
Immune Cells Profiling in ANCA-Associated Vasculitis Patients—Relation to Disease Activity

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Flow Cytometry
2.2. ELISA Assay
2.3. Statistical Analysis
3. Results
3.1. Flow Cytometry—General Lymphocytes Phenotyping
3.2. T Cell Subpopulations

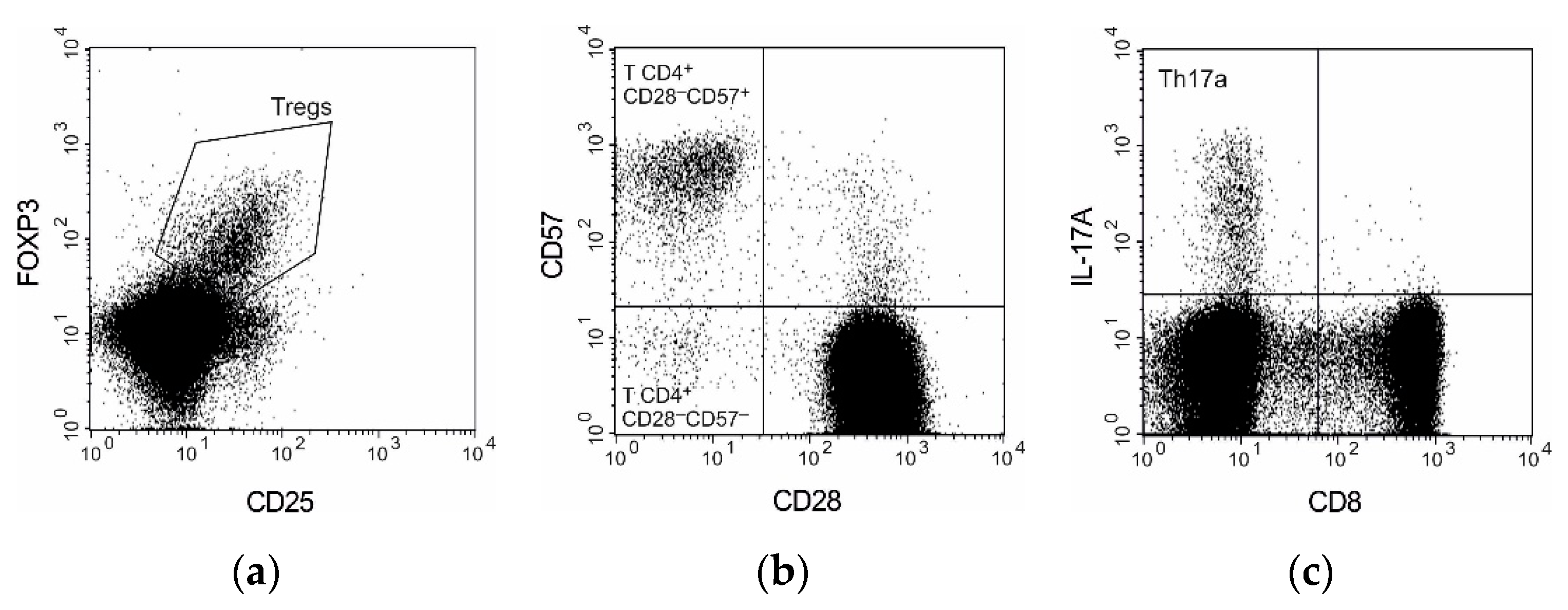{kind=link}
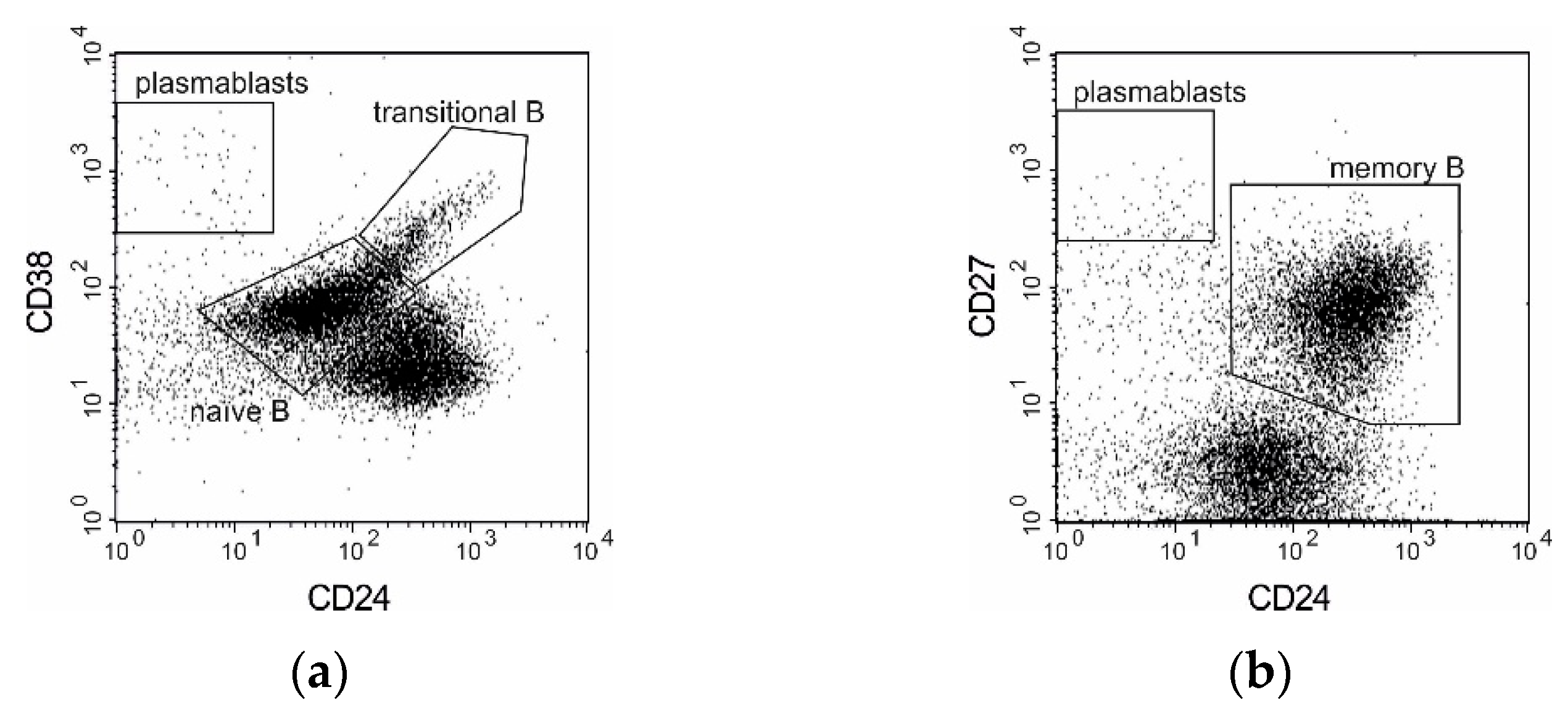{kind=link}
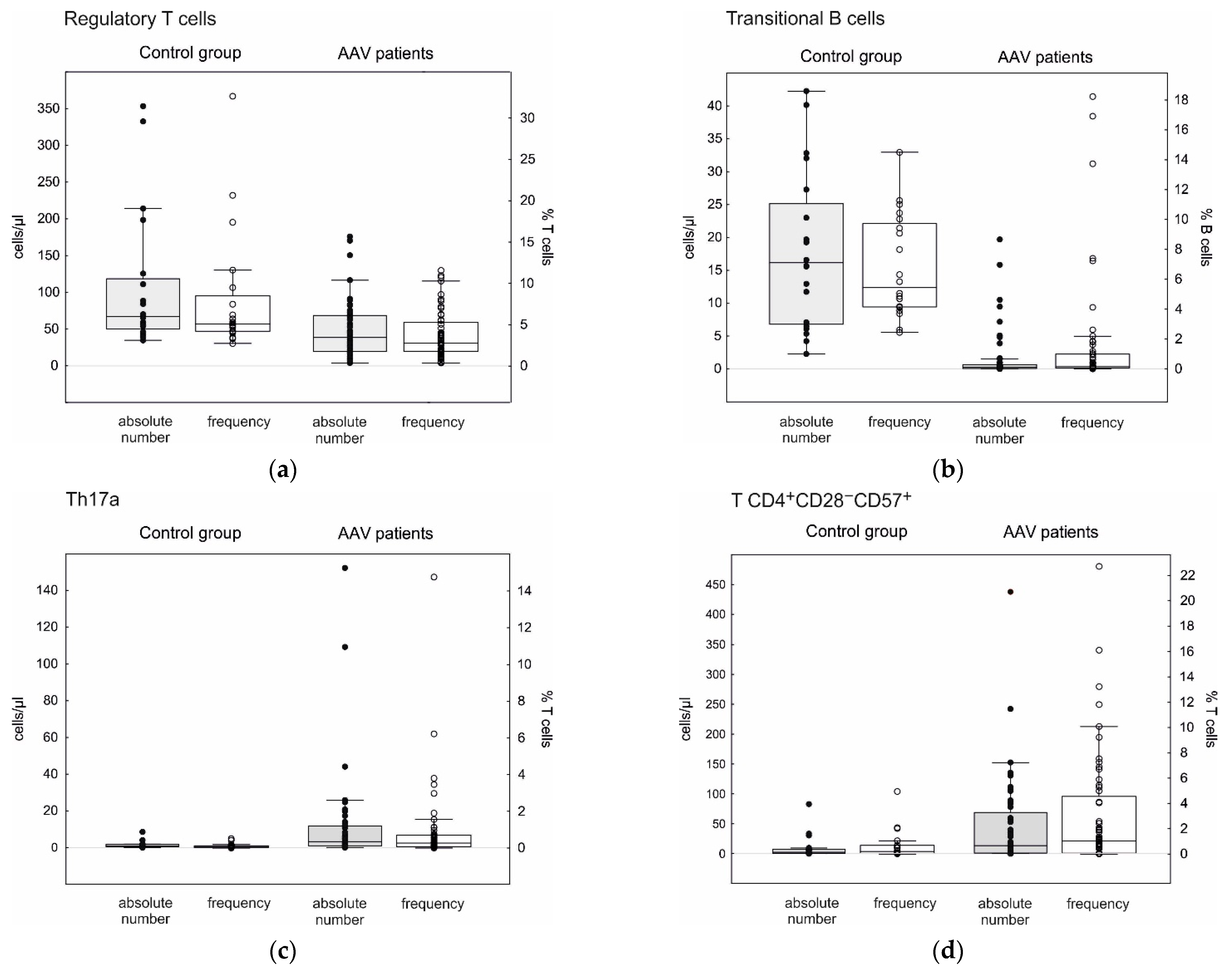{kind=link}
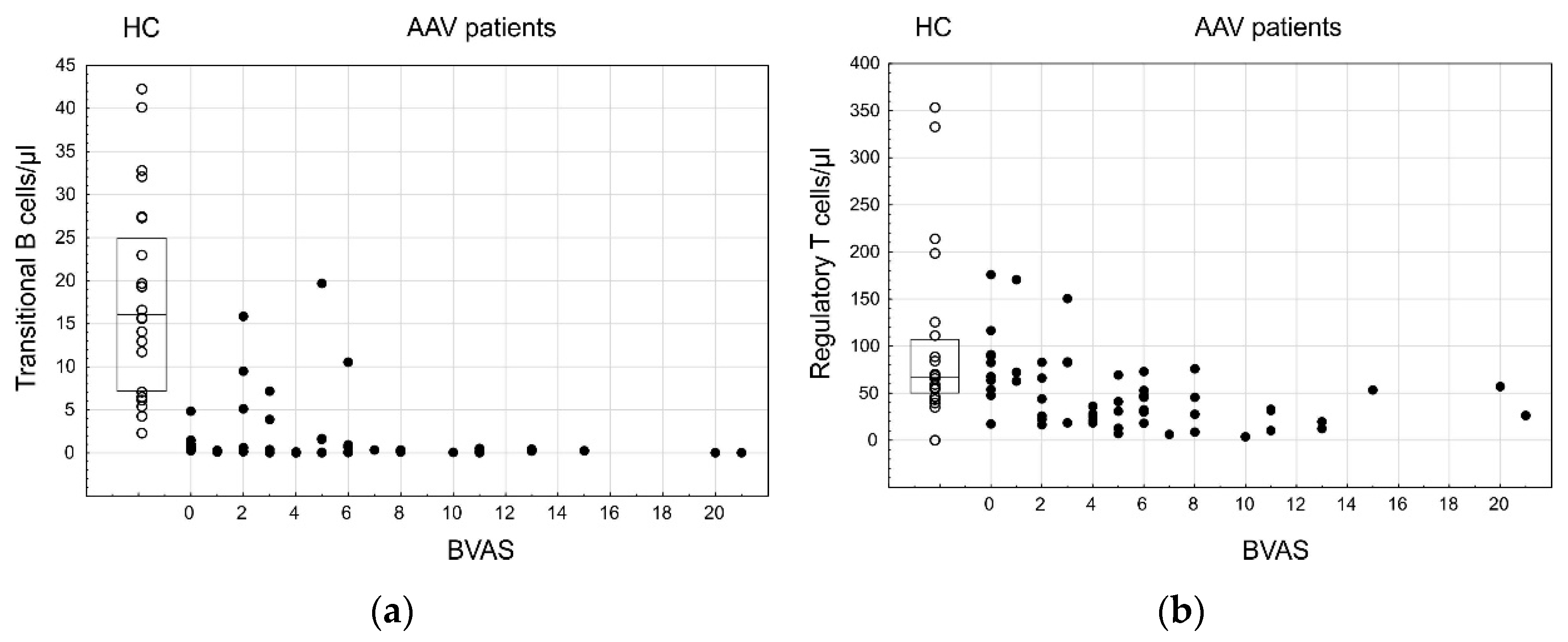{kind=link}
{kind=link}
| Cell Population | AAV Patients | Healthy Controls | p-Value | |||
|---|---|---|---|---|---|---|
| Mean ± SD | Median, IQ Range | Mean ± SD | Median, IQ Range | |||
| Th17a cells | Cells/µL | 11.23 ± 25.10 | 3.3, 0.8–11.7 | 1.42 ± 1.95 | 0.7, 0.4–1.8 | <0.001 |
| % T | 1.117 ± 2.675 | 0.29, 0.08–0.74 | 0.108 ± 0.134 | 0.05, 0.03–0.13 | <0.001 | |
| Th17f cells | Cells/µL | 1.11 ± 1.79 | 0.5, 0.1–1.2 | 0.53 ± 1.43 | 0.1, 0.0–0.3 | <0.001 |
| % T | 0.279 ± 1.364 | 0.04, 0.01–0.07 | 0.043 ± 0.123 | 0.01, 0.00–0.02 | <0.001 | |
| Treg cells | Cells/µL | 55.5 ± 61.8 | 40, 19–71 | 107.3 ± 94.3 | 67, 50–118 | 0.001 |
| % T | 9.19 ± 34.29 | 2.8, 1.8–5.5 | 8.16 ± 7.43 | 5.1, 4.2–8.5 | <0.001 | |
| T CD4+CD28−CD57+ | Cells/µL | 44.78 ± 74.25 | 13.5, 1.1–69.1 | 10.03 ± 19.60 | 2.5, 0.6–7.6 | 0.023 |
| % T | 3.09 ± 4.56 | 1.1, 0.3–4.6 | 0.69 ± 1.18 | 0.20, 0.05–0.68 | 0.010 | |
| T CD4+CD28−CD57− | Cells/µL | 12.29 ± 24.87 | 4.6, 1.0–12.3 | 80.63 ± 243.95 | 0.7, 0.3–2.6 | 0.016 |
| % T | 0.95 ± 1.73 | 0.4, 0.1–1.1 | 5.58 ± 16.83 | 0.07, 0.02–0.16 | 0.013 | |
| Plasmablasts | Cells/µL | 2.27 ± 5.58 | 0.8, 0.4–1.9 | 1.06 ± 0.69 | 1.0, 0.6–1.3 | 0.773 |
| % B | 5.51 ± 22.10 | 0.6, 0.2–1.3 | 0.53 ± 0.41 | 0.4, 0.2–0.8 | 0.222 | |
| Naïve B cells | Cells/µL | 114.9 ± 133.7 | 72, 27–153 | 153.4 ± 94.5 | 121, 80–227 | 0.017 |
| % B | 49.0 ± 21.4 | 55, 30–67 | 56.9 ± 11.1 | 55, 46–67 | 0.339 | |
| Transitional B cells | Cells/µL | 1.66 ± 3.85 | 0.3, 0.1–0.7 | 18.00 ± 11.88 | 16.1, 6.8–25.1 | <0.001 |
| % B | 1.59 ± 3.86 | 0.2, 0.1–1.0 | 6.81 ± 3.40 | 5.4, 4.2–9.7 | <0.001 | |
| Memory B cells | Cells/µL | 59.6 ± 53.4 | 44, 17–90 | 73.7 ± 48.4 | 60, 44–97 | 0.110 |
| % B | 37.8 ± 24.4 | 29, 19–56 | 29.4 ± 11.5 | 30, 21–37 | 0.544 | |
3.3. B Cell Subpopulations

3.4. Lymphocyte Subpopulations and AAV Disease Activity
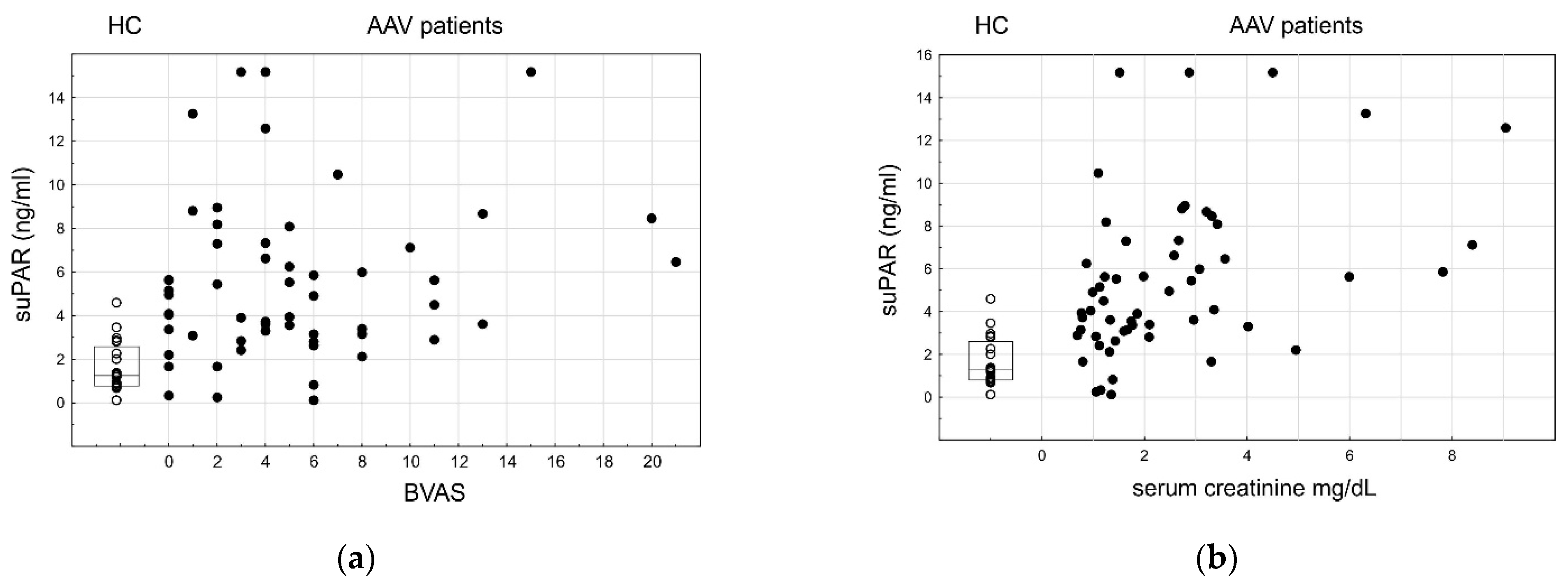
3.5. SuPAR
4. Discussion
Limitations of the Study
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Harper, L.; Savage, C.O. ANCA-associated renal vasculitis at the end of the twentieth century—A disease of older patients. Rheumatology 2005, 44, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Watts, R.A.; Lane, S.E.; Bentham, G.; Scott, D.G. Epidemiology of systemic vasculitis: A ten-year study in the United Kingdom. Arthritis Rheum. 2000, 43, 414–419. [Google Scholar] [CrossRef]
- Kronbichler, A.; Lee, K.H.; Denicolò, S.; Choi, D.; Lee, H.; Ahn, D.; Kim, K.H.; Lee, J.H.; Kim, H.; Hwang, M.; et al. Immunopathogenesis of ANCA-Associated Vasculitis. Int. J. Mol. Sci. 2020, 21, 7319. [Google Scholar] [CrossRef]
- Tervaert, J.W.C.; Damoiseaux, J. Antineutrophil cytoplasmic autoantibodies: How are they detected and what is their use for diagnosis, classification and follow-up? Clin. Rev. Allergy Immunol. 2012, 43, 211–219. [Google Scholar] [CrossRef]
- Radice, A.; Sinico, R.A. Antineutrophil cytoplasmic antibodies ({ANCA}). Autoimmunity 2005, 38, 93–103. [Google Scholar] [CrossRef]
- Jarrot, P.-A.; Kaplanski, G. Pathogenesis of ANCA-associated vasculitis: An update. Autoimmun. Rev. 2016, 15, 704–713. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-S.; Wang, S.-L.; Zhang, H.-J.; He, S.-H.; Liang, X.; Li, T.-F. The pathogenesis and treatment in antineutrophil cytoplasmic antibody associated vasculitis. Am. J. Transl. Res. 2020, 12, 4094–4107. [Google Scholar] [PubMed]
- Merkt, W.; Claus, M.; Blank, N.; Hundemer, M.; Cerwenka, A.; Lorenz, H.-M.; Watzl, C. Active but not inactive granulomatosis with polyangiitis is associated with decreased and phenotypically and functionally altered CD56dim natural killer cells. Arthritis Res. Ther. 2016, 18, 204. [Google Scholar] [CrossRef] [Green Version]
- Carvajal Alegria, G.; Gazeau, P.; Hillion, S.; Daïen, C.I.; Cornec, D.Y.K. Could Lymphocyte Profiling be Useful to Diagnose Systemic Autoimmune Diseases? Clin. Rev. Allergy Immunol. 2017, 53, 219–236. [Google Scholar] [CrossRef]
- Thiel, J.; Salzer, U.; Hässler, F.; Effelsberg, N.M.; Hentze, C.; Sic, H.; Bartsch, M.; Miehle, N.; Peter, H.H.; Warnatz, K.; et al. B cell homeostasis is disturbed by immunosuppressive therapies in patients with ANCA-associated vasculitides. Autoimmunity 2013, 46, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Hua, F.; Wilde, B.; Dolff, S.; Witzke, O. T-Lymphocytes and Disease Mechanisms in Wegener’s Granulomatosis. Kidney Blood Press. Res. 2009, 32, 389–398. [Google Scholar] [CrossRef]
- Merkt, W.; Sturm, P.; Lasitschka, F.; Tretter, T.; Watzl, C.; Saure, D.; Hundemer, M.; Schwenger, V.; Blank, N.; Lorenz, H.-M.; et al. Peripheral blood natural killer cell percentages in granulomatosis with polyangiitis correlate with disease inactivity and stage. Arthritis Res. Ther. 2015, 17, 337. [Google Scholar] [CrossRef] [Green Version]
- Blair, P.A.; Noreña, L.Y.; Flores-Borja, F.; Rawlings, D.J.; Isenberg, D.A.; Ehrenstein, M.R.; Mauri, C. CD19+CD24hiCD38hi B Cells Exhibit Regulatory Capacity in Healthy Individuals but Are Functionally Impaired in Systemic Lupus Erythematosus Patients. Immunity 2010, 32, 129–140. [Google Scholar] [CrossRef] [Green Version]
- De Masson, A.; Le Buanec, H.; Bouaziz, J.-D. Purification and Immunophenotypic Characterization of Human B Cells with Regulatory Functions. In Regulatory B Cells. Methods in Molecular Biology (Methods and Protocols); Vitale, G., Mion, F., Eds.; Humana Press: New York, NY, USA, 2014; Volume 1190, pp. 45–52. [Google Scholar]
- DiLillo, D.J.; Matsushita, T.; Tedder, T.F. B10 cells and regulatory B cells balance immune responses during inflammation, autoimmunity, and cancer. Ann. N. Y. Acad. Sci. 2010, 1183, 38–57. [Google Scholar] [CrossRef]
- Hoffman, W.; Lakkis, F.G.; Chalasani, G. B Cells, Antibodies, and More. Clin. J. Am. Soc. Nephrol. 2016, 11, 137–154. [Google Scholar] [CrossRef]
- Blum, L.K.; Cao, R.R.L.; Sweatt, A.J.; Bill, M.; Lahey, L.J.; Hsi, A.C.; Lee, C.S.; Kongpachith, S.; Ju, C.-H.; Mao, R.; et al. Circulating plasmablasts are elevated and produce pathogenic anti-endothelial cell autoantibodies in idiopathic pulmonary arterial hypertension. Eur. J. Immunol. 2018, 48, 874–884. [Google Scholar] [CrossRef] [PubMed]
- Odendahl, M.; Keitzer, R.; Wahn, U.; Hiepe, F.; Radbruch, A.; Dörner, T.; Bunikowski, R. Perturbations of peripheral B lymphocyte homoeostasis in children with systemic lupus erythematosus. Ann. Rheum. Dis. 2003, 62, 851–858. [Google Scholar] [CrossRef]
- Jacobi, A.M.; Odendahl, M.; Reiter, K.; Bruns, A.; Burmester, G.R.; Radbruch, A.; Valet, G.; Lipsky, P.E.; Dörner, T. Correlation between circulating CD27high plasma cells and disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. 2003, 48, 1332–1342. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.-C.; Kongpachith, S.; Blum, L.K.; Ju, C.-H.; Lahey, L.J.; Lu, D.R.; Cai, X.; Wagner, C.A.; Lindstrom, T.M.; Sokolove, J.; et al. Barcode-enabled sequencing of plasmablast antibody repertoires in rheumatoid arthritis. Arthritis Rheumatol. 2014, 66, 2706–2715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabó, K.; Papp, G.; Szántó, A.; Tarr, T.; Zeher, M. A comprehensive investigation on the distribution of circulating follicular T helper cells and B cell subsets in primary Sjögren’s syndrome and systemic lupus erythematosus. Clin. Exp. Immunol. 2016, 183, 76–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaczorowski, M.; Jutel, M. Human T regulatory cells: On the way to cognition. Arch. Immunol. Ther. Exp. 2013, 61, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Kitching, A.R.; Anders, H.-J.; Basu, N.; Brouwer, E.; Gordon, J.; Jayne, D.R.; Kullman, J.; Lyons, P.A.; Merkel, P.A.; Savage, C.O.S.; et al. ANCA-associated vasculitis. Nat. Rev. Dis. Prim. 2020, 6, 71. [Google Scholar] [CrossRef]
- Happel, K.I.; Dubin, P.J.; Zheng, M.; Ghilardi, N.; Lockhart, C.; Quinton, L.J.; Odden, A.R.; Shellito, J.E.; Bagby, G.J.; Nelson, S.; et al. Divergent roles of IL-23 and IL-12 in host defense against Klebsiella pneumoniae. J. Exp. Med. 2005, 202, 761–769. [Google Scholar] [CrossRef]
- Wu, Q.; Martin, R.J.; Rino, J.G.; Breed, R.; Torres, R.M.; Chu, H.W. IL-23-dependent IL-17 production is essential in neutrophil recruitment and activity in mouse lung defense against respiratory Mycoplasma pneumoniae infection. Microbes Infect. 2007, 9, 78–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weyand, C.M.; Goronzy, J.J.; Liuzzo, G.; Kopecky, S.L.; Holmes, D.R.; Frye, R.L. T-cell immunity in acute coronary syndromes. Mayo Clin. Proc. 2001, 76, 1011–1020. [Google Scholar] [CrossRef] [Green Version]
- Baeten, D.; Louis, S.; Braud, C.; Braudeau, C.; Ballet, C.; Moizant, F.; Pallier, A.; Giral, M.; Brouard, S.; Soulillou, J.-P. Phenotypically and functionally distinct CD8+ lymphocyte populations in long-term drug-free tolerance and chronic rejection in human kidney graft recipients. JASN 2006, 17, 294–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thewissen, M.; Somers, V.; Hellings, N.; Fraussen, J.; Damoiseaux, J.; Stinissen, P. CD4+CD28null T cells in autoimmune disease: Pathogenic features and decreased susceptibility to immunoregulation. J. Immunol. 2007, 179, 6514–6523. [Google Scholar] [CrossRef] [Green Version]
- Ugarte-Gil, M.F.; Sánchez-Zúñiga, C.; Gamboa-Cárdenas, R.V.; Aliaga-Zamudio, M.; Zevallos, F.; Tineo-Pozo, G.; Cucho-Venegas, J.M.; Mosqueira-Riveros, A.; Medina, M.; Perich-Campos, R.A.; et al. Circulating CD4+CD28null and extra-thymic CD+CD8+ double positive T cells are independently associated with disease damage in systemic lupus erythematosus patients. Lupus 2016, 25, 233–240. [Google Scholar] [CrossRef]
- Schmidt, D.; Goronzy, J.J.; Weyand, C.M. CD4+ CD7− CD28− T cells are expanded in rheumatoid arthritis and are characterized by autoreactivity. J. Clin. Investig. 1996, 97, 2027–2037. [Google Scholar] [CrossRef] [PubMed]
- Liuzzo, G.; Goronzy, J.J.; Yang, H.; Kopecky, S.L.; Holmes, D.R.; Frye, R.L.; Weyand, C.M. Monoclonal T-cell proliferation and plaque instability in acute coronary syndromes. Circulation 2000, 101, 2883–2888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Menthon, M.; Lambert, M.; Guiard, E.; Tognarelli, S.; Bienvenu, B.; Karras, A.; Guillevin, L.; Caillat-Zucman, S. Excessive interleukin-15 transpresentation endows NKG2D+CD4+ T cells with innate-like capacity to lyse vascular endothelium in granulomatosis with polyangiitis (Wegener’s). Arthritis Rheum. 2011, 63, 2116–2126. [Google Scholar] [CrossRef]
- Suppiah, R.; Mukhtyar, C.; Flossmann, O.; Alberici, F.; Baslund, B.; Batra, R.; Brown, D.; Holle, J.; Hruskova, Z.; Jayne, D.R.W.; et al. A cross-sectional study of the Birmingham Vasculitis Activity Score version 3 in systemic vasculitis. Rheumatology 2011, 50, 899–905. [Google Scholar] [CrossRef] [Green Version]
- Miloslavsky, E.M.; Specks, U.; Merkel, P.A.; Seo, P.; Spiera, R.; Langford, C.A.; Hoffman, G.S.; Kallenberg, C.G.M.; St Clair, E.W.; Tchao, N.K.; et al. Clinical outcomes of remission induction therapy for severe antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum. 2013, 65, 2441–2449. [Google Scholar] [CrossRef] [PubMed]
- Thai, L.-H.; Charles, P.; Resche-Rigon, M.; Desseaux, K.; Guillevin, L. Are anti-proteinase-3 ANCA a useful marker of granulomatosis with polyangiitis (Wegener’s) relapses? Results of a retrospective study on 126 patients. Autoimmun. Rev. 2014, 13, 313–318. [Google Scholar] [CrossRef]
- Boomsma, M.M.; Stegeman, C.A.; Van Der Leij, M.J.; Oost, W.; Hermans, J.; Kallenberg, C.G.M.; Limburg, P.C.; Tervaert, J.W.C. Prediction of relapses in Wegener’s granulomatosis by measurement of antineutrophil cytoplasmic antibody levels: A prospective study. Arthritis Rheum. 2000, 43, 2025–2033. [Google Scholar] [CrossRef]
- Kerr, G.S.; Fleisher, T.A.; Hallahan, C.W.; Leavitt, R.Y.; Fauci, A.S.; Hoffman, G.S. Limited prognostic value of changes in antineutrophil cytoplasmic antibody titer in patients with wegener’s granulomatosis. Arthritis Rheum. 1993, 36, 365–371. [Google Scholar] [CrossRef]
- Park, H.J.; Jung, S.M.; Song, J.J.; Park, Y.-B.; Lee, S.-W. Platelet to lymphocyte ratio is associated with the current activity of {ANCA}-associated vasculitis at diagnosis: A retrospective monocentric study. Rheumatol. Int. 2018, 38, 1865–1871. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Nagao, T.; Itabashi, M.; Hamano, Y.; Sugamata, R.; Yamazaki, Y.; Yumura, W.; Tsukita, S.; Wang, P.-C.; Nakayama, T.; et al. A novel autoantibody against moesin in the serum of patients with MPO-ANCA-associated vasculitis. Nephrol. Dial. Transplant. 2014, 29, 1168–1177. [Google Scholar] [CrossRef] [Green Version]
- Ahn, S.S.; Jung, S.M.; Song, J.J.; Park, Y.-B.; Lee, S.-W. Neutrophil to lymphocyte ratio at diagnosis can estimate vasculitis activity and poor prognosis in patients with ANCA-associated vasculitis: A retrospective study. BMC Nephrol. 2018, 19, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thunø, M.; Macho, B.; Eugen-Olsen, J. suPAR: The Molecular Crystal Ball. Dis. Markers 2009, 27, 157–172. [Google Scholar] [CrossRef]
- Sehestedt, T.; Lyngbæk, S.; Eugen-Olsen, J.; Jeppesen, J.; Andersen, O.; Hansen, T.W.; Linneberg, A.; Jørgensen, T.; Haugaard, S.B.; Olsen, M.H. Soluble urokinase plasminogen activator receptor is associated with subclinical organ damage and cardiovascular events. Atherosclerosis 2011, 216, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Burcsár, S.; Toldi, G.; Kovács, L.; Szalay, B.; Vásárhelyi, B.; Balog, A. Urine soluble urokinase plasminogen activator receptor as a potential biomarker of lupus nephritis activity. Biomarkers 2021, 26, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Slot, O.; Brünner, N.; Locht, H.; Oxholm, P.; Stephens, R.W. Soluble urokinase plasminogen activator receptor in plasma of patients with inflammatory rheumatic disorders: Increased concentrations in rheumatoid arthritis. Ann. Rheum. Dis. 1999, 58, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Möller, C.C.; Altintas, M.M.; Li, J.; Schwarz, K.; Zacchigna, S.; Xie, L.; Henger, A.; Schmid, H.; Rastaldi, M.P.; et al. Modification of kidney barrier function by the urokinase receptor. Nat. Med. 2008, 14, 55–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, I.; Watts, R.A. Classification of ANCA-associated vasculitis. Curr. Rheumatol. Rep. 2013, 15, 383. [Google Scholar] [CrossRef]
- Bloch, D.A.; Michel, B.A.; Hunder, G.G.; McShane, D.J.; Arend, W.P.; Calabrese, L.H.; Edworthy, S.M.; Fauci, A.S.; Fries, J.F.; Leavitt, R.Y.; et al. The American College of Rheumatology 1990 criteria for the classification of vasculitis: Patients and methods. Arthritis Rheum. 1990, 33, 1068–1073. [Google Scholar] [CrossRef] [PubMed]
- Watts, R.; Lane, S.; Hanslik, T.; Hauser, T.; Hellmich, B.; Koldingsnes, W.; Mahr, A.; Segelmark, M.; Cohen-Tervaert, J.W.; Scott, D. Development and validation of a consensus methodology for the classification of the ANCA-associated vasculitides and polyarteritis nodosa for epidemiological studies. Ann. Rheum. Dis. 2007, 66, 222–227. [Google Scholar] [CrossRef]
- Schlesier, M.; Kaspar, T.; Gutfleisch, J.; Wolff-Vorbeck, G.; Peter, H.H. Activated CD4+ and CD8+ T-cell subsets in Wegener’s granulomatosis. Rheumatol. Int. 1995, 14, 213–219. [Google Scholar] [CrossRef]
- Moosig, F.; Csernok, E.; Wang, G.; Gross, W.L. Costimulatory molecules in Wegener’s granulomatosis (WG): Lack of expression of CD28 and preferential up-regulation of its ligands B7-1 (CD80) and B7-2 (CD86) on T cells. Clin. Exp. Immunol. 1998, 114, 113–118. [Google Scholar] [CrossRef]
- Klapa, S.; Mueller, A.; Csernok, E.; Fagin, U.; Klenerman, P.; Holl-Ulrich, K.; Gross, W.L.; Lamprecht, P. Lower numbers of FoxP3 and CCR4 co-expressing cells in an elevated subpopulation of CD4+CD25high regulatory T cells from Wegener’s granulomatosis. Clin. Exp. Rheumatol. 2010, 28, 72–80. [Google Scholar] [PubMed]
- Xu, Y.; Xu, H.; Zhen, Y.; Sang, X.; Wu, H.; Hu, C.; Ma, Z.; Yu, M.; Yi, H. Imbalance of Circulatory T Follicular Helper and T Follicular Regulatory Cells in Patients with ANCA-Associated Vasculitis. Mediat. Inflamm. 2019, 2019. [Google Scholar] [CrossRef]
- Tsurikisawa, N.; Saito, H.; Oshikata, C.; Tsuburai, T.; Akiyama, K. Decreases in the Numbers of Peripheral Blood Regulatory T Cells, and Increases in the Levels of Memory and Activated B Cells, in Patients with Active Eosinophilic Granulomatosis and Polyangiitis. J. Clin. Immunol. 2013, 33, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Szczeklik, W.; Jakieła, B.; Wawrzycka-Adamczyk, K.; Sanak, M.; Hubalewska-Mazgaj, M.; Padjas, A.; Surmiak, M.; Szczeklik, K.; Sznajd, J.; Musiał, J. Skewing toward Treg and Th2 responses is a characteristic feature of sustained remission in ANCA-positive granulomatosis with polyangiitis. Eur. J. Immunol. 2017, 47, 724–733. [Google Scholar] [CrossRef] [Green Version]
- Reijnders, T.D.Y.; Stegeman, C.A.; Huitema, M.G.; Rutgers, A.; Heeringa, P.; Abdulahad, W.H. Unraveling the identity of FoxP3+ regulatory T cells in Granulomatosis with Polyangiitis patients. Sci. Rep. 2019, 9, 8273. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Su, R.; Ding, T.; Xue, H.; An, J.; LI, X.; Wang, C. THU0325 Reduced of Treg Cells Associated with the Disease Activity of ANCA-Associated Vasculitis. Ann. Rheum. Dis. 2020, 79, 392. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, S.; Zhang, N.; Feng, M.; Liang, Z.; Zhao, X.; Gao, C.; Qin, Y.; Wu, Y.; Liu, G.; et al. Reduced activated regulatory T cells and imbalance of Th17/activated Treg cells marks renal involvement in ANCA-associated vasculitis. Mol. Immunol. 2020, 118, 19–29. [Google Scholar] [CrossRef]
- Free, M.E.; Bunch, D.O.; McGregor, J.A.; Jones, B.E.; Berg, E.A.; Hogan, S.L.; Hu, Y.; Preston, G.A.; Jennette, J.C.; Falk, R.J.; et al. Patients with antineutrophil cytoplasmic antibody-associated vasculitis have defective Treg cell function exacerbated by the presence of a suppression-resistant effector cell population. Arthritis Rheum. 2013, 65, 1922–1933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakiela, B.; Sanak, M.; Szczeklik, W.; Sokolowska, B.; Plutecka, H.; Mastalerz, L.; Musial, J.; Szczeklik, A. Both Th2 and Th17 responses are involved in the pathogenesis of Churg-Strauss syndrome. Clin. Exp. Rheumatol. 2011, 29, S23–S34. [Google Scholar] [PubMed]
- Nogueira, E.; Hamour, S.; Sawant, D.; Henderson, S.; Mansfield, N.; Chavele, K.-M.; Pusey, C.; Salama, A. Serum IL-17 and IL-23 levels and autoantigen-specific Th17 cells are elevated in patients with ANCA-associated vasculitis. Nephrol. Dial. Transplant. 2010, 25, 2209–2217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilde, B.; Thewissen, M.; Damoiseaux, J.; Hilhorst, M.; van Paassen, P.; Witzke, O.; Cohen Tervaert, J.W. Th17 expansion in granulomatosis with polyangiitis (Wegener’s): The role of disease activity, immune regulation and therapy. Arthritis Res. Ther. 2012, 14, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Lamprecht, P.; Moosig, F.; Csernok, E.; Seitzer, U.; Schnabel, A.; Mueller, A.; Gross, W.L. CD28 negative T cells are enriched in granulomatous lesions of the respiratory tract in Wegener’s granulomatosis. Thorax 2001, 56, 751–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komocsi, A.; Lamprecht, P.; Csernok, E.; Mueller, A.; Holl-Ulrich, K.; Seitzer, U.; Moosig, F.; Schnabel, A.; Gross, W.L. Peripheral blood and granuloma CD4+CD28− T cells are a major source of interferon-gamma and tumor necrosis factor-alpha in Wegener’s granulomatosis. Am. J. Pathol. 2002, 160, 1717–1724. [Google Scholar] [CrossRef]
- Lamprecht, P. Off balance: T-cells in antineutrophil cytoplasmic antibody (ANCA)-associated vasculitides. Clin. Exp. Immunol. 2005, 141, 201–210. [Google Scholar] [CrossRef]
- Hänsch, G.M. Shortening of telomeres: Evidence for replicative senescence of T cells derived from patients with Wegener’s granulomatosis. Kidney Int. 2003, 63, 2144–2151. [Google Scholar]
- Specks, U.; Merkel, P.A.; Seo, P.; Spiera, R.; Langford, C.A.; Hoffman, G.S.; Kallenberg, C.G.M.; St Clair, E.W.; Fessler, B.J.; Ding, L.; et al. Efficacy of remission-induction regimens for ANCA-associated vasculitis. N. Engl. J. Med. 2013, 369, 417–427. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.B.; Furuta, S.; Tervaert, J.W.C.; Hauser, T.; Luqmani, R.; Morgan, M.D.; Peh, C.A.; Savage, C.O.; Segelmark, M.; Tesar, V.; et al. Rituximab versus cyclophosphamide in ANCAassociated renal vasculitis: 2-year results of a randomised trial. Ann. Rheum. Dis. 2015, 74, 1178–1182. [Google Scholar] [CrossRef]
- Iwata, Y.; Matsushita, T.; Horikawa, M.; Dilillo, D.J.; Yanaba, K.; Venturi, G.M.; Szabolcs, P.M.; Bernstein, S.H.; Magro, C.M.; Williams, A.D.; et al. Characterization of a rare IL-10-competent B-cell subset in humans that parallels mouse regulatory B10 cells. Blood 2011, 117, 530–541. [Google Scholar] [CrossRef] [Green Version]
- Wilde, B.; Thewissen, M.; Damoiseaux, J.; Knippenberg, S.; Hilhorst, M.; van Paassen, P.; Witzke, O.; Cohen Tervaert, J.W. Regulatory {B} cells in {ANCA}-associated vasculitis. Ann. Rheum. Dis. 2013, 72, 1416–1419. [Google Scholar] [CrossRef]
- Todd, S.K.; Pepper, R.J.; Draibe, J.; Tanna, A.; Pusey, C.D.; Mauri, C.; Salama, A.D. Regulatory B cells are numerically but not functionally deficient in anti-neutrophil cytoplasm antibody-associated vasculitis. Rheumatology 2014, 53, 1693–1703. [Google Scholar] [CrossRef] [Green Version]
- Lepse, N.; Abdulahad, W.H.; Rutgers, A.; Kallenberg, C.G.M.; Stegeman, C.A.; Heeringa, P. Altered B cell balance, but unaffected B cell capacity to limit monocyte activation in anti-neutrophil cytoplasmic antibody-associated vasculitis in remission. Rheumatology 2014, 53, 1683–1692. [Google Scholar] [CrossRef] [Green Version]
- Aybar, L.T.; McGregor, J.G.; Hogan, S.L.; Hu, Y.; Mendoza, C.E.; Brant, E.J.; Poulton, C.J.; Henderson, C.D.; Falk, R.J.; Bunch, D.O. Reduced CD5+ CD24hi CD38hi and interleukin-10+ regulatory B cells in active anti-neutrophil cytoplasmic autoantibody-associated vasculitis permit increased circulating autoantibodies. Clin. Exp. Immunol. 2015, 180, 178–188. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhang, Y.; Han, J.; Yang, M.; Zhu, J.; Jin, T. Transitional B cells involved in autoimmunity and their impact on neuroimmunological diseases. J. Transl. Med. 2020, 18, 131. [Google Scholar] [CrossRef]
- Yanaba, K.; Bouaziz, J.-D.; Haas, K.M.; Poe, J.C.; Fujimoto, M.; Tedder, T.F. A regulatory {B} cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity 2008, 28, 639–650. [Google Scholar] [CrossRef] [Green Version]
- Flores-Borja, F.; Bosma, A.; Ng, D.; Reddy, V.; Ehrenstein, M.R.; Isenberg, D.A.; Mauri, C. CD19+CD24hiCD38hi B cells maintain regulatory T cells while limiting TH1 and TH17 differentiation. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef]
- Von Borstel, A.; Lintermans, L.L.; Heeringa, P.; Rutgers, A.; Stegeman, C.A.; Sanders, J.S.; Abdulahad, W.H. Circulating CD24hiCD38hi regulatory B cells correlate inversely with the ThEM17 cell frequency in granulomatosis with polyangiitis patients. Rheumatology 2019, 58, 1361–1366. [Google Scholar] [CrossRef] [PubMed]
- Tognarelli, S.; Gayet, J.; Lambert, M.; Dupuy, S.; Karras, A.; Cohen, P.; Guillevin, L.; de Menthon, M.; Caillat-Zucman, S. Tissue-Specific Microvascular Endothelial Cells Show Distinct Capacity To Activate NK Cells: Implications for the Pathophysiology of Granulomatosis with Polyangiitis. J. Immunol. 2014, 192, 3399–3408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, F.; Li, Y.; Xu, R.; Cheng, A.; Lv, Y.; Liu, Q. The Plasma Soluble Urokinase Plasminogen Activator Receptor Is Related to Disease Activity of Patients with ANCA-Associated Vasculitis. Mediat. Inflamm. 2020, 2020. [Google Scholar] [CrossRef] [PubMed]




| Patients’ Characteristics | GPA Patients | MPA Patients | ||
|---|---|---|---|---|
| N | 40 | 16 | ||
| Sex (M/F) | 18/22 | 9/7 | ||
| ANCA-positive/negative | 33/7 | 16/0 | ||
| Mean ± SD | Median, IQR | Mean ± SD | Median, IQR | |
| Age (years) | 54.6 ± 17.8 | 58, 43–66 | 61.8 ± 10.8 | 62, 59–69 |
| MPO-ANCA, RU/ml | 130.9 ± 235.9 | 40, 13–119 | – | – |
| PR3-ANCA, RU/ml | – | – | 83.0 ± 43.6 | 80, 64–117 |
| Creatinine | 1.91 ± 1.24 | 1.4, 1.1–2.4 | 3.92 ± 2.53 | 3.1, 2.5–5.0 |
| CRP | 29.06 ± 32.53 | 16.8, 3.5–46 | 12.1 ± 19.7 | 4.3, 2.7–7.2 |
| BVAS | 5.5 ± 4.7 | 5, 0–20 | 4.3 ± 5.1 | 4, 0–21 |
| Cell Population Cells/µL | AAV Patients | Healthy Controls | p-Value | ||
|---|---|---|---|---|---|
| Mean ± SD | Median, IQ Range | Mean ± SD | Median, IQ Range | ||
| Total lymphocytes | 1766.0 ± 775.9 | 1725, 1244–2176 | 1870.3 ± 434.3 | 1821, 1504–2222 | 0.219 |
| T lymphocytes | 1336.4 ± 609.2 | 1316, 946–1678 | 1338.7 ± 349.9 | 1267, 1113–1680 | 0.686 |
| CD8+ T lymphocytes | 492.5 ± 277.1 | 409, 263–691 | 500.7 ± 205.1 | 514, 326–658 | 0.703 |
| CD4+ T lymphocytes | 854.8 ± 442.6 | 836, 560–1061 | 826.1 ± 223.4 | 847, 668–982 | 0.949 |
| CD4/CD8 | 2.06 ± 1.22 | 1.7, 1.2–2.6 | 1.86 ± 0.76 | 1.7, 1.4–2.1 | 0.925 |
| B lymphocytes | 198.2 ± 176.5 | 160, 65–254 | 261.5 ± 133.6 | 211, 169–376 | 0.035 |
| NK cells | 177.3 ± 176.3 | 134, 89–198 | 252.5 ± 106.4 | 227, 164–330 | <0.001 |
| NKT cells | 121.9 ± 116.9 | 88, 47–154 | 105.5 ± 70.8 | 94, 54–138 | 0.995 |
| NKbright cells | 10.0 ± 7.5 | 8, 5–15 | 22.0 ± 9.7 | 21, 15–27 | <0.001 |
| Cell Population Cells/µL | GPA Patients | MPA Patients | p-Value | ||
|---|---|---|---|---|---|
| Mean | Median, IQ Range | Mean | Median, IQ Range | ||
| Total lymphocytes | 1904.9 ± 824.7 | 1809, 1307–2359 | 1419.6 ± 509.6 | 1505, 969–1801 | 0.050 |
| T lymphocytes | 1414.5 ± 661.1 | 1334, 975–1790 | 1140.9 ± 409.0 | 1231, 785–1441 | 0.140 |
| CD8+ T lymphocytes | 538.6 ± 296.9 | 472, 301–733 | 377.4 ± 180.7 | 344, 246–564 | 0.050 |
| CD4+ T lymphocytes | 897.2 ± 489.6 | 885, 565–1115 | 748.6 ± 279.6 | 738, 524–967 | 0.363 |
| CD4/CD8 | 1.99 ± 1.32 | 1.5, 1.1–2.4 | 2.25 ± 0.93 | 2.1, 1.7–3.0 | 0.144 |
| B lymphocytes | 220.2 ± 190.4 | 188, 85–264 | 143.9 ± 123.6 | 101, 51–239 | 0.178 |
| NK lymphocytes | 199.3 ± 199.5 | 155, 97–212 | 122.3 ± 77.4 | 112, 78–186 | 0.197 |
| NKbright lymphocytes | 10.3 ± 8.17 | 8, 5–15 | 9.31 ± 5.66 | 8, 5–15 | 0.847 |
| Cell Population | GPA Patients | MPA Patients | p-Value | |||
|---|---|---|---|---|---|---|
| Mean | Median, IQ Range | Mean | Median, IQ Range | |||
| Th17a cells | Cells/µL | 13.41 ± 28.77 | 4.7, 0.8–13.6 | 5.79 ± 10.68 | 2.3, 0.8–5.6 | 0.314 |
| % T | 1.379 ± 3.105 | 0.30, 0.08–0.93 | 0.461 ± 0.726 | 0.22, 0.09–0.51 | 0.508 | |
| Th17f cells | Cells/µL | 1.34 ± 2.05 | 0.6, 0.2–1.5 | 0.54 ± 0.63 | 0.3, 0.1–0.7 | 0.145 |
| % T | 0.370 ± 1.610 | 0.05, 0.01–0.09 | 0.053 ± 0.072 | 0.02, 0.01–0.06 | 0.257 | |
| Treg cells | Cells/µL | 58.4 ± 67.6 | 47, 20–71 | 48.4 ± 44.9 | 27, 20–68 | 0.571 |
| % T | 11.24 ± 40.49 | 2.8, 1.8–5.9 | 4.09 ± 3.26 | 3.3, 1.9–4.7 | 0.942 | |
| T CD4+CD28−CD57+ | Cells/µL | 38.88 ± 47.76 | 14.0, 1.1–81.1 | 59.54 ± 118.14 | 8.8, 2.3–58.8 | 0.596 |
| % T | 2.80 ± 3.58 | 1.0, 0.3–5.1 | 3.82 ± 6.49 | 1.2, 0.2–4.1 | 0.828 | |
| T CD4+CD28−CD57− | Cells/µL | 12.7 ± 28.0 | 5, 2–12 | 11.2 ± 15.2 | 3, 0.3–20 | 0.686 |
| % T | 1.03 ± 1.98 | 0.4, 0.2–1.0 | 0.76 ± 0.90 | 0.4, 0.0–1.5 | 0.618 | |
| Naïve B cells | Cells/µL | 133.4 ± 147.4 | 100, 26–159 | 68.8 ± 76.7 | 45, 27–89 | 0.145 |
| % B | 50.7 ± 22.2 | 58, 29–68 | 44.7 ± 19.3 | 46, 34–58 | 0.235 | |
| Memory B cells | Cells/µL | 59.9 ± 44.2 | 57, 21–95 | 58.8 ± 73.2 | 23, 15–77 | 0.344 |
| % B | 37.0 ± 25.1 | 28, 18–58 | 39.6 ± 23.3 | 31, 20–56 | 0.496 | |
| Transitional B cells | Cells/µL | 1.61 ± 3.66 | 0.3, 0.1–0.9 | 1.77 ± 4.42 | 0.1, 0.1–0.6 | 0.145 |
| % B | 1.25 ± 2.66 | 0.2, 0.1–1.1 | 2.43 ± 5.93 | 0.1, 0.0–0.5 | 0.502 | |
| Plasmablasts | Cells/µL | 2.89 ± 6.51 | 1.0, 0.4–2.5 | 0.71 ± 0.47 | 0.1, 0.4–1.0 | 0.184 |
| % B | 7.09 ± 26.00 | 0.5, 0.2–1.3 | 1.54 ± 2.82 | 0.9, 0.2–1.3 | 0.827 | |
| Cell Population | Association with the BVAS | ||
|---|---|---|---|
| Correlation Coefficient, rs | p-Value | ||
| Th17a cells | Cells/µL | −0.17 | 0.213 |
| % T | −0.18 | 0.190 | |
| Th17f cells | Cells/µL | −0.23 | 0.094 |
| % T | −0.18 | 0.188 | |
| Treg cells | Cells/µL | −0.43 | < 0.001 |
| % T | −0.36 | 0.009 | |
| T CD4+CD28−CD57+ | Cells/µL | −0.07 | 0.588 |
| % T | −0.03 | 0.800 | |
| T CD4+CD28−CD57− | Cells/µL | 0.05 | 0.712 |
| % T | 0.10 | 0.467 | |
| Transitional B cells | Cells/µL | −0.35 | 0.009 |
| % B | −0.23 | 0.089 | |
| NK cells | Cells/µL | −0.19 | 0.159 |
| NKbright cells | Cells/µL | 0.00 | 0.959 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Żabińska, M.; Kościelska-Kasprzak, K.; Krajewska, J.; Bartoszek, D.; Augustyniak-Bartosik, H.; Krajewska, M. Immune Cells Profiling in ANCA-Associated Vasculitis Patients—Relation to Disease Activity. Cells 2021, 10, 1773. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10071773
Żabińska M, Kościelska-Kasprzak K, Krajewska J, Bartoszek D, Augustyniak-Bartosik H, Krajewska M. Immune Cells Profiling in ANCA-Associated Vasculitis Patients—Relation to Disease Activity. Cells. 2021; 10(7):1773. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10071773
Chicago/Turabian StyleŻabińska, Marcelina, Katarzyna Kościelska-Kasprzak, Joanna Krajewska, Dorota Bartoszek, Hanna Augustyniak-Bartosik, and Magdalena Krajewska. 2021. "Immune Cells Profiling in ANCA-Associated Vasculitis Patients—Relation to Disease Activity" Cells 10, no. 7: 1773. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10071773