
Potential Therapies Targeting Metabolic Pathways in Cancer Stem Cells

 , , and
, , and 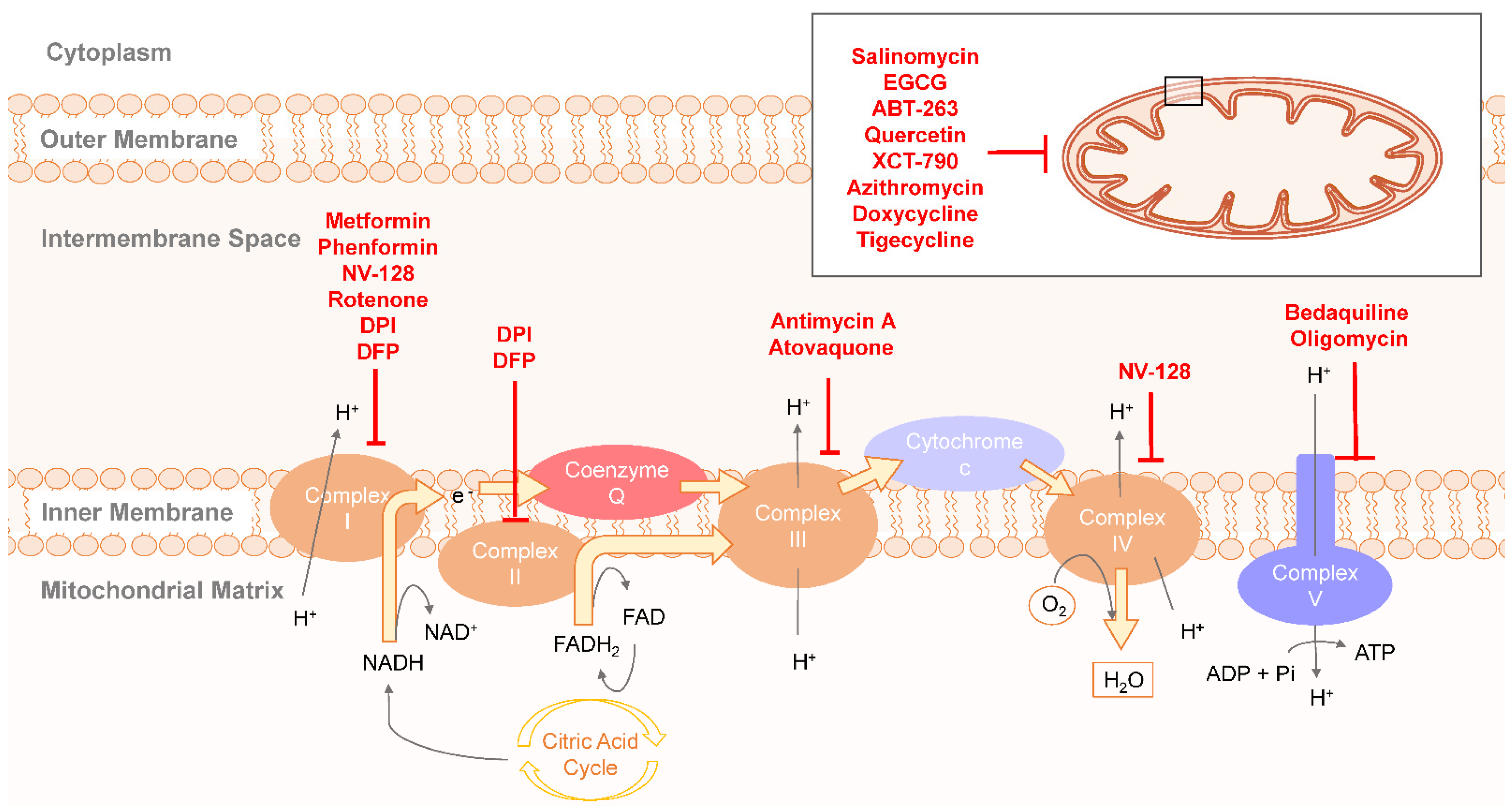{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mitochondrial Respiration and Oxidative Phosphorylation in CSCs
2.1. CSCs Mainly Use OXPHOS for Energy Production
2.2. Regulation of Mitochondrial Biogenesis in CSCs
2.3. Therapeutic Targeting OXPHOS in CSCs Metabolism
3. Glycolytic Switch in CSCs
3.1. Glycolysis in CSCs
3.2. Therapy Targeting Glycolysis in CSCs
4. Glutamine Metabolism in CSCs
4.1. Glutaminolysis in CSCs
4.2. Therapy Targeting Glutaminolysis in CSCs
5. Fatty Acid Metabolism in CSCs
5.1. The Importance of Fatty Acid Metabolism in CSCs
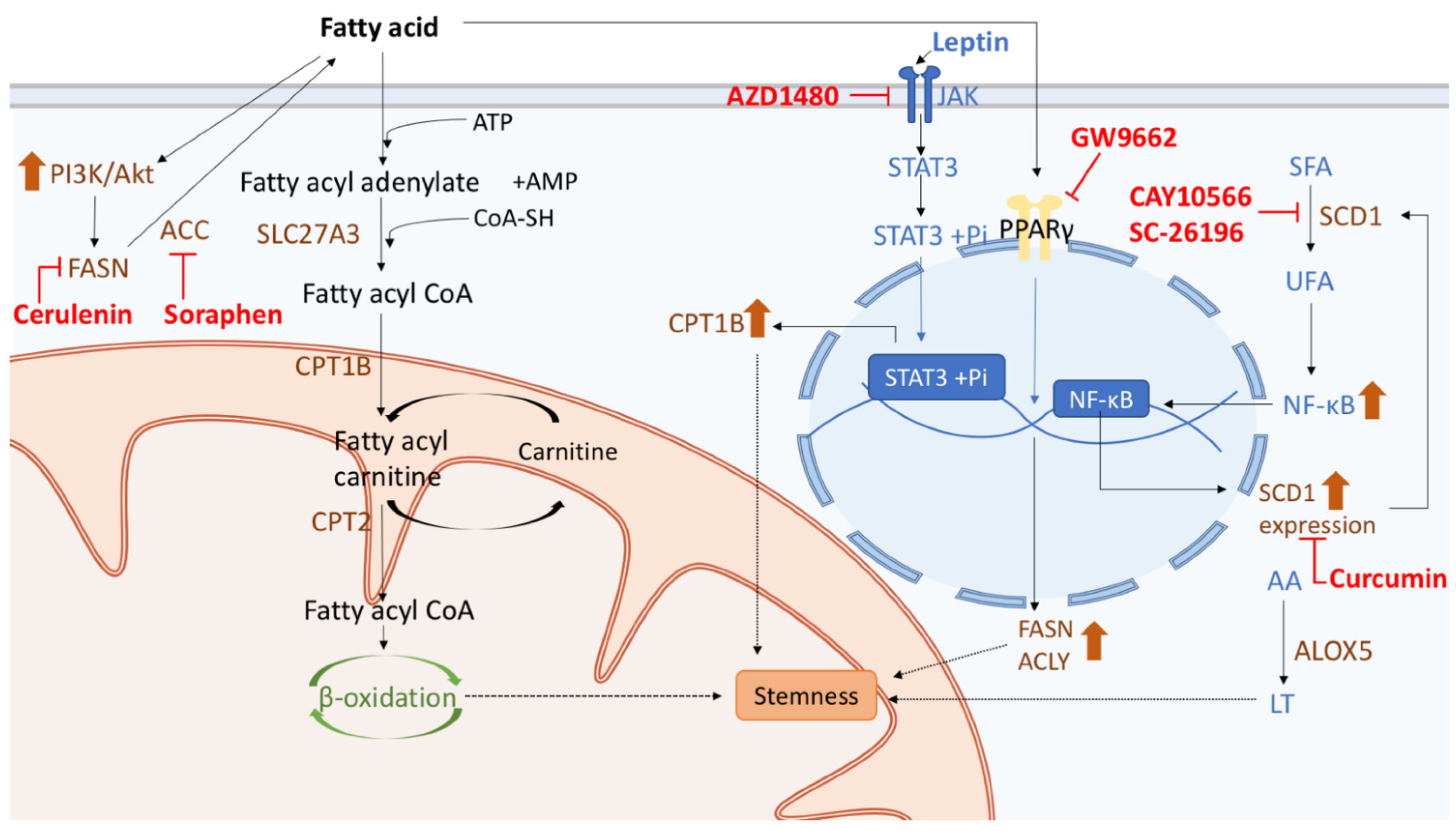
5.2. Therapy Targeting Fatty Acid Metabolism in CSCs
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aguilar-Gallardo, C.; Simon, C. Cells, stem cells, and cancer stem cells. Semin. Reprod. Med. 2013, 31, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef]
- Phillips, T.M.; McBride, W.H.; Pajonk, F. The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. J. Natl. Cancer Inst. 2006, 98, 1777–1785. [Google Scholar] [CrossRef] [Green Version]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Ghisolfi, L.; Keates, A.C.; Zhang, J.; Xiang, S.; Lee, D.K.; Li, C.J. Induction of cancer cell stemness by chemotherapy. Cell Cycle 2012, 11, 2691–2698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touil, Y.; Igoudjil, W.; Corvaisier, M.; Dessein, A.F.; Vandomme, J.; Monte, D.; Stechly, L.; Skrypek, N.; Langlois, C.; Grard, G.; et al. Colon cancer cells escape 5FU chemotherapy-induced cell death by entering stemness and quiescence associated with the c-Yes/YAP axis. Clin. Cancer Res. 2014, 20, 837–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Rogoff, H.A.; Keates, S.; Gao, Y.; Murikipudi, S.; Mikule, K.; Leggett, D.; Li, W.; Pardee, A.B.; Li, C.J. Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc. Natl. Acad. Sci. USA 2015, 112, 1839–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobo, N.A.; Shimono, Y.; Qian, D.; Clarke, M.F. The biology of cancer stem cells. Annu. Rev. Cell Dev. Biol. 2007, 23, 675–699. [Google Scholar] [CrossRef] [Green Version]
- Hadnagy, A.; Gaboury, L.; Beaulieu, R.; Balicki, D. SP analysis may be used to identify cancer stem cell populations. Exp. Cell Res. 2006, 312, 3701–3710. [Google Scholar] [CrossRef] [PubMed]
- Dawood, S.; Austin, L.; Cristofanilli, M. Cancer stem cells: Implications for cancer therapy. Oncology 2014, 28, 1101–1107, 1110. [Google Scholar] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Dou, J.; Gu, N. Emerging strategies for the identification and targeting of cancer stem cells. Tumour Biol. 2010, 31, 243–253. [Google Scholar] [CrossRef]
- Li, W.; Ma, H.; Zhang, J.; Zhu, L.; Wang, C.; Yang, Y. Unraveling the roles of CD44/CD24 and ALDH1 as cancer stem cell markers in tumorigenesis and metastasis. Sci. Rep. 2017, 7, 13856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, M.; Brooks, M.; Wicha, M.S. Epithelial-mesenchymal plasticity of breast cancer stem cells: Implications for metastasis and therapeutic resistance. Curr. Pharm. Des. 2015, 21, 1301–1310. [Google Scholar] [CrossRef] [Green Version]
- Bahmad, H.F.; Cheaito, K.; Chalhoub, R.M.; Hadadeh, O.; Monzer, A.; Ballout, F.; El-Hajj, A.; Mukherji, D.; Liu, Y.N.; Daoud, G.; et al. Sphere-formation assay: Three-dimensional in vitro culturing of prostate cancer stem/progenitor sphere-forming Cells. Front. Oncol. 2018, 8, 347. [Google Scholar] [CrossRef] [Green Version]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells—Perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.A.; Pan, S.C.; Chu, I.; Lai, R.Y.; Wei, Y.H. Targeting cancer stem cells from a metabolic perspective. Exp. Biol. Med. 2020, 245, 465–476. [Google Scholar] [CrossRef] [Green Version]
- El-Sahli, S.; Wang, L. Cancer stem cell-associated pathways in the metabolic reprogramming of breast cancer. Int. J. Mol. Sci. 2020, 21, 9125. [Google Scholar] [CrossRef]
- Ahmed, N.; Escalona, R.; Leung, D.; Chan, E.; Kannourakis, G. Tumour microenvironment and metabolic plasticity in cancer and cancer stem cells: Perspectives on metabolic and immune regulatory signatures in chemoresistant ovarian cancer stem cells. Semin. Cancer Biol. 2018, 53, 265–281. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Sancho, P.; Barneda, D.; Heeschen, C. Hallmarks of cancer stem cell metabolism. Br. J. Cancer 2016, 114, 1305–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantor, J.R.; Sabatini, D.M. Cancer cell metabolism: One hallmark, many faces. Cancer Discov. 2012, 2, 881–898. [Google Scholar] [CrossRef] [Green Version]
- Peixoto, J.; Lima, J. Metabolic traits of cancer stem cells. Dis. Model. Mech 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenda Suster, N.; Virant-Klun, I. Presence and role of stem cells in ovarian cancer. World J. Stem Cells 2019, 11, 383–397. [Google Scholar] [CrossRef]
- Pasto, A.; Bellio, C.; Pilotto, G.; Ciminale, V.; Silic-Benussi, M.; Guzzo, G.; Rasola, A.; Frasson, C.; Nardo, G.; Zulato, E.; et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget 2014, 5, 4305–4319. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Kawana, K.; Adachi, K.; Fujimoto, A.; Yoshida, M.; Nakamura, H.; Nishida, H.; Inoue, T.; Taguchi, A.; Takahashi, J.; et al. Spheroid cancer stem cells display reprogrammed metabolism and obtain energy by actively running the tricarboxylic acid (TCA) cycle. Oncotarget 2016, 7, 33297–33305. [Google Scholar] [CrossRef] [Green Version]
- Courtois, S.; de Luxan-Delgado, B.; Penin-Peyta, L.; Royo-Garcia, A.; Parejo-Alonso, B.; Jagust, P.; Alcala, S.; Rubiolo, J.A.; Sanchez, L.; Sainz, B., Jr.; et al. Inhibition of mitochondrial dynamics preferentially targets pancreatic cancer cells with enhanced tumorigenic and invasive potential. Cancers 2021, 13, 698. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.; Swargiary, G.; Singh, K.K. Natural agents targeting mitochondria in cancer. Int. J. Mol. Sci. 2020, 21, 6992. [Google Scholar] [CrossRef] [PubMed]
- Walsh, H.R.; Cruickshank, B.M.; Brown, J.M.; Marcato, P. The flick of a switch: Conferring survival advantage to breast cancer stem cells through metabolic plasticity. Front. Oncol. 2019, 9, 753. [Google Scholar] [CrossRef]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Janiszewska, M.; Suva, M.L.; Riggi, N.; Houtkooper, R.H.; Auwerx, J.; Clement-Schatlo, V.; Radovanovic, I.; Rheinbay, E.; Provero, P.; Stamenkovic, I. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012, 26, 1926–1944. [Google Scholar] [CrossRef] [Green Version]
- Snyder, V.; Reed-Newman, T.C.; Arnold, L.; Thomas, S.M.; Anant, S. Cancer stem cell metabolism and potential therapeutic targets. Front. Oncol. 2018, 8, 203. [Google Scholar] [CrossRef]
- Valle, S.; Alcala, S.; Martin-Hijano, L.; Cabezas-Sainz, P.; Navarro, D.; Munoz, E.R.; Yuste, L.; Tiwary, K.; Walter, K.; Ruiz-Canas, L.; et al. Exploiting oxidative phosphorylation to promote the stem and immunoevasive properties of pancreatic cancer stem cells. Nat. Commun. 2020, 11, 5265. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, Y.; Shingu, T.; Feng, L.; Chen, Z.; Ogasawara, M.; Keating, M.J.; Kondo, S.; Huang, P. Metabolic alterations in highly tumorigenic glioblastoma cells: Preference for hypoxia and high dependency on glycolysis. J. Biol. Chem. 2011, 286, 32843–32853. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Shen, Y.; Jin, F.; Miao, Y.; Qiu, X. Cancer stem cells in small cell Lung cancer cell line H446: Higher dependency on oxidative phosphorylation and mitochondrial substrate-level phosphorylation than non-stem cancer cells. PLoS ONE 2016, 11, e0154576. [Google Scholar] [CrossRef] [Green Version]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Grana, O.; et al. MYC/PGC-1alpha balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.Q.; Li, Q.; Wang, G.H.; Sun, F.F.; Huang, G.J.; Bian, X.W.; Yu, S.C.; Qian, G.S. Mitochondrial and energy metabolism-related properties as novel indicators of lung cancer stem cells. Int. J. Cancer 2011, 129, 820–831. [Google Scholar] [CrossRef]
- Ciavardelli, D.; Rossi, C.; Barcaroli, D.; Volpe, S.; Consalvo, A.; Zucchelli, M.; De Cola, A.; Scavo, E.; Carollo, R.; D’Agostino, D.; et al. Breast cancer stem cells rely on fermentative glycolysis and are sensitive to 2-deoxyglucose treatment. Cell Death Dis. 2014, 5, e1336. [Google Scholar] [CrossRef] [Green Version]
- Palorini, R.; Votta, G.; Balestrieri, C.; Monestiroli, A.; Olivieri, S.; Vento, R.; Chiaradonna, F. Energy metabolism characterization of a novel cancer stem cell-like line 3AB-OS. J. Cell. Biochem. 2014, 115, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.F.; Chao, K.S.; Chen, Y.J.; Chang, M.S. Complementary and alternative medicine in cancer stem cells. Evid. Based Complement. Altern. Med. 2013, 2013, 560274. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Fiorillo, M.; Peiris-Pages, M.; Ozsvari, B.; Smith, D.L.; Sanchez-Alvarez, R.; Martinez-Outschoorn, U.E.; Cappello, A.R.; Pezzi, V.; Lisanti, M.P.; et al. Mitochondrial biogenesis is required for the anchorage-independent survival and propagation of stem-like cancer cells. Oncotarget 2015, 6, 14777–14795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, R.; Bonuccelli, G.; Ozsvari, B.; Peiris-Pages, M.; Fiorillo, M.; Smith, D.L.; Bevilacqua, G.; Mazzanti, C.M.; McDonnell, L.A.; Naccarato, A.G.; et al. Mitochondrial mass, a new metabolic biomarker for stem-like cancer cells: Understanding WNT/FGF-driven anabolic signaling. Oncotarget 2015, 6, 30453–30471. [Google Scholar] [CrossRef] [Green Version]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Reue, K.; Frohnen, P.; Chan, M.; Alhiyari, Y.; Dratver, M.B.; Pajonk, F. Metabolic differences in breast cancer stem cells and differentiated progeny. Breast Cancer Res. Treat. 2014, 146, 525–534. [Google Scholar] [CrossRef] [Green Version]
- Lamb, R.; Harrison, H.; Hulit, J.; Smith, D.L.; Lisanti, M.P.; Sotgia, F. Mitochondria as new therapeutic targets for eradicating cancer stem cells: Quantitative proteomics and functional validation via MCT1/2 inhibition. Oncotarget 2014, 5, 11029–11037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farnie, G.; Sotgia, F.; Lisanti, M.P. High mitochondrial mass identifies a sub-population of stem-like cancer cells that are chemo-resistant. Oncotarget 2015, 6, 30472–30486. [Google Scholar] [CrossRef] [Green Version]
- Vazquez, F.; Lim, J.H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.G.; Douglas-Jones, A.; Mansel, R.E. Expression of peroxisome-proliferator activated receptor-gamma (PPARgamma) and the PPARgamma co-activator, PGC-1, in human breast cancer correlates with clinical outcomes. Int. J. Cancer 2003, 106, 752–757. [Google Scholar] [CrossRef]
- Mayer, M.J.; Klotz, L.H.; Venkateswaran, V. Metformin and prostate cancer stem cells: A novel therapeutic target. Prostate Cancer Prostatic Dis. 2015, 18, 303–309. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.W.; Park, S.B.; Lee, S.J.; Seo, M.S.; Trosko, J.E.; Kang, K.S. Metformin represses self-renewal of the human breast carcinoma stem cells via inhibition of estrogen receptor-mediated OCT4 expression. PLoS ONE 2011, 6, e28068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009, 69, 7507–7511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syahrani, R.A.; Yunita, E.; Wanandi, S.I. Suppression of rotenone-treated human breast cancer stem cell survival using surviving inhibitor YM155 is associated to oxidative stress modulation. Asian Pac. J. Cancer Prev. 2020, 21, 2631–2637. [Google Scholar] [CrossRef] [PubMed]
- Heinz, S.; Freyberger, A.; Lawrenz, B.; Schladt, L.; Schmuck, G.; Ellinger-Ziegelbauer, H. Mechanistic investigations of the mitochondrial complex I inhibitor rotenone in the context of pharmacological and safety Evaluation. Sci. Rep. 2017, 7, 45465. [Google Scholar] [CrossRef] [Green Version]
- Alvero, A.B.; Montagna, M.K.; Holmberg, J.C.; Craveiro, V.; Brown, D.; Mor, G. Targeting the mitochondria activates two independent cell death pathways in ovarian cancer stem cells. Mol. Cancer Ther. 2011, 10, 1385–1393. [Google Scholar] [CrossRef] [Green Version]
- Song, I.S.; Jeong, J.Y.; Jeong, S.H.; Kim, H.K.; Ko, K.S.; Rhee, B.D.; Kim, N.; Han, J. Mitochondria as therapeutic targets for cancer stem cells. World J. Stem Cells 2015, 7, 418–427. [Google Scholar] [CrossRef]
- Ozsvari, B.; Bonuccelli, G.; Sanchez-Alvarez, R.; Foster, R.; Sotgia, F.; Lisanti, M.P. Targeting flavin-containing enzymes eliminates cancer stem cells (CSCs), by inhibiting mitochondrial respiration: Vitamin B2 (Riboflavin) in cancer therapy. Aging (Albany NY) 2017, 9, 2610–2628. [Google Scholar] [CrossRef] [Green Version]
- Fiorillo, M.; Toth, F.; Brindisi, M.; Sotgia, F.; Lisanti, M.P. Deferiprone (DFP) targets cancer stem cell (CSC) propagation by inhibiting mitochondrial metabolism and inducing ROS production. Cells 2020, 9, 1529. [Google Scholar] [CrossRef]
- Fiorillo, M.; Lamb, R.; Tanowitz, H.B.; Mutti, L.; Krstic-Demonacos, M.; Cappello, A.R.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Repurposing atovaquone: Targeting mitochondrial complex III and OXPHOS to eradicate cancer stem cells. Oncotarget 2016, 7, 34084–34099. [Google Scholar] [CrossRef] [Green Version]
- Yeh, C.T.; Su, C.L.; Huang, C.Y.; Lin, J.K.; Lee, W.H.; Chang, P.M.; Kuo, Y.L.; Liu, Y.W.; Wang, L.S.; Wu, C.H.; et al. A preclinical evaluation of antimycin a as a potential antilung cancer stem cell agent. Evid. Based Complement. Altern. Med. 2013, 2013, 910451. [Google Scholar] [CrossRef] [Green Version]
- Fiorillo, M.; Lamb, R.; Tanowitz, H.B.; Cappello, A.R.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Bedaquiline, an FDA-approved antibiotic, inhibits mitochondrial function and potently blocks the proliferative expansion of stem-like cancer cells (CSCs). Aging (Albany NY) 2016, 8, 1593–1607. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, C.R.; Tilkens, S.B.; Guan, H.; Garner, J.A.; Or, P.M.; Chan, A.M. Differential sensitivities of glioblastoma cell lines towards metabolic and signaling pathway inhibitions. Cancer Lett. 2013, 336, 299–306. [Google Scholar] [CrossRef]
- Okamoto, Y.; Musha, T.; Harumi, K. Estimation of the omitted-lead potentials with the temporal and spatial correlations. Iyodenshi Seitai Kogaku 1988, 26, 146–151. [Google Scholar]
- Jiang, J.; Li, H.; Qaed, E.; Zhang, J.; Song, Y.; Wu, R.; Bu, X.; Wang, Q.; Tang, Z. Salinomycin, as an autophagy modulator—A new avenue to anticancer: A review. J. Exp. Clin. Cancer Res. 2018, 37, 26. [Google Scholar] [CrossRef] [Green Version]
- Klose, J.; Trefz, S.; Wagner, T.; Steffen, L.; Preissendorfer Charrier, A.; Radhakrishnan, P.; Volz, C.; Schmidt, T.; Ulrich, A.; Dieter, S.M.; et al. Salinomycin: Anti-tumor activity in a pre-clinical colorectal cancer model. PLoS ONE 2019, 14, e0211916. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Wang, X.Q.; Zhang, Q.; Zhu, J.Y.; Li, Y.; Xie, C.F.; Li, X.T.; Wu, J.S.; Geng, S.S.; Zhong, C.Y.; et al. (-)-Epigallocatechin-3-Gallate inhibits colorectal cancer stem cells by suppressing Wnt/beta-Catenin pathway. Nutrients 2017, 9, 572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Jiang, Y.; Yang, X.; Wang, S.; Xie, C.; Li, X.; Li, Y.; Chen, Y.; Wang, X.; Meng, Y.; et al. Wnt/beta-catenin pathway mediates (-)-Epigallocatechin-3-gallate (EGCG) inhibition of lung cancer stem cells. Biochem. Biophys. Res. Commun. 2017, 482, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Si, Y.; Wang, Z.; Wang, J.; Guo, Y.; Zhang, X. Quercetin inhibits the growth of human gastric cancer stem cells by inducing mitochondrial-dependent apoptosis through the inhibition of PI3K/Akt signaling. Int. J. Mol. Med. 2016, 38, 619–626. [Google Scholar] [CrossRef]
- Bonuccelli, G.; Peiris-Pages, M.; Ozsvari, B.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Targeting cancer stem cell propagation with palbociclib, a CDK4/6 inhibitor: Telomerase drives tumor cell heterogeneity. Oncotarget 2017, 8, 9868–9884. [Google Scholar] [CrossRef] [Green Version]
- Gargini, R.; Garcia-Escudero, V.; Izquierdo, M. Therapy mediated by mitophagy abrogates tumor progression. Autophagy 2011, 7, 466–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, R.; Ozsvari, B.; Lisanti, C.L.; Tanowitz, H.B.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: Treating cancer like an infectious disease. Oncotarget 2015, 6, 4569–4584. [Google Scholar] [CrossRef] [Green Version]
- Skoda, J.; Borankova, K.; Jansson, P.J.; Huang, M.L.; Veselska, R.; Richardson, D.R. Pharmacological targeting of mitochondria in cancer stem cells: An ancient organelle at the crossroad of novel anti-cancer therapies. Pharmacol. Res. 2019, 139, 298–313. [Google Scholar] [CrossRef]
- Zhao, Y.; Dong, Q.; Li, J.; Zhang, K.; Qin, J.; Zhao, J.; Sun, Q.; Wang, Z.; Wartmann, T.; Jauch, K.W.; et al. Targeting cancer stem cells and their niche: Perspectives for future therapeutic targets and strategies. Semin. Cancer Biol. 2018, 53, 139–155. [Google Scholar] [CrossRef]
- Cole, A.; Wang, Z.; Coyaud, E.; Voisin, V.; Gronda, M.; Jitkova, Y.; Mattson, R.; Hurren, R.; Babovic, S.; Maclean, N.; et al. Inhibition of the mitochondrial protease ClpP as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 2015, 27, 864–876. [Google Scholar] [CrossRef] [Green Version]
- Emmink, B.L.; Verheem, A.; Van Houdt, W.J.; Steller, E.J.; Govaert, K.M.; Pham, T.V.; Piersma, S.R.; Borel Rinkes, I.H.; Jimenez, C.R.; Kranenburg, O. The secretome of colon cancer stem cells contains drug-metabolizing enzymes. J. Proteom. 2013, 91, 84–96. [Google Scholar] [CrossRef]
- Liao, J.; Qian, F.; Tchabo, N.; Mhawech-Fauceglia, P.; Beck, A.; Qian, Z.; Wang, X.; Huss, W.J.; Lele, S.B.; Morrison, C.D.; et al. Ovarian cancer spheroid cells with stem cell-like properties contribute to tumor generation, metastasis and chemotherapy resistance through hypoxia-resistant metabolism. PLoS ONE 2014, 9, e84941. [Google Scholar] [CrossRef] [Green Version]
- Folmes, C.D.; Nelson, T.J.; Martinez-Fernandez, A.; Arrell, D.K.; Lindor, J.Z.; Dzeja, P.P.; Ikeda, Y.; Perez-Terzic, C.; Terzic, A. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab. 2011, 14, 264–271. [Google Scholar] [CrossRef] [Green Version]
- Panopoulos, A.D.; Yanes, O.; Ruiz, S.; Kida, Y.S.; Diep, D.; Tautenhahn, R.; Herrerias, A.; Batchelder, E.M.; Plongthongkum, N.; Lutz, M.; et al. The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res. 2012, 22, 168–177. [Google Scholar] [CrossRef] [Green Version]
- Mori, Y.; Yamawaki, K.; Ishiguro, T.; Yoshihara, K.; Ueda, H.; Sato, A.; Ohata, H.; Yoshida, Y.; Minamino, T.; Okamoto, K.; et al. ALDH-dependent glycolytic activation mediates stemness and paclitaxel resistance in patient-derived spheroid models of uterine endometrial cancer. Stem Cell Rep. 2019, 13, 730–746. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Yuan, T.; Wu, Y.; Wang, Y.; Fan, T.W.; Miriyala, S.; Lin, Y.; Yao, J.; Shi, J.; Kang, T.; et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 2013, 23, 316–331. [Google Scholar] [CrossRef] [Green Version]
- Schieber, M.S.; Chandel, N.S. ROS links glucose metabolism to breast cancer stem cell and EMT phenotype. Cancer Cell 2013, 23, 265–267. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Fu, Z.; Zhou, S.; Gong, J.; Liu, C.A.; Qiao, Z.; Li, S. HIF-1alpha and HIF-2alpha: Siblings in promoting angiogenesis of residual hepatocellular carcinoma after high-intensity focused ultrasound ablation. PLoS ONE 2014, 9, e88913. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Wu, Q.; Hitomi, M.; Rahim, N.; Kim, Y.; Sloan, A.E.; Weil, R.J.; Nakano, I.; Sarkaria, J.N.; Stringer, B.W.; et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat. Neurosci. 2013, 16, 1373–1382. [Google Scholar] [CrossRef]
- Goidts, V.; Bageritz, J.; Puccio, L.; Nakata, S.; Zapatka, M.; Barbus, S.; Toedt, G.; Campos, B.; Korshunov, A.; Momma, S.; et al. RNAi screening in glioma stem-like cells identifies PFKFB4 as a key molecule important for cancer cell survival. Oncogene 2012, 31, 3235–3243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, P.; Joshi, K.; Li, J.; Kim, S.H.; Li, P.; Santana-Santos, L.; Luthra, S.; Chandran, U.R.; Benos, P.V.; Smith, L.; et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc. Natl. Acad. Sci. USA 2013, 110, 8644–8649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.A.; Wang, C.Y.; Hsieh, Y.T.; Chen, Y.J.; Wei, Y.H. Metabolic reprogramming orchestrates cancer stem cell properties in nasopharyngeal carcinoma. Cell Cycle 2015, 14, 86–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuen, C.A.; Asuthkar, S.; Guda, M.R.; Tsung, A.J.; Velpula, K.K. Cancer stem cell molecular reprogramming of the Warburg effect in glioblastomas: A new target gleaned from an old concept. CNS Oncol. 2016, 5, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Kocabas, F.; Zheng, J.; Thet, S.; Copeland, N.G.; Jenkins, N.A.; DeBerardinis, R.J.; Zhang, C.; Sadek, H.A. Meis1 regulates the metabolic phenotype and oxidant defense of hematopoietic stem cells. Blood 2012, 120, 4963–4972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordan, J.D.; Thompson, C.B.; Simon, M.C. HIF and c-Myc: Sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell 2007, 12, 108–113. [Google Scholar] [CrossRef] [Green Version]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med. 2014, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.L.; Wang, P.; Lu, M.Z.; Zhang, S.D.; Zheng, L. c-Myc maintains the self-renewal and chemoresistance properties of colon cancer stem cells. Oncol. Lett. 2019, 17, 4487–4493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Jang, H.; Kim, T.W.; Kang, B.H.; Lee, S.E.; Jeon, Y.K.; Chung, D.H.; Choi, J.; Shin, J.; Cho, E.J.; et al. Core pluripotency factors directly regulate metabolism in embryonic stem cell to maintain pluripotency. Stem Cells 2015, 33, 2699–2711. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.; Markowitz, S.; Zhou, S.; et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009, 325, 1555–1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blum, R.; Kloog, Y. Metabolism addiction in pancreatic cancer. Cell Death Dis. 2014, 5, e1065. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, M.; Tanaka, N. IL-8-induced O-GlcNAc modification via GLUT3 and GFAT regulates cancer stem cell-like properties in colon and lung cancer cells. Oncogene 2019, 38, 1520–1533. [Google Scholar] [CrossRef]
- Vander Heiden, M.G. Targeting cancer metabolism: A therapeutic window opens. Nat. Rev. Drug Discov. 2011, 10, 671–684. [Google Scholar] [CrossRef] [Green Version]
- Flaig, T.W.; Gustafson, D.L.; Su, L.J.; Zirrolli, J.A.; Crighton, F.; Harrison, G.S.; Pierson, A.S.; Agarwal, R.; Glode, L.M. A phase I and pharmacokinetic study of silybin-phytosome in prostate cancer patients. Invest. New Drugs 2007, 25, 139–146. [Google Scholar] [CrossRef]
- Gadducci, A.; Brunetti, I.; Muttini, M.P.; Fanucchi, A.; Dargenio, F.; Giannessi, P.G.; Conte, P.F. Epidoxorubicin and lonidamine in refractory or recurrent epithelial ovarian cancer. Eur. J. Cancer 1994, 30A, 1432–1435. [Google Scholar] [CrossRef]
- De Lena, M.; Lorusso, V.; Bottalico, C.; Brandi, M.; De Mitrio, A.; Catino, A.; Guida, M.; Latorre, A.; Leone, B.; Vallejo, C.; et al. Revertant and potentiating activity of lonidamine in patients with ovarian cancer previously treated with platinum. J. Clin. Oncol. 1997, 15, 3208–3213. [Google Scholar] [CrossRef]
- De Marinis, F.; Rinaldi, M.; Ardizzoni, A.; Bruzzi, P.; Pennucci, M.C.; Portalone, L.; D’Aprile, M.; Ripanti, P.; Romano, F.; Belli, M.; et al. The role of vindesine and lonidamine in the treatment of elderly patients with advanced non-small cell lung cancer: A phase III randomized FONICAP trial. Italian Lung Cancer Task Force. Tumori 1999, 85, 177–182. [Google Scholar] [CrossRef]
- Berruti, A.; Bitossi, R.; Gorzegno, G.; Bottini, A.; Alquati, P.; De Matteis, A.; Nuzzo, F.; Giardina, G.; Danese, S.; De Lena, M.; et al. Time to progression in metastatic breast cancer patients treated with epirubicin is not improved by the addition of either cisplatin or lonidamine: Final results of a phase III study with a factorial design. J. Clin. Oncol. 2002, 20, 4150–4159. [Google Scholar] [CrossRef] [PubMed]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [Green Version]
- Mazur, P.K.; Herner, A.; Mello, S.S.; Wirth, M.; Hausmann, S.; Sanchez-Rivera, F.J.; Lofgren, S.M.; Kuschma, T.; Hahn, S.A.; Vangala, D.; et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat. Med. 2015, 21, 1163–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, P.L.; Miller, A.L.; Kreitzburg, K.M.; Council, L.N.; Gamblin, T.L.; Christein, J.D.; Heslin, M.J.; Arnoletti, J.P.; Richardson, J.H.; Chen, D.; et al. The BET bromodomain inhibitor JQ1 suppresses growth of pancreatic ductal adenocarcinoma in patient-derived xenograft models. Oncogene 2016, 35, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef]
- Le Grand, M.; Mukha, A.; Püschel, J.; Valli, E.; Kamili, A.; Vittorio, O.; Dubrovska, A.; Kavallaris, M. Interplay between MycN and c-Myc regulates radioresistance and cancer stem cell phenotype in neuroblastoma upon glutamine deprivation. Theranostics 2020, 10, 6411–6429. [Google Scholar] [CrossRef]
- Li, D.; Fu, Z.; Chen, R.; Zhao, X.; Zhou, Y.; Zeng, B.; Yu, M.; Zhou, Q.; Lin, Q.; Gao, W.; et al. Inhibition of glutamine metabolism counteracts pancreatic cancer stem cell features and sensitizes cells to radiotherapy. Oncotarget 2015, 6, 31151–31163. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A hallmark of cancer metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 163–194. [Google Scholar] [CrossRef] [PubMed]
- El Hout, M.; Cosialls, E.; Mehrpour, M.; Hamaï, A. Crosstalk between autophagy and metabolic regulation of cancer stem cells. Mol. Cancer 2020, 19, 27. [Google Scholar] [CrossRef]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, G.J. Emerging roles of Myc in stem cell biology and novel tumor therapies. J. Exp. Clin. Cancer Res. 2018, 37, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Lee, K.J.; Seo, Y.; Kwon, J.H.; Yoon, J.P.; Kang, J.Y.; Lee, H.J.; Park, S.J.; Hong, S.P.; Cheon, J.H.; et al. Publisher Correction: Effects of metformin on colorectal cancer stem cells depend on alterations in glutamine metabolism. Sci. Rep. 2018, 8, 13111. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Choi, J.H.; Nam, J.S. Targeting cancer stem cells in triple-negative breast cancer. Cancers 2019, 11, 965. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Kawana, K.; Adachi, K.; Fujimoto, A.; Taguchi, A.; Fujikawa, T.; Yoshida, M.; Nakamura, H.; Nishida, H.; Inoue, T.; et al. Low uptake of fluorodeoxyglucose in positron emission tomography/computed tomography in ovarian clear cell carcinoma may reflect glutaminolysis of its cancer stem cell-like properties. Oncol. Rep. 2017, 37, 1883–1888. [Google Scholar] [CrossRef]
- Mukha, A.; Kahya, U.; Linge, A.; Chen, O.; Löck, S.; Lukiyanchuk, V.; Richter, S.; Alves, T.C.; Peitzsch, M.; Telychko, V.; et al. GLS-driven glutamine catabolism contributes to prostate cancer radiosensitivity by regulating the redox state, stemness and ATG5-mediated autophagy. bioRxiv 2021. [Google Scholar] [CrossRef]
- Hayes, J.D.; McMahon, M. NRF2 and KEAP1 mutations: Permanent activation of an adaptive response in cancer. Trends Biochem. Sci. 2009, 34, 176–188. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Wang, H.; Sun, Q.; Ji, X.; Zhu, L.; Cong, Z.; Zhou, Y.; Liu, H.; Zhou, M. Nrf2 is required to maintain the self-renewal of glioma stem cells. BMC Cancer 2013, 13, 380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryoo, I.G.; Choi, B.H.; Kwak, M.K. Activation of NRF2 by p62 and proteasome reduction in sphere-forming breast carcinoma cells. Oncotarget 2015, 6, 8167–8184. [Google Scholar] [CrossRef] [Green Version]
- Ryoo, I.G.; Kim, G.; Choi, B.H.; Lee, S.H.; Kwak, M.K. Involvement of NRF2 signaling in doxorubicin resistance of cancer stem cell-enriched colonospheres. Biomol. Ther. (Seoul) 2016, 24, 482–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, K.; Hartmann, R.; Tsiampali, J.; Uhlmann, C.; Nickel, A.C.; He, X.; Kamp, M.A.; Sabel, M.; Barker, R.A.; Steiger, H.J.; et al. A comparative pharmaco-metabolomic study of glutaminase inhibitors in glioma stem-like cells confirms biological effectiveness but reveals differences in target-specificity. Cell Death Discov. 2020, 6, 20. [Google Scholar] [CrossRef] [Green Version]
- Elhammali, A.; Ippolito, J.E.; Collins, L.; Crowley, J.; Marasa, J.; Piwnica-Worms, D. A high-throughput fluorimetric assay for 2-hydroxyglutarate identifies Zaprinast as a glutaminase inhibitor. Cancer Discov. 2014, 4, 828–839. [Google Scholar] [CrossRef] [Green Version]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J. Clin. Investig. 2013, 123, 3678–3684. [Google Scholar] [CrossRef] [Green Version]
- Jariyal, H.; Gupta, C.; Andhale, S.; Gadge, S.; Srivastava, A. Comparative stemness and differentiation of luminal and basal breast cancer stem cell type under glutamine-deprivation. J. Cell Commun. Signal. 2021, 15, 207–222. [Google Scholar] [CrossRef]
- Shukla, K.; Ferraris, D.V.; Thomas, A.G.; Stathis, M.; Duvall, B.; Delahanty, G.; Alt, J.; Rais, R.; Rojas, C.; Gao, P.; et al. Design, synthesis, and pharmacological evaluation of bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide 3 (BPTES) analogs as glutaminase inhibitors. J. Med. Chem. 2012, 55, 10551–10563. [Google Scholar] [CrossRef] [Green Version]
- Ryoo, I.G.; Lee, S.H.; Kwak, M.K. Redox modulating NRF2: A potential mediator of cancer stem cell resistance. Oxid. Med. Cell. Longev. 2016, 2016, 2428153. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, W.C.; Liao, Y.W.; Chen, P.N.; Lu, K.H.; Yu, C.C.; Hsieh, P.L. Hinokitiol suppresses cancer stemness and oncogenicity in glioma stem cells by Nrf2 regulation. Cancer Chemother. Pharmacol. 2017, 80, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.; Oh, J.; Kim, J.S. Suppression of Nrf2 activity by chestnut leaf extract increases chemosensitivity of breast cancer stem cells to Paclitaxel. Nutrients 2017, 9, 760. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Li, J.; Chen, S.; Cai, J.; Ban, Y.; Peng, Q.; Zhou, Y.; Zeng, Z.; Peng, S.; Li, X.; et al. Emerging role of lipid metabolism alterations in Cancer stem cells. J. Exp. Clin. Cancer Res. 2018, 37, 118. [Google Scholar] [CrossRef]
- Hedvat, M.; Huszar, D.; Herrmann, A.; Gozgit, J.M.; Schroeder, A.; Sheehy, A.; Buettner, R.; Proia, D.; Kowolik, C.M.; Xin, H.; et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell 2009, 16, 487–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Fahrmann, J.F.; Lee, H.; Li, Y.J.; Tripathi, S.C.; Yue, C.; Zhang, C.; Lifshitz, V.; Song, J.; Yuan, Y.; et al. JAK/STAT3-regulated fatty acid beta-oxidation is critical for breast cancer stem cell self-renewal and chemoresistance. Cell Metab. 2018, 27, 136–150.e135. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.; Yoo, Y.J.; Kim, H.; Lee, H.; Chung, H.; Nam, M.H.; Moon, J.Y.; Lee, H.S.; Yoon, S.; Kim, W.Y. Clinical and biochemical relevance of monounsaturated fatty acid metabolism targeting strategy for cancer stem cell elimination in colon cancer. Biochem. Biophys. Res. Commun. 2019, 519, 100–105. [Google Scholar] [CrossRef]
- Jagust, P.; de Luxan-Delgado, B.; Parejo-Alonso, B.; Sancho, P. Metabolism-based therapeutic strategies targeting cancer stem cells. Front. Pharmacol. 2019, 10, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, A.; Kenny, H.A.; Lengyel, E. Unsaturated Fatty Acids Maintain Cancer Cell Stemness. Cell Stem Cell 2017, 20, 291–292. [Google Scholar] [CrossRef] [Green Version]
- Van de Sande, T.; De Schrijver, E.; Heyns, W.; Verhoeven, G.; Swinnen, J.V. Role of the phosphatidylinositol 3’-kinase/PTEN/Akt kinase pathway in the overexpression of fatty acid synthase in LNCaP prostate cancer cells. Cancer Res. 2002, 62, 642–646. [Google Scholar]
- Wang, X.; Sun, Y.; Wong, J.; Conklin, D.S. PPARgamma maintains ERBB2-positive breast cancer stem cells. Oncogene 2013, 32, 5512–5521. [Google Scholar] [CrossRef] [Green Version]
- Corominas-Faja, B.; Cuyas, E.; Gumuzio, J.; Bosch-Barrera, J.; Leis, O.; Martin, A.G.; Menendez, J.A. Chemical inhibition of acetyl-CoA carboxylase suppresses self-renewal growth of cancer stem cells. Oncotarget 2014, 5, 8306–8316. [Google Scholar] [CrossRef]
- Colacino, J.A.; McDermott, S.P.; Sartor, M.A.; Wicha, M.S.; Rozek, L.S. Transcriptomic profiling of curcumin-treated human breast stem cells identifies a role for stearoyl-coa desaturase in breast cancer prevention. Breast Cancer Res. Treat. 2016, 158, 29–41. [Google Scholar] [CrossRef]
- Li, J.; Condello, S.; Thomes-Pepin, J.; Ma, X.; Xia, Y.; Hurley, T.D.; Matei, D.; Cheng, J.X. Lipid desaturation is a metabolic marker and therapeutic target of ovarian cancer stem cells. Cell Stem Cell 2017, 20, 303–314 e305. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Wang, Y.; Shi, Z.; Liu, J.; Sun, P.; Hou, X.; Zhang, J.; Zhao, S.; Zhou, B.P.; Mi, J. Metabolic reprogramming of cancer-associated fibroblasts by IDH3alpha downregulation. Cell Rep. 2015, 10, 1335–1348. [Google Scholar] [CrossRef] [Green Version]
- Keith, B.; Simon, M.C. Hypoxia-inducible factors, stem cells, and cancer. Cell 2007, 129, 465–472. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Liu, V.W.; Chan, D.W.; Yao, K.M.; Ngan, H.Y. LY294002 and metformin cooperatively enhance the inhibition of growth and the induction of apoptosis of ovarian cancer cells. Int. J. Gynecol. Cancer 2012, 22, 15–22. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, Y.-A.; Chen, C.-C.; Chen, B.-J.; Wu, Y.-T.; Juan, J.-R.; Chen, L.-Y.; Teng, Y.-C.; Wei, Y.-H. Potential Therapies Targeting Metabolic Pathways in Cancer Stem Cells. Cells 2021, 10, 1772. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10071772
Shen Y-A, Chen C-C, Chen B-J, Wu Y-T, Juan J-R, Chen L-Y, Teng Y-C, Wei Y-H. Potential Therapies Targeting Metabolic Pathways in Cancer Stem Cells. Cells. 2021; 10(7):1772. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10071772
Chicago/Turabian StyleShen, Yao-An, Chang-Cyuan Chen, Bo-Jung Chen, Yu-Ting Wu, Jiun-Ru Juan, Liang-Yun Chen, Yueh-Chun Teng, and Yau-Huei Wei. 2021. "Potential Therapies Targeting Metabolic Pathways in Cancer Stem Cells" Cells 10, no. 7: 1772. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10071772