar] [CrossRef]
- Roy, S.K.; Srivastava, R.K.; Shankar, S. Inhibition of PI3K/AKT and MAPK/ERK Pathways Causes Activation of FOXO Transcription Factor, Leading to Cell Cycle Arrest and Apoptosis in Pancreatic Cancer. J. Mol. Signal. 2010, 5, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, C.H.; Wu, G.; Cai, Q.; Gao, X.C.; Tong, F.; Zhou, R.; Zhang, R.G.; Dong, J.H.; Hu, Y.; Dong, X.R. MicroRNA-330-3p Promotes Cell Invasion and Metastasis in Non-Small Cell Lung Cancer through GRIA3 by Activating MAPK/ERK Signaling Pathway. J. Hematol. Oncol. 2017, 10. [Google Scholar] [CrossRef]
- He, L.; Zhou, H.; Zeng, Z.; Yao, H.; Jiang, W.; Qu, H. Wnt/β-Catenin Signaling Cascade: A Promising Target for Glioma Therapy. J. Cell. Physiol. 2019, 234, 2217–2228. [Google Scholar] [CrossRef] [PubMed]
- Razak, S.; Afsar, T.; Ullah, A.; Almajwal, A.; Alkholief, M.; Alshamsan, A.; Jahan, S. Taxifolin, a Natural Flavonoid Interacts with Cell Cycle Regulators Causes Cell Cycle Arrest and Causes Tumor Regression by Activating Wnt/β-Catenin Signaling Pathway 06 Biological Sciences 0601 Biochemistry and Cell Biology 11 Medical and Health Sciences 1112 Oncology and Carcinogenesis. BMC Cancer 2018, 18. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Li, X.; Xiao, Q.; Wang, Z. MiR-574-5p Mediates the Cell Cycle and Apoptosis in Thyroid Cancer Cells via Wnt/β-Catenin Signaling by Repressing the Expression of Quaking Proteins. Oncol. Lett. 2018, 15, 5841–5848. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wei, W.; Zhu, S.; Zhu, J.; Shi, Y.; Lin, T.; Hao, E.; Hayek, A.; Deng, H.; Ding, S. Generation of Rat and Human Induced Pluripotent Stem Cells by Combining Genetic Reprogramming and Chemical Inhibitors. Cell Stem Cell 2009, 4, 16–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteban, M.A.; Wang, T.; Qin, B.; Yang, J.; Qin, D.; Cai, J.; Li, W.; Weng, Z.; Chen, J.; Ni, S.; et al. Vitamin C Enhances the Generation of Mouse and Human Induced Pluripotent Stem Cells. Cell Stem Cell 2010, 6, 71–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.J.; Park, Y.I.; So, B.; Kang, H.G. Sodium Butyrate Efficiently Converts Fully Reprogrammed Induced Pluripotent Stem Cells from Mouse Partially Reprogrammed Cells. Cell. Reprogram. 2014, 16, 345–354. [Google Scholar] [CrossRef]
- Huangfu, D.; Maehr, R.; Guo, W.; Eijkelenboom, A.; Snitow, M.; Chen, A.E.; Melton, D.A. Induction of Pluripotent Stem Cells by Defined Factors Is Greatly Improved by Small-Molecule Compounds. Nat. Biotechnol. 2008, 26, 795–797. [Google Scholar] [CrossRef]
- Hou, P.; Li, Y.; Zhang, X.; Liu, C.; Guan, J.; Li, H.; Zhao, T.; Ye, J.; Yang, W.; Liu, K.; et al. Pluripotent Stem Cells Induced from Mouse Somatic Cells by Small-Molecule Compounds. Science 2013, 341, 651–654. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, T.; Guan, J.; Zhang, X.; Fu, Y.; Ye, J.; Zhu, J.; Meng, G.; Ge, J.; Yang, S.; et al. A XEN-like State Bridges Somatic Cells to Pluripotency during Chemical Reprogramming. Cell 2015, 163, 1678–1691. [Google Scholar] [CrossRef] [Green Version]
- Pagliuca, F.W.; Millman, J.R.; Gürtler, M.; Segel, M.; van Dervort, A.; Ryu, J.H.; Peterson, Q.P.; Greiner, D.; Melton, D.A. Generation of Functional Human Pancreatic β Cells In Vitro. Cell 2014, 159, 428–439. [Google Scholar] [CrossRef] [Green Version]
- Russ, H.A.; Parent, A.V.; Ringler, J.J.; Hennings, T.G.; Nair, G.G.; Shveygert, M.; Guo, T.; Puri, S.; Haataja, L.; Cirulli, V.; et al. Controlled Induction of Human Pancreatic Progenitors Produces Functional Beta-like Cells In Vitro. EMBO J. 2015, 34, 1759–1772. [Google Scholar] [CrossRef] [Green Version]
- Yoshioka, M.; Kayo, T.; Ikeda, T.; Koizumi, A. A Novel Locus, Mody4, Distal to D7Mit189 on Chromosome 7 Determines Early-Onset NIDDM in Nonobese C57BL/6 (Akita) Mutant Mice. Diabetes 1997, 46, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Zhu, S. Chemical Strategies for Pancreatic β Cell Differentiation, Reprogramming, and Regeneration. Acta Biochim. Biophys. Sin. 2017, 49, 298–301. [Google Scholar] [CrossRef] [Green Version]
- Yabe, S.G.; Fukuda, S.; Takeda, F.; Nashiro, K.; Shimoda, M.; Okochi, H. Efficient Generation of Functional Pancreatic β-Cells from Human Induced Pluripotent Stem Cells. J. Diabetes 2017, 9, 168–179. [Google Scholar] [CrossRef]
- Memon, B.; Karam, M.; Al-Khawaga, S.; Abdelalim, E.M. Enhanced Differentiation of Human Pluripotent Stem Cells into Pancreatic Progenitors Co-Expressing PDX1 and NKX6.1. Stem Cell Res. Ther. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Shahjalal, H.M.; Shiraki, N.; Sakano, D.; Kikawa, K.; Ogaki, S.; Baba, H.; Kume, K.; Kume, S. Generation of Insulin-Producing β-like Cells from Human IPS Cells in a Defined and Completely Xeno-Free Culture System. J. Mol. Cell Biol. 2014, 6, 394–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Jiang, W.; Liu, M.; Sui, X.; Yin, X.; Chen, S.; Shi, Y.; Deng, H. Highly Efficient Differentiation of Human ES Cells and IPS Cells into Mature Pancreatic Insulin-Producing Cells. Cell Res. 2009, 19, 429–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezania, A.; Bruin, J.E.; Arora, P.; Rubin, A.; Batushansky, I.; Asadi, A.; O’Dwyer, S.; Quiskamp, N.; Mojibian, M.; Albrecht, T.; et al. Reversal of Diabetes with Insulin-Producing Cells Derived In Vitro from Human Pluripotent Stem Cells. Nat. Biotechnol. 2014, 32, 1121–1133. [Google Scholar] [CrossRef]
- Nair, G.G.; Liu, J.S.; Russ, H.A.; Tran, S.; Saxton, M.S.; Chen, R.; Juang, C.; Li, M.I.; Nguyen, V.Q.; Giacometti, S.; et al. Recapitulating Endocrine Cell Clustering in Culture Promotes Maturation of Human Stem-Cell-Derived β Cells. Nat. Cell Biol. 2019, 21, 263–274. [Google Scholar] [CrossRef]
- Li, X.; Yang, K.Y.; Chan, V.W.; Leung, K.T.; Zhang, X.B.; Wong, A.S.; Chong, C.C.N.; Wang, C.C.; Ku, M.; Lui, K.O. Single-Cell RNA-Seq Reveals That CD9 Is a Negative Marker of Glucose-Responsive Pancreatic β-like Cells Derived from Human Pluripotent Stem Cells. Stem Cell Rep. 2020, 15, 1111–1126. [Google Scholar] [CrossRef]
- Yoshihara, E.; O’Connor, C.; Gasser, E.; Wei, Z.; Oh, T.G.; Tseng, T.W.; Wang, D.; Cayabyab, F.; Dai, Y.; Yu, R.T.; et al. Immune-Evasive Human Islet-like Organoids Ameliorate Diabetes. Nature 2020, 586, 606–611. [Google Scholar] [CrossRef]
- Petersen, M.B.K.; Azad, A.; Ingvorsen, C.; Hess, K.; Hansson, M.; Grapin-Botton, A.; Honoré, C. Single-Cell Gene Expression Analysis of a Human ESC Model of Pancreatic Endocrine Development Reveals Different Paths to β-Cell Differentiation. Stem Cell Rep. 2017, 9, 1246–1261. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Donelan, W.; Ye, H.; Jin, Y.; Lin, Y.; Wu, X.; Wang, Y.; Xi, Y. Real-Time Observation of Pancreatic Beta Cell Differentiation from Human Induced Pluripotent Stem Cells. Am. J. Transl. Res. 2019, 11, 3490–3504. [Google Scholar]
- Velazco-Cruz, L.; Song, J.; Maxwell, K.G.; Goedegebuure, M.M.; Augsornworawat, P.; Hogrebe, N.J.; Millman, J.R. Acquisition of Dynamic Function in Human Stem Cell-Derived β Cells. Stem Cell Rep. 2019, 12, 351–365. [Google Scholar] [CrossRef] [Green Version]
- Sharon, N.; Vanderhooft, J.; Straubhaar, J.; Mueller, J.; Chawla, R.; Zhou, Q.; Engquist, E.N.; Trapnell, C.; Gifford, D.K.; Melton, D.A. Wnt Signaling Separates the Progenitor and Endocrine Compartments during Pancreas Development. Cell Rep. 2019, 27, 2281–2291.e5. [Google Scholar] [CrossRef] [Green Version]
- D’Amour, K.A.; Bang, A.G.; Eliazer, S.; Kelly, O.G.; Agulnick, A.D.; Smart, N.G.; Moorman, M.A.; Kroon, E.; Carpenter, M.K.; Baetge, E.E. Production of Pancreatic Hormone-Expressing Endocrine Cells from Human Embryonic Stem Cells. Nat. Biotechnol. 2006, 24, 1392–1401. [Google Scholar] [CrossRef]
- Jiang, J.; Au, M.; Lu, K.; Eshpeter, A.; Korbutt, G.; Fisk, G.; Majumdar, A.S. Generation of Insulin-Producing Islet-Like Clusters from Human Embryonic Stem Cells. Stem Cells 2007, 25, 1940–1953. [Google Scholar] [CrossRef]
- Bruin, J.E.; Erener, S.; Vela, J.; Hu, X.; Johnson, J.D.; Kurata, H.T.; Lynn, F.C.; Piret, J.M.; Asadi, A.; Rezania, A.; et al. Characterization of Polyhormonal Insulin-Producing Cells Derived In Vitro from Human Embryonic Stem Cells. Stem Cell Res. 2014, 12, 194–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.; Shi, Y.; Zhao, D.; Chen, S.; Yong, J.; Zhang, J.; Qing, T.; Sun, X.; Zhang, P.; Ding, M.; et al. In Vitro Derivation of Functional Insulin-Producing Cells from Human Embryonic Stem Cells. Cell Res. 2007, 17, 333–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, B.W.; Hentze, H.; Rust, W.L.; Chen, Q.P.; Chipperfield, H.; Tan, E.K.; Abraham, S.; Sadasivam, A.; Poh, L.S.; Siew, T.W.; et al. Directed Differentiation of Human Embryonic Stem Cells into the Pancreatic Endocrine Lineage. Stem Cells Dev. 2007, 16, 561–578. [Google Scholar] [CrossRef] [PubMed]
- Kroon, E.; Martinson, L.A.; Kadoya, K.; Bang, A.G.; Kelly, O.G.; Eliazer, S.; Young, H.; Richardson, M.; Smart, N.G.; Cunningham, J.; et al. Pancreatic Endoderm Derived from Human Embryonic Stem Cells Generates Glucose-Responsive Insulin-Secreting Cells In Vivo. Nat. Biotechnol. 2008, 26, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Rezania, A.; Bruin, J.E.; Riedel, M.J.; Mojibian, M.; Asadi, A.; Xu, J.; Gauvin, R.; Narayan, K.; Karanu, F.; O’Neil, J.J.; et al. Maturation of Human Embryonic Stem Cell-Derived Pancreatic Progenitors into Functional Islets Capable of Treating Pre-Existing Diabetes in Mice. Diabetes 2012, 61, 2016–2029. [Google Scholar] [CrossRef] [Green Version]
- Bruin, J.E.; Asadi, A.; Fox, J.K.; Erener, S.; Rezania, A.; Kieffer, T.J. Accelerated Maturation of Human Stem Cell-Derived Pancreatic Progenitor Cells into Insulin-Secreting Cells in Immunodeficient Rats Relative to Mice. Stem Cell Rep. 2015, 5, 1081–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, T.C. Enabling Technologies for Cell-Based Clinical Translation Concise Review: Manufacturing of Pancreatic Endoderm Cells for Clinical Trials in Type 1 Diabetes. Stem Cells Transl. Med. 2015, 4, 927–931. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.J.; Thompson, D.; Donner, T.W.; Bellin, M.D.; Hsueh, W. Insulin Expression and Glucose-Responsive Circulating C-Peptide in Type 1 Diabetes Patients Implanted Subcutaneously with Pluripotent Stem Cell-Derived Pancreatic Endoderm Cells in a Macro-Device. Lancet 2019. [Google Scholar] [CrossRef]
- Wang, H.; Brun, T.; Kataoka, K.; Sharma, A.J.; Wollheim, C.B. MAFA Controls Genes Implicated in Insulin Biosynthesis and Secretion. Diabetologia 2007, 50, 348–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, J.; Correa-Medina, M.; Ricordi, C.; Edlund, H.; Diez, J.A. Endocrine Cell Clustering during Human Pancreas Development. J. Histochem. Cytochem. 2009, 57, 811–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stock, A.A.; Manzoli, V.; de Toni, T.; Abreu, M.M.; Poh, Y.C.; Ye, L.; Roose, A.; Pagliuca, F.W.; Thanos, C.; Ricordi, C.; et al. Conformal Coating of Stem Cell-Derived Islets for β Cell Replacement in Type 1 Diabetes. Stem Cell Rep. 2020, 14, 91–104. [Google Scholar] [CrossRef] [Green Version]
- Vegas, A.J.; Veiseh, O.; Gürtler, M.; Millman, J.R.; Pagliuca, F.W.; Bader, A.R.; Doloff, J.C.; Li, J.; Chen, M.; Olejnik, K.; et al. Long-Term Glycemic Control Using Polymer-Encapsulated Human Stem Cell-Derived Beta Cells in Immune-Competent Mice. Nat. Med. 2016, 22, 306–311. [Google Scholar] [CrossRef]
- de Klerk, E.; Hebrok, M. Stem Cell-Based Clinical Trials for Diabetes Mellitus. Front. Endocrinol. 2021, 12. [Google Scholar] [CrossRef]
- Brissova, M.; Haliyur, R.; Saunders, D.; Shrestha, S.; Dai, C.; Blodgett, D.M.; Bottino, R.; Campbell-Thompson, M.; Aramandla, R.; Poffenberger, G.; et al. α Cell Function and Gene Expression Are Compromised in Type 1 Diabetes. Cell Rep. 2018, 22, 2667–2676. [Google Scholar] [CrossRef] [Green Version]
- Siafarikas, A.; Johnston, R.J.; Bulsara, M.K.; O’Leary, P.; Jones, T.W.; Davis, E.A. Early Loss of the Glucagon Response to Hypoglycemia in Adolescents with Type 1 Diabetes. Diabetes Care 2012, 35, 1757–1762. [Google Scholar] [CrossRef] [Green Version]
- Rezania, A.; Riedel, M.J.; Wideman, R.D.; Karanu, F.; Ao, Z.; Warnock, G.L.; Kieffer, T.J. Production of Functional Glucagon-Secreting α-Cells from Human Embryonic Stem Cells. Diabetes 2011, 60, 239–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, Q.P.; Veres, A.; Chen, L.; Slama, M.Q.; Kenty, J.H.R.; Hassoun, S.; Brown, M.R.; Dou, H.; Duffy, C.D.; Zhou, Q.; et al. A Method for the Generation of Human Stem Cell-Derived Alpha Cells. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.A.; Redick, S.D.; Blodgett, D.M.; Richardson, S.J.; Leete, P.; Krogvold, L.; Dahl-Jørgensen, K.; Bottino, R.; Brissova, M.; Spaeth, J.M.; et al. HLA Class II Antigen Processing and Presentation Pathway Components Demonstrated by Transcriptome and Protein Analyses of Islet β-Cells from Donors with Type 1 Diabetes. Diabetes 2019, 68, 988–1001. [Google Scholar] [CrossRef] [Green Version]
- Pugliese, A. Autoreactive T Cells in Type 1 Diabetes. J. Clin. Investig. 2017, 127, 2881–2891. [Google Scholar] [CrossRef]
- Choo, S.Y. The HLA System: Genetics, Immunology, Clinical Testing, and Clinical Implications. Yonsei Med. J. 2007, 48, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumura, M.; Fremont, D.H.; Peterson, P.A.; Wilson, I.A. Emerging Principles for the Recognition of Peptide Antigens by MHC Class I Molecules. Science 1992, 257, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Shiina, T.; Inoko, H.; Kulski, J.K. An Update of the HLA Genomic Region, Locus Information and Disease Associations: 2004. Tissue Antigens 2004, 64, 631–649. [Google Scholar] [CrossRef]
- Hu, X.; Deutsch, A.J.; Lenz, T.L.; Onengut-Gumuscu, S.; Han, B.; Chen, W.M.; Howson, J.M.M.; Todd, J.A.; De Bakker, P.I.W.; Rich, S.S.; et al. Additive and Interaction Effects at Three Amino Acid Positions in HLA-DQ and HLA-DR Molecules Drive Type 1 Diabetes Risk. Nat. Genet. 2015, 47, 898–905. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Abbas, A.K.; Fausto, N.; Mitchell, R.S. Robbins Basic Pathology, 8th ed.; Saunders: Philadelphia, PA, USA, 2010; Available online: https://medbook.com.pl/ksiazka/pokaz/id/30048/tytul/robbins-basic-pathology-kumar-abbas-fausto-mitchell-saunders (accessed on 17 August 2020).
- Margaritte-Jeannin, P.; Babron, M.C.; Bourgey, M.; Louka, A.S.; Clot, F.; Percopo, S.; Coto, I.; Hugot, J.P.; Ascher, H.; Sollid, L.M.; et al. HLA-DQ Relative Risks for Coeliac Disease in European Populations: A Study of the European Genetics Cluster on Coeliac Disease. Tissue Antigens 2004, 63, 562–567. [Google Scholar] [CrossRef]
- Wicker, L.S.; Leiter, E.H.; Todd, J.A.; Renjilian, R.J.; Peterson, E.; Fischer, P.A.; Podolin, P.L.; Zijlstra, M.; Jaenisch, R.; Peterson, L.B. Β2-Microglobulin-Deficient NOD Mice Do Not Develop Insulitis or Diabetes. Diabetes 1994, 43, 500–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serreze, D.V.; Leiter, E.H.; Christianson, G.J.; Greiner, D.; Roopenian, D.C. Major Histocompatibility Complex Class I-Deficient N0D-E2/n. Diabetes 1994, 43, 505–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Challa-Malladi, M.; Lieu, Y.K.; Califano, O.; Holmes, A.B.; Bhagat, G.; Murty, V.V.; Dominguez-Sola, D.; Pasqualucci, L.; Dalla-Favera, R. Combined Genetic Inactivation of Β2-Microglobulin and CD58 Reveals Frequent Escape from Immune Recognition in Diffuse Large B Cell Lymphoma. Cancer Cell 2011, 20, 728–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nedelkovska, H.; Edholm, E.S.; Haynes, N.; Robert, J. Effective RNAi-Mediated Β2-Microglobulin Loss of Function by Transgenesis in Xenopus laevis. Biol. Open 2013, 2, 335–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, P.K.; Ferreira, L.M.R.; Collins, R.; Meissner, T.B.; Boutwell, C.L.; Friesen, M.; Vrbanac, V.; Garrison, B.S.; Stortchevoi, A.; Bryder, D.; et al. Efficient Ablation of Genes in Human Hematopoietic Stem and Effector Cells Using CRISPR/Cas9. Cell Stem Cell 2014, 15, 643–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Quan, Y.; Qing, Y.; Morales, J.E.; Wetsel, R.A. Targeted Disruption of the B2-Microglobulin Gene Minimizes the Immunogenicity of Human Embryonic Stem Cells. Stem Cells Transl. Med. 2014, 4, 1–10. [Google Scholar]
- Chen, H.; Li, Y.; Lin, X.; Cui, D.; Cui, C.; Li, H.; Xiao, L. Functional Disruption of Human Leukocyte Antigen II in Human Embryonic Stem Cell. Biol. Res. 2015, 48, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to Checkpoint Blockade Therapy through Inactivation of Antigen Presentation. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Mattapally, S.; Pawlik, K.M.; Fast, V.G.; Zumaquero, E.; Lund, F.E.; Randall, T.D.; Townes, T.M.; Zhang, J. Human Leukocyte Antigen Class I and II Knockout Human Induced Pluripotent Stem Cell–Derived Cells: Universal Donor for Cell Therapy. J. Am. Heart Assoc. 2018, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Zijlstra, M.; Bix, M.; Simister, N.E.; Loring, J.M.; Raulet, D.H.; Jaenisch, R. Β2-Microglobulin Deficient Mice Lack CD4−8+ Cytolytic T Cells. Nature 1990, 344, 742–746. [Google Scholar] [CrossRef]
- Bjorkman, P.J.; Saper, M.A.; Samraoui, B.; Bennett, W.S.; Strominger, J.L.; Wiley, D.C. Structure of the Human Class I Histocompatibility Antigen, HLA-A2. Nature 1987, 329, 506–512. [Google Scholar] [CrossRef]
- Chang, C.H.; Fontes, J.D.; Peterlin, M.; Flavell, R.A. Class II Transactivator (CIITA) Is Sufficient for the Inducible Expression of Major Histocompatibility Complex Class II Genes. J. Exp. Med. 1994, 180, 1367–1374. [Google Scholar] [CrossRef]
- Chang, C.H.; Flavell, R.A. Class II Transactivator Regulates the Expression of Multiple Genes Involved in Antigen Presentation. J. Exp. Med. 1995, 181, 765–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buch, T.; Polic, B.; Clausen, B.E.; Weiss, S.; Akilli-Ozturk, O.; Chang, C.-H.; Flavell, R.; Schulz, A.; Jonjic, S.; Waisman, A.; et al. MHC Class II Expression through a Hitherto Unknown Pathway Supports T Helper Cell-Dependent Immune Responses: Implications for MHC Class II Deficiency. Blood 2006, 107, 1434–1444. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.H.; Sohn, H.J.; Lee, H.J.; Cho, H.I.; Kim, T.G. Antigen Presentation by Individually Transferred HLA Class i Genes in HLA-A, HLA-B, HLA-C Null Human Cell Line Generated Using the Multiplex CRISPR-Cas9 System. J. Immunother. 2017, 40, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.; Choi, J.; Park, N.; Kang, J.; Kim, M.; Kim, Y.; Ju, J.H. Development of Immunocompatible Pluripotent Stem Cells via CRISPR-Based Human Leukocyte Antigen Engineering. Exp. Mol. Med. 2019, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gornalusse, G.G.; Hirata, R.K.; Funk, S.E.; Riolobos, L.; Lopes, V.S.; Manske, G.; Prunkard, D.; Colunga, A.G.; Hanafi, L.A.; Clegg, D.O.; et al. HLA-E-Expressing Pluripotent Stem Cells Escape Allogeneic Responses and Lysis by NK Cells. Nat. Biotechnol. 2017, 35, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Deuse, T.; Hu, X.; Gravina, A.; Wang, D.; Tediashvili, G.; De, C.; Thayer, W.O.; Wahl, A.; Garcia, J.V.; Reichenspurner, H.; et al. Hypoimmunogenic Derivatives of Induced Pluripotent Stem Cells Evade Immune Rejection in Fully Immunocompetent Allogeneic Recipients. Nat. Biotechnol. 2019, 37, 252–258. [Google Scholar] [CrossRef]
- Han, X.; Wang, M.; Duan, S.; Franco, P.J.; Kenty, J.H.R.; Hedrick, P.; Xia, Y.; Allen, A.; Ferreira, L.M.R.; Strominger, J.L.; et al. Generation of Hypoimmunogenic Human Pluripotent Stem Cells. Proc. Natl. Acad. Sci. USA 2019, 116, 10441–10446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Wang, B.; Ono, M.; Kagita, A.; Fujii, K.; Sasakawa, N.; Ueda, T.; Gee, P.; Nishikawa, M.; Nomura, M.; et al. Targeted Disruption of HLA Genes via CRISPR-Cas9 Generates IPSCs with Enhanced Immune Compatibility. Cell Stem Cell 2019, 24, 566–578.e7. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Li, W.; Liu, Y.; Chen, Z.; Hui, Y.; Hao, P.; Xu, X.; Zhang, S.; Feng, H.; Zhang, B.; et al. Generation of Hypoimmunogenic Human Pluripotent Stem Cells via Expression of Membrane-Bound and Secreted Β2m-HLA-G Fusion Proteins. Stem Cells 2020, 38, 1423–1437. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M.; Kida, Y.S.; Hawkins, R.D.; Nery, J.R.; Hon, G.; Antosiewicz-Bourget, J.; Ogmalley, R.; Castanon, R.; Klugman, S.; et al. Hotspots of Aberrant Epigenomic Reprogramming in Human Induced Pluripotent Stem Cells. Nature 2011, 471, 68–73. [Google Scholar] [CrossRef] [Green Version]
- Gore, A.; Li, Z.; Fung, H.L.; Young, J.E.; Agarwal, S.; Antosiewicz-Bourget, J.; Canto, I.; Giorgetti, A.; Israel, M.A.; Kiskinis, E.; et al. Somatic Coding Mutations in Human Induced Pluripotent Stem Cells. Nature 2011, 471, 63–67. [Google Scholar] [CrossRef]
- Miura, K.; Okada, Y.; Aoi, T.; Okada, A.; Takahashi, K.; Okita, K.; Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Ohnuki, M.; et al. Variation in the Safety of Induced Pluripotent Stem Cell Lines. Nat. Biotechnol. 2009, 27, 743–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okano, H.; Nakamura, M.; Yoshida, K.; Okada, Y.; Tsuji, O.; Nori, S.; Ikeda, E.; Yamanaka, S.; Miura, K. Steps toward Safe Cell Therapy Using Induced Pluripotent Stem Cells. Circ. Res. 2013, 112, 523–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Introna, M.; Barbui, A.M.; Bambacioni, F.; Casati, C.; Gaipa, G.; Borleri, G.; Bernasconi, S.; Barbui, T.; Golay, J.; Biondi, A.; et al. Genetic Modification of Human T Cells with CD20: A Strategy to Purify and Lyse Transduced Cells with Anti-CD20 Antibodies. Hum. Gene Ther. 2000, 11, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, T.T.; Pedersen, N.; Juel, H.; Poulsen, H.S. A Chimeric Fusion of the HASH1 and EZH2 Promoters Mediates High and Specific Reporter and Suicide Gene Expression and Cytotoxicity in Small Cell Lung Cancer Cells. Cancer Gene Ther. 2008, 15, 563–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, L.; Freeman, K.W.; Khan, T.; Pham, E.; Spencer, D.M. Improved Artificial Death Switches Based on Caspases and FADD. Hum. Gene Ther. 1999, 10, 2273–2285. [Google Scholar] [CrossRef] [PubMed]
- Clackson, T.; Yang, W.; Rozamus, L.W.; Hatada, M.; Amara, J.F.; Rollins, C.T.; Stevenson, L.F.; Magari, S.R.; Wood, S.A.; Courage, N.L.; et al. Redesigning an FKBP-Ligand Interface to Generate Chemical Dimerizers with Novel Specificity. Proc. Natl. Acad. Sci. USA 1998, 95, 10437–10442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Naik, S.; Dakhova, O.; Dotti, G.; Heslop, H.E.; Brenner, M.K. Serial Activation of the Inducible Caspase 9 Safety Switch after Human Stem Cell Transplantation. Mol. Ther. 2016, 24, 823–831. [Google Scholar] [CrossRef] [Green Version]
- Di Stasi, A.; Tey, S.K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible Apoptosis as a Safety Switch for Adoptive Cell Therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef] [Green Version]
- Straathof, K.C.; Pulè, M.A.; Yotnda, P.; Dotti, G.; Vanin, E.F.; Brenner, M.K.; Heslop, H.E.; Spencer, D.M.; Rooney, C.M. An Inducible Caspase 9 Safety Switch for T-Cell Therapy. Blood 2005, 105, 4247–4254. [Google Scholar] [CrossRef]
- Shi, Z.D.; Tchao, J.; Wu, L.; Carman, A.J. Precision Installation of a Highly Efficient Suicide Gene Safety Switch in Human Induced Pluripotent Stem Cells. Stem Cells Transl. Med. 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- González, F.; Zhu, Z.; Shi, Z.D.; Lelli, K.; Verma, N.; Li, Q.V.; Huangfu, D. An ICRISPR Platform for Rapid, Multiplexable, and Inducible Genome Editing in Human Pluripotent Stem Cells. Cell Stem Cell 2014, 15, 215–226. [Google Scholar] [CrossRef] [Green Version]
- DeKelver, R.C.; Choi, V.M.; Moehle, E.A.; Paschon, D.E.; Hockemeyer, D.; Meijsing, S.H.; Sancak, Y.; Cui, X.; Steine, E.J.; Miller, J.C.; et al. Functional Genomics, Proteomics, and Regulatory DNA Analysis in Isogenic Settings Using Zinc Finger Nuclease-Driven Transgenesis into a Safe Harbor Locus in the Human Genome. Genome Res. 2010, 20, 1133–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadelain, M.; Papapetrou, E.P.; Bushman, F.D. Safe Harbours for the Integration of New DNA in the Human Genome. Nat. Rev. Cancer 2012, 12, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Moede, T.; Leibiger, I.B.; Berggren, P.O. Alpha Cell Regulation of Beta Cell Function. Diabetologia 2020, 63, 2064–2075. [Google Scholar] [CrossRef]
- Adams, M.T.; Gilbert, J.M.; Hinojosa Paiz, J.; Bowman, F.M.; Blum, B. Endocrine Cell Type Sorting and Mature Architecture in the Islets of Langerhans Require Expression of Roundabout Receptors in β Cells. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitsou-Mylona, I.; Burns, C.J.; Squires, P.E.; Persaud, S.J.; Jones, P.M. A Role for the Extracellular Calcium-Sensing Receptor in Cell-Cell Communication in Pancreatic Islets of Langerhans. Cell. Physiol. Biochem. 2008, 22, 557–566. [Google Scholar] [CrossRef] [Green Version]
- Aamodt, K.I.; Powers, A.C. Signals in the Pancreatic Islet Microenvironment Influence β-Cell Proliferation. Diabetes Obes. Metab. 2017, 19, 124–136. [Google Scholar] [CrossRef] [Green Version]
- Abdulreda, M.H.; Rodriguez-Diaz, R.; Cabrera, O.; Caicedo, A.; Berggren, P.O. The different faces of the pancreatic islet. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2016; Volume 938, pp. 11–24. [Google Scholar]
- Röder, P.V.; Wu, B.; Liu, Y.; Han, W. Pancreatic Regulation of Glucose Homeostasis. Exp. Mol. Med. 2016, 48, e219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang Do, O.; Thorn, P. Insulin Secretion from Beta Cells within Intact Islets: Location Matters. Clin. Exp. Pharmacol. Physiol. 2015, 42, 406–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, Y.; Lee, J.S.; Choi, S.; Yang, J.H.; Sander, M.; Chung, S.; Lee, S.H. In Vivo–Mimicking Microfluidic Perfusion Culture of Pancreatic Islet Spheroids. Sci. Adv. 2019, 5, eaax4520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napierala, H.; Hillebrandt, K.H.; Haep, N.; Tang, P.; Tintemann, M.; Gassner, J.; Noesser, M.; Everwien, H.; Seiffert, N.; Kluge, M.; et al. Engineering an Endocrine Neo-Pancreas by Repopulation of a Decellularized Rat Pancreas with Islets of Langerhans. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Everwien, H.; Keshi, E.; Hillebrandt, K.H.; Ludwig, B.; Weinhart, M.; Tang, P.; Beierle, A.S.; Napierala, H.; Gassner, J.M.; Seiffert, N.; et al. Engineering an Endothelialized, Endocrine Neo-Pancreas: Evaluation of Islet Functionality in an Ex Vivo Model. Acta Biomater. 2020, 117. [Google Scholar] [CrossRef]
- Ribeiro, D.; Kvist, A.J.; Wittung-Stafshede, P.; Hicks, R.; Forslöw, A. 3D-Models of Insulin-Producing β-Cells: From Primary Islet Cells to Stem Cell-Derived Islets. Stem Cell Rev. Rep. 2018, 14, 177–188. [Google Scholar] [CrossRef]
- Caicedo, A. Paracrine and Autocrine Interactions in the Human Islet: More than Meets the Eye. Semin. Cell Dev. Biol. 2013, 24, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Eberhard, D.; Kragl, M.; Lammert, E. “Giving and Taking”: Endothelial and β-Cells in the Islets of Langerhans. Trends Endocrinol. Metab. 2010, 21, 457–463. [Google Scholar] [CrossRef]
- Tomei, A.A.; Manzoli, V.; Fraker, C.A.; Giraldo, J.; Velluto, D.; Najjar, M.; Pileggi, A.; Molano, R.D.; Ricordi, C.; Stabler, C.L.; et al. Device Design and Materials Optimization of Conformal Coating for Islets of Langerhans. Proc. Natl. Acad. Sci. USA 2014, 111, 10514–10519. [Google Scholar] [CrossRef] [Green Version]
- Qi, M.; Strand, B.L.; Mørch, Y.; Lacík, I.; Wang, Y.; Salehi, P.; Barbaro, B.; Gangemi, A.; Kuechle, J.; Romagnoli, T.; et al. Encapsulation of Human Islets in Novel Inhomogeneous Alginate-Ca2+/Ba2+ Microbeads: In Vitro and In Vivo Function. Artif. Cells Blood Substit. Biotechnol. 2008, 36, 403–420. [Google Scholar] [CrossRef] [Green Version]
- Strand, B.L.; Gåserød, O.; Kulseng, B.; Espevik, T.; Skjåk-Bræk, G. Alginate-Polylysine-Alginate Microcapsules: Effect of Size Reduction on Capsule Properties. J. Microencapsul. 2002, 19, 615–630. [Google Scholar] [CrossRef] [PubMed]
- Buchwald, P.; Tamayo-Garcia, A.; Manzoli, V.; Tomei, A.A.; Stabler, C.L. Glucose-Stimulated Insulin Release: Parallel Perifusion Studies of Free and Hydrogel Encapsulated Human Pancreatic Islets. Biotechnol. Bioeng. 2018, 115, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Novosel, E.C.; Kleinhans, C.; Kluger, P.J. Vascularization Is the Key Challenge in Tissue Engineering. Adv. Drug Deliv. Rev. 2011, 63, 300–311. [Google Scholar] [CrossRef] [PubMed]
- Shor, L.; Güçeri, S.; Chang, R.; Gordon, J.; Kang, Q.; Hartsock, L.; An, Y.; Sun, W. Precision Extruding Deposition (PED) Fabrication of Polycaprolactone (PCL) Scaffolds for Bone Tissue Engineering. Biofabrication 2009, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, T.; Zhao, W.; Zhu, J.M.; Albanna, M.Z.; Yoo, J.J.; Atala, A. Complex Heterogeneous Tissue Constructs Containing Multiple Cell Types Prepared by Inkjet Printing Technology. Biomaterials 2013, 34, 130–139. [Google Scholar] [CrossRef]
- Barron, J.A.; Wu, P.; Ladouceur, H.D.; Ringeisen, B.R. Biological Laser Printing: A Novel Technique for Creating Heterogeneous 3-Dimensional Cell Patterns. Biomed. Microdevices 2004, 6, 139–147. [Google Scholar] [CrossRef]
- Wang, Z.; Tian, Z.; Jin, X.; Holzman, J.F.; Menard, F.; Kim, K. Visible Light-Based Stereolithography Bioprinting of Cell-Adhesive Gelatin Hydrogels. In Proceedings of the 39th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), Jeju, Korea, 11–15 July 2017; pp. 1599–1602. [Google Scholar] [CrossRef]
- Klak, M.; Bryniarski, T.; Kowalska, P.; Gomolka, M.; Tymicki, G.; Kosowska, K.; Cywoniuk, P.; Dobrzanski, T.; Turowski, P.; Wszola, M. Novel Strategies in Artificial Organ Development: What Is the Future of Medicine? Micromachines 2020, 11, 646. [Google Scholar] [CrossRef] [PubMed]
- Marchioli, G.; van Gurp, L.; van Krieken, P.P.; Stamatialis, D.; Engelse, M.; van Blitterswijk, C.A.; Karperien, M.B.J.; de Koning, E.; Alblas, J.; Moroni, L.; et al. Fabrication of Three-Dimensional Bioplotted Hydrogel Scaffolds for Islets of Langerhans Transplantation. Biofabrication 2015, 7, 25009. [Google Scholar] [CrossRef] [PubMed]
- Duin, S.; Schütz, K.; Ahlfeld, T.; Lehmann, S.; Lode, A.; Ludwig, B.; Gelinsky, M. 3D Bioprinting of Functional Islets of Langerhans in an Alginate/Methylcellulose Hydrogel Blend. Adv. Healthc. Mater. 2019, 8, 1–14. [Google Scholar] [CrossRef]
- Berman, A.; Klak, M.; Adamiok, A.; Kaczyński, Ł.; Tymicki, G.; Gomółka, M.; Kowalska, P.; Kosowska, K.; Cywoniuk, P.; Turowski, P.; et al. The Influence of the Flow of Detergent and Donor Characteristics on the Extracellular Matrix Composition After Human Pancreas Decellularization. Transplant. Proc. 2020, 52, 2043–2049. [Google Scholar] [CrossRef]
- Klak, M.; Kosowska, K.; Majdanska, E.; Dobrzanski, T.; Berman, A.; Kaczynski, L.; Kowalska, P.; Gomolka, M.; Wszoła, M. Towards 3D-Bioprinting of Bionic Pancreas: Effect of Pressure on the Viability of Pancreatic Islets. In Proceedings of the 2019 American Transplant Congress, Hoboken, NJ, USA, 3 June 2019. [Google Scholar]
- Guzowski, J.; Korczyk, P.M.; Jakiela, S.; Garstecki, P. Automated High-Throughput Generation of Droplets. Lab Chip 2011, 11, 3593–3595. [Google Scholar] [CrossRef] [PubMed]
- Guzowski, J.; Garstecki, P. Droplet Clusters: Exploring the Phase Space of Soft Mesoscale Atoms. Phys. Rev. Lett. 2015, 114. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Authors | Growth Factors and Small Molecules Used in the Study | Outcome |
|---|---|---|
| Zhang et al., 2009 [140] | Act A, Wort, RA, Noggin, FGF7, EGF, Nico, bFGF, Ex-4, BMP4 | Successful differentiation into mature insulin-positive cells |
| Rezania et al., 2014 [141] | GDF8, GSK3β inh, FGF7, Vit C, RA, SANT, TPB, LDN, SANT, Alk5 inh, T3, LDN, GS inh XX, N-cys, AXL inh | Approximately 50% of cells were insulin-positive |
| Pagliuca et al., 2014 [133] | Act A CHIR, KGF, RA, SANT1 LDN, PdbU, T3, XXI, Alk5 inh, Heparin, Betacelluin, CMRL | Around 33% of cells were C-peptide-positive |
| Russ et al., 2015 [134] | WNT3a, Act A, TGFb inh, KGF, RA, Cyclopamine, Noggin, EGF, TBP, Alk5 inh | Simplified protocol, around 23% C-peptide-positive cells |
| Nair et al., 2019 [142] | Wnt3a, Act A, TGB inh, KGF, RA, EGF, ALK5 inh, XX inh, LDN, Vit C | Clustering of immature β-like cells as a critical step in gaining full functionality |
| Li et al., 2020 [143] | Act A, Chir99021, FgF-β, Vit C, KGF, Sant1, RA, Noggin, EGF, RepSox, GC1, LDN, CoE, Y-27632, R428, Trolox, N-cys | Generation of pancreatic like islets that contained approximately 30–40% of β-like cells |
| Yoshihara et al., 2020 [144] | Act A, GSK3β inh, FGF7, Vit C, RA, TGFβ inh, BMPR inh, Hedgehog inh, PKC, T3, Alk5 inh, Notch inh, Vit C, Vit E, cAMP, WNT4 | 50–60% of human islets-like organoids cells expressed insulin and β-cells markers |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wszoła, M.; Nitarska, D.; Cywoniuk, P.; Gomółka, M.; Klak, M. Stem Cells as a Source of Pancreatic Cells for Production of 3D Bioprinted Bionic Pancreas in the Treatment of Type 1 Diabetes. Cells 2021, 10, 1544. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10061544
Wszoła M, Nitarska D, Cywoniuk P, Gomółka M, Klak M. Stem Cells as a Source of Pancreatic Cells for Production of 3D Bioprinted Bionic Pancreas in the Treatment of Type 1 Diabetes. Cells. 2021; 10(6):1544. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10061544
Chicago/Turabian StyleWszoła, Michał, Daria Nitarska, Piotr Cywoniuk, Magdalena Gomółka, and Marta Klak. 2021. "Stem Cells as a Source of Pancreatic Cells for Production of 3D Bioprinted Bionic Pancreas in the Treatment of Type 1 Diabetes" Cells 10, no. 6: 1544. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10061544