
Heat Shock Factor 1 Prevents Age-Related Hearing Loss by Decreasing Endoplasmic Reticulum Stress

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Experimental Procedures
2.3. Auditory Brainstem Response Analysis
2.4. IHC, OHC, and SP Counts
2.5. House Ear Institute-Organ of Corti 1 (HEI-OC1) Cell Culture
2.6. Immunoblotting
2.7. Immunohistochemistry
2.8. Cell Viability Assay
2.9. Doxorubicin (DOXO) Treatment
2.10. Senescence-Associated Β-Gal Assay
2.11. Brdu Incorporation Assay
2.12. Cell Cycle Analysis
2.13. Real-Time PCR
2.14. siRNA Transfection
2.15. HSF1 Viral Production and Titration
2.16. Antibodies and Reagents
2.17. Statistical Analyses
3. Results
3.1. Auditory Hair Cell Loss in Aged Cochlea Was Accompanied by a Decrease in HSF1 Expression and Increased ER Stress and Apoptosis
3.2. During Cellular Senescence, the Expression of HSF1 and HSP Decreases, While the Expression of ER Stress and Apoptosis Markers Increases
3.3. HSF1 Overexpression Increased Cell Viability by Inhibiting ER Stress and Apoptosis via HSP Expression Regulation
3.4. Cell Viability and ER Stress Were Regulated by HSF1 Protein Expression under Heat Shock Stress
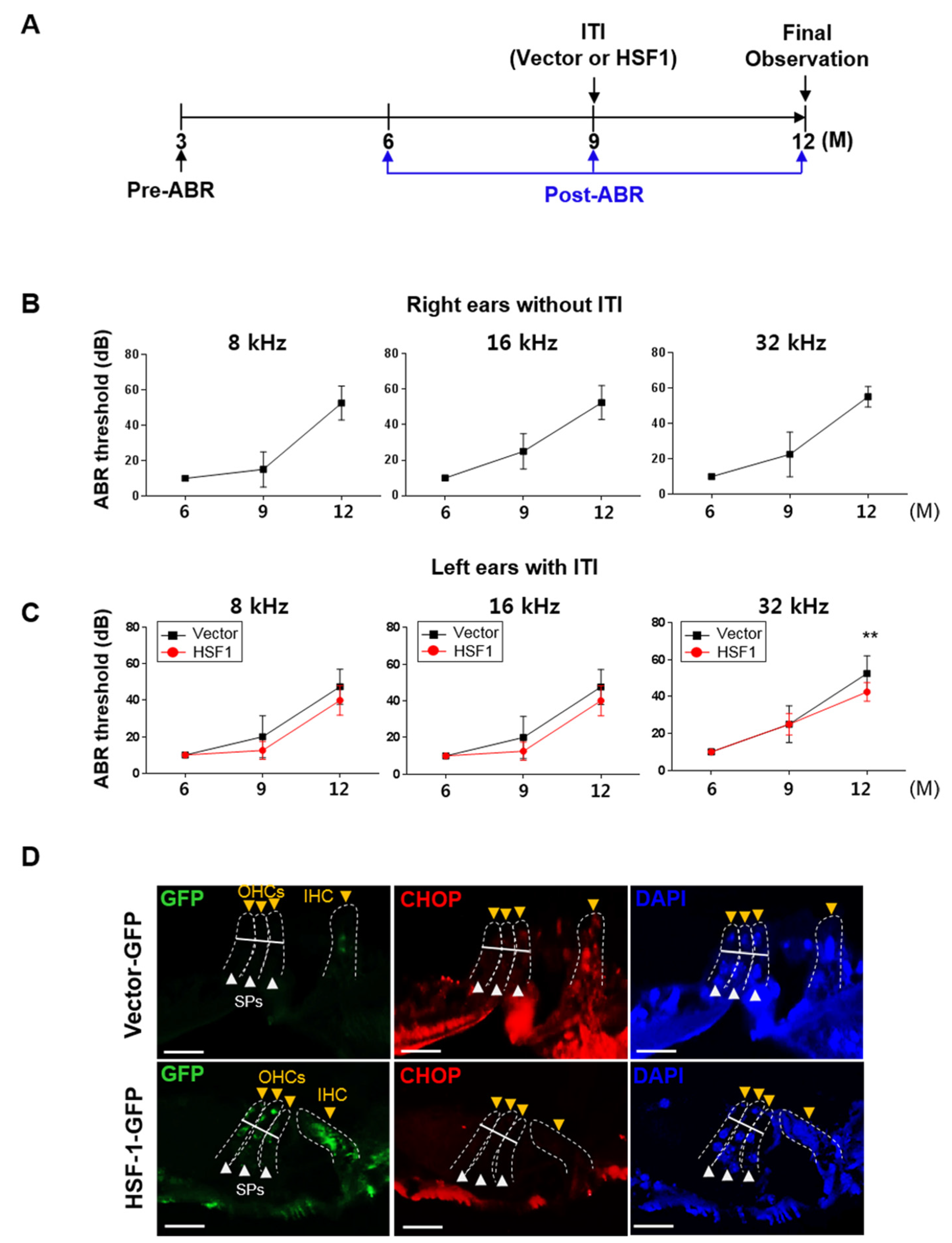
3.5. HSF1 Overexpression Inhibited ER Stress-Mediated UPR Branch Apoptosis in the OC of Aging Mice, Suppressing High-Frequency Hearing Loss
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bowl, M.R.; Dawson, S.J. Age-Related Hearing Loss. Cold Spring Harb. Perspect. Med. 2019, 9, a033217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamogashira, T.; Fujimoto, C.; Yamasoba, T. Reactive oxygen species, apoptosis, and mitochondrial dysfunction in hearing loss. Biomed. Res. Int. 2015, 2015, 617207. [Google Scholar] [CrossRef] [Green Version]
- Someya, S.; Prolla, T.A. Mitochondrial oxidative damage and apoptosis in age-related hearing loss. Mech. Ageing Dev. 2010, 131, 480–486. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Mollereau, B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 2014, 15, 233–249. [Google Scholar] [CrossRef]
- Wang, X.Z.; Lawson, B.; Brewer, J.W.; Zinszner, H.; Sanjay, A.; Mi, L.J.; Boorstein, R.; Kreibich, G.; Hendershot, L.M.; Ron, D. Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153). Mol. Cell. Biol. 1996, 16, 4273–4280. [Google Scholar] [CrossRef] [Green Version]
- Zinszner, H.; Kuroda, M.; Wang, X.; Batchvarova, N.; Lightfoot, R.T.; Remotti, H.; Stevens, J.L.; Ron, D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes. Dev. 1998, 12, 982–995. [Google Scholar] [CrossRef]
- Shone, G.; Raphael, Y.; Miller, J.M. Hereditary deafness occurring in cd/1 mice. Hear. Res. 1991, 57, 153–156. [Google Scholar] [CrossRef] [Green Version]
- Fujinami, Y.; Mutai, H.; Mizutari, K.; Nakagawa, S.; Matsunaga, T. A novel animal model of hearing loss caused by acute endoplasmic reticulum stress in the cochlea. J. Pharmacol. Sci. 2012, 118, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Akil, O.; Rouse, S.L.; McLaughlin, C.W.; Matthews, I.R.; Lustig, L.R.; Chan, D.K.; Sherr, E.H. Deletion of Tmtc4 activates the unfolded protein response and causes postnatal hearing loss. J. Clin. Investig. 2018, 128, 5150–5162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, D.; Sun, G.W.; Cui, Y.; Mita, S.; Otsuki, N.; Kanzaki, S.; Nibu, K.; Ogawa, K.; Matsunaga, T. Neuroprotective effects of cutamesine, a ligand of the sigma-1 receptor chaperone, against noise-induced hearing loss. J. Neurosci. Res. 2015, 93, 788–795. [Google Scholar] [CrossRef]
- Akerfelt, M.; Morimoto, R.I.; Sistonen, L. Heat shock factors: Integrators of cell stress, development and lifespan. Nat. Rev. Mol. Cell. Biol. 2010, 11, 545–555. [Google Scholar] [CrossRef]
- Barna, J.; Csermely, P.; Vellai, T. Roles of heat shock factor 1 beyond the heat shock response. Cell Mol. Life Sci. 2018, 75, 2897–2916. [Google Scholar] [CrossRef]
- Li, J.; Labbadia, J.; Morimoto, R.I. Rethinking HSF1 in Stress, Development, and Organismal Health. Trends Cell Biol. 2017, 27, 895–905. [Google Scholar] [CrossRef]
- Carnemolla, A.; Labbadia, J.P.; Lazell, H.; Neueder, A.; Moussaoui, S.; Bates, G.P. Contesting the dogma of an age-related heat shock response impairment: Implications for cardiac-specific age-related disorders. Hum. Mol. Genet. 2014, 23, 3641–3656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morimoto, R.I. Cells in stress: Transcriptional activation of heat shock genes. Science 1993, 259, 1409–1410. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Pastor, R.; Burchfiel, E.T.; Thiele, D.J. Regulation of heat shock transcription factors and their roles in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 4–19. [Google Scholar] [CrossRef]
- Pelham, H.R. A regulatory upstream promoter element in the Drosophila hsp 70 heat-shock gene. Cell 1982, 30, 517–528. [Google Scholar] [CrossRef]
- Sakurai, H.; Enoki, Y. Novel aspects of heat shock factors: DNA recognition, chromatin modulation and gene expression. FEBS J. 2010, 277, 4140–4149. [Google Scholar] [CrossRef]
- Kovacs, D.; Sigmond, T.; Hotzi, B.; Bohar, B.; Fazekas, D.; Deak, V.; Vellai, T.; Barna, J. HSF1Base: A Comprehensive Database of HSF1 (Heat Shock Factor 1) Target Genes. Int. J. Mol. Sci. 2019, 20, 5815. [Google Scholar] [CrossRef] [Green Version]
- Fairfield, D.A.; Kanicki, A.C.; Lomax, M.I.; Altschuler, R.A. Expression and localization of heat shock factor (Hsf) 1 in the rodent cochlea. Hear. Res. 2002, 173, 109–118. [Google Scholar] [CrossRef]
- Yoshida, N.; Kristiansen, A.; Liberman, M.C. Heat stress and protection from permanent acoustic injury in mice. J. Neurosci. 1999, 19, 10116–10124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choung, Y.H.; Kim, S.W.; Tian, C.; Min, J.Y.; Lee, H.K.; Park, S.N.; Lee, J.B.; Park, K. Korean red ginseng prevents gentamicin-induced hearing loss in rats. Laryngoscope 2011, 121, 1294–1302. [Google Scholar] [CrossRef]
- Chu, C.; Xin, A.; Zhou, Y.; Zhang, Y. A simple protocol for producing high-titer lentivirus. Acta Biochim. Biophys. Sin. 2013, 45, 1079–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuknecht, H.F. Further Observations on the Pathology of Presbycusis. Arch. Otolaryngol. 1964, 80, 369–382. [Google Scholar] [CrossRef]
- Schuknecht, H.F.; Gacek, M.R. Cochlear pathology in presbycusis. Ann. Otol. Rhinol. Laryngol. 1993, 102, 1–16. [Google Scholar] [CrossRef]
- Wu, P.Z.; O’Malley, J.T.; de Gruttola, V.; Liberman, M.C. Age-Related Hearing Loss Is Dominated by Damage to Inner Ear Sensory Cells, Not the Cellular Battery That Powers Them. J. Neurosci. 2020, 40, 6357–6366. [Google Scholar] [CrossRef]
- Iurlaro, R.; Munoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef] [Green Version]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [Green Version]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Shaw, P.H. The role of p53 in cell cycle regulation. Pathol. Res. Pract. 1996, 192, 669–675. [Google Scholar] [CrossRef]
- Lee, Y.Y.; Choi, Y.S.; Kim, D.W.; Cheong, J.Y.; Song, K.Y.; Ryu, M.S.; Lim, I.K. Mitochondrial nucleoid remodeling and biogenesis are regulated by the p53-p21(WAF1)-PKCzeta pathway in p16(INK4a)-silenced cells. Aging 2020, 12, 6700–6732. [Google Scholar] [CrossRef]
- Anckar, J.; Sistonen, L. Regulation of HSF1 function in the heat stress response: Implications in aging and disease. Annu. Rev. Biochem. 2011, 80, 1089–1115. [Google Scholar] [CrossRef]
- Huth, M.E.; Ricci, A.J.; Cheng, A.G. Mechanisms of aminoglycoside ototoxicity and targets of hair cell protection. Int. J. Otolaryngol. 2011, 2011, 937861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gates, G.A.; Mills, J.H. Presbycusis. Lancet 2005, 366, 1111–1120. [Google Scholar] [CrossRef]
- Matthews, L.J.; Lee, F.S.; Mills, J.H.; Dubno, J.R. Extended high-frequency thresholds in older adults. J. Speech Lang. Hear. Res. 1997, 40, 208–214. [Google Scholar] [CrossRef]
- Schacht, J.; Talaska, A.E.; Rybak, L.P. Cisplatin and aminoglycoside antibiotics: Hearing loss and its prevention. Anat. Rec. (Hoboken) 2012, 295, 1837–1850. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Guo, Y.; Tang, J.; Jiang, J.; Chen, Z. New insights into the roles of CHOP-induced apoptosis in ER stress. Acta Biochim. Biophys. Sin. 2014, 46, 629–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.Z.; Yan, D. Ageing and hearing loss. J. Pathol. 2007, 211, 188–197. [Google Scholar] [CrossRef]
- Pickles, J.O. Mutation in mitochondrial DNA as a cause of presbyacusis. Audiol. Neurootol. 2004, 9, 23–33. [Google Scholar] [CrossRef]
- Lim, H.H.; Jenkins, O.H.; Myers, M.W.; Miller, J.M.; Altschuler, R.A. Detection of HSP 72 synthesis after acoustic overstimulation in rat cochlea. Hear. Res. 1993, 69, 146–150. [Google Scholar] [CrossRef] [Green Version]
- Taleb, M.; Brandon, C.S.; Lee, F.S.; Lomax, M.I.; Dillmann, W.H.; Cunningham, L.L. Hsp70 inhibits aminoglycoside-induced hair cell death and is necessary for the protective effect of heat shock. J. Assoc. Res. Otolaryngol. 2008, 9, 277–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taleb, M.; Brandon, C.S.; Lee, F.S.; Harris, K.C.; Dillmann, W.H.; Cunningham, L.L. Hsp70 inhibits aminoglycoside-induced hearing loss and cochlear hair cell death. Cell Stress Chaperones 2009, 14, 427–437. [Google Scholar] [CrossRef] [Green Version]
- May, L.A.; Kramarenko, I.I.; Brandon, C.S.; Voelkel-Johnson, C.; Roy, S.; Truong, K.; Francis, S.P.; Monzack, E.L.; Lee, F.S.; Cunningham, L.L. Inner ear supporting cells protect hair cells by secreting HSP70. J. Clin. Investig. 2013, 123, 3577–3587. [Google Scholar] [CrossRef]
- Caplan, A.J.; Cyr, D.M.; Douglas, M.G. YDJ1p facilitates polypeptide translocation across different intracellular membranes by a conserved mechanism. Cell 1992, 71, 1143–1155. [Google Scholar] [CrossRef]
- Ungermann, C.; Neupert, W.; Cyr, D.M. The role of Hsp70 in conferring unidirectionality on protein translocation into mitochondria. Science 1994, 266, 1250–1253. [Google Scholar] [CrossRef]
- Dey, B.; Caplan, A.J.; Boschelli, F. The Ydj1 molecular chaperone facilitates formation of active p60v-src in yeast. Mol. Biol. Cell 1996, 7, 91–100. [Google Scholar] [CrossRef]
- Liu, J.S.; Kuo, S.R.; Makhov, A.M.; Cyr, D.M.; Griffith, J.D.; Broker, T.R.; Chow, L.T. Human Hsp70 and Hsp40 chaperone proteins facilitate human papillomavirus-11 E1 protein binding to the origin and stimulate cell-free DNA replication. J. Biol. Chem. 1998, 273, 30704–30712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meacham, G.C.; Lu, Z.; King, S.; Sorscher, E.; Tousson, A.; Cyr, D.M. The Hdj-2/Hsc70 chaperone pair facilitates early steps in CFTR biogenesis. EMBO J. 1999, 18, 1492–1505. [Google Scholar] [CrossRef]
- Oda, T.; Sekimoto, T.; Kurashima, K.; Fujimoto, M.; Nakai, A.; Yamashita, T. Acute HSF1 depletion induces cellular senescence through the MDM2-p53-p21 pathway in human diploid fibroblasts. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.; Meriin, A.B.; Gabai, V.L.; Christians, E.; Benjamin, I.; Wilson, A.; Wolozin, B.; Sherman, M.Y. The heat shock transcription factor Hsf1 is downregulated in DNA damage-associated senescence, contributing to the maintenance of senescence phenotype. Aging Cell 2012, 11, 617–627. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Sakata, K.; Liao, F.F. Bidirectional interplay of HSF1 degradation and UPR activation promotes tau hyperphosphorylation. PLoS Genet. 2017, 13, e1006849. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Grammatikakis, N.; Siganou, A.; Calderwood, S.K. Regulation of molecular chaperone gene transcription involves the serine phosphorylation, 14-3-3 epsilon binding, and cytoplasmic sequestration of heat shock factor 1. Mol. Cell. Biol. 2003, 23, 6013–6026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, B.; Soncin, F.; Price, B.D.; Stevenson, M.A.; Calderwood, S.K. Sequential phosphorylation by mitogen-activated protein kinase and glycogen synthase kinase 3 represses transcriptional activation by heat shock factor-1. J. Biol. Chem. 1996, 271, 30847–30857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, R.V.; Budd Haeberlein, S.L.; Avila, J. Glycogen synthase kinase 3: A drug target for CNS therapies. J. Neurochem. 2004, 89, 1313–1317. [Google Scholar] [CrossRef] [PubMed]
- Nyberg, S.; Abbott, N.J.; Shi, X.; Steyger, P.S.; Dabdoub, A. Delivery of therapeutics to the inner ear: The challenge of the blood-labyrinth barrier. Sci. Transl. Med. 2019, 11, eaao0935. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.Y.; Gil, E.S.; Jeong, I.H.; Kim, H.; Jang, J.H.; Choung, Y.-H. Heat Shock Factor 1 Prevents Age-Related Hearing Loss by Decreasing Endoplasmic Reticulum Stress. Cells 2021, 10, 2454. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10092454
Lee YY, Gil ES, Jeong IH, Kim H, Jang JH, Choung Y-H. Heat Shock Factor 1 Prevents Age-Related Hearing Loss by Decreasing Endoplasmic Reticulum Stress. Cells. 2021; 10(9):2454. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10092454
Chicago/Turabian StyleLee, Yun Yeong, Eun Sol Gil, In Hye Jeong, Hantai Kim, Jeong Hun Jang, and Yun-Hoon Choung. 2021. "Heat Shock Factor 1 Prevents Age-Related Hearing Loss by Decreasing Endoplasmic Reticulum Stress" Cells 10, no. 9: 2454. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10092454