

Deficient Sarcolemma Repair in ALS: A Novel Mechanism with Therapeutic Potential
by
 ,
,
Ang Li
1,
Jianxun Yi
1,
Xuejun Li
1,
Li Dong
1,
Lyle W. Ostrow
2,*,
Jianjie Ma
3,* and
Jingsong Zhou
1,* 1
Department of Kinesiology, College of Nursing and Health Innovation, University of Texas at Arlington, Arlington, TX 76019, USA
2
Department of Neurology, Lewis Katz School of Medicine at Temple University, Philadelphia, PA 19122, USA
3
Department of Surgery, University of Virginia, Charlottesville, VA 22903, USA
*
Authors to whom correspondence should be addressed.
Cells 2022, 11(20), 3263; https://0-doi-org.brum.beds.ac.uk/10.3390/cells11203263
Submission received: 6 September 2022
/
Revised: 7 October 2022
/
Accepted: 14 October 2022
/
Published: 17 October 2022
(This article belongs to the Special Issue Redox Control of Cell Signaling in Cardiac and Skeletal Muscle)
{kind=link}
Abstract
:The plasma membrane (sarcolemma) of skeletal muscle myofibers is susceptible to injury caused by physical and chemical stresses during normal daily movement and/or under disease conditions. These acute plasma membrane disruptions are normally compensated by an intrinsic membrane resealing process involving interactions of multiple intracellular proteins including dysferlin, annexin, caveolin, and Mitsugumin 53 (MG53)/TRIM72. There is new evidence for compromised muscle sarcolemma repair mechanisms in Amyotrophic Lateral Sclerosis (ALS). Mitochondrial dysfunction in proximity to neuromuscular junctions (NMJs) increases oxidative stress, triggering MG53 aggregation and loss of its function. Compromised membrane repair further worsens sarcolemma fragility and amplifies oxidative stress in a vicious cycle. This article is to review existing literature supporting the concept that ALS is a disease of oxidative-stress induced disruption of muscle membrane repair that compromise the integrity of the NMJs and hence augmenting muscle membrane repair mechanisms could represent a viable therapeutic strategy for ALS.
1. Introduction
ALS is a fatal neuromuscular disease characterized by progressive motor neuron loss and muscle wasting. Riluzole (Rilutek) and Edaravone (Radicava), the two FDA approved treatments for ALS, demonstrate only limited efficacy to slow disease progression [1]. In most cases, the disease may be the product of multiple inter-related factors, with many efforts focused on identifying distinct patient subsets using various integrated-omics approaches [2]. While diverse cell types, biological mechanisms, and genetic factors are implicated in sporadic and familial ALS pathogenesis, there are also commonly shared pathological and clinical features [3]. Progressive respiratory muscle weakness is a main cause of morbidity and eventually death in all forms of ALS [4,5,6].
Motor neurons communicate with individual muscle fibers at neuromuscular junctions (NMJs), and retrograde signals are also conducted from muscle back to nerve [7]. While ALS is classically considered a “dying-forward” process starting in motor neurons, accumulating evidence [8,9,10,11,12,13,14,15,16,17] also implicates early muscle cell dysfunction in ALS pathophysiology. The degree to which a “dying-back” process [18,19,20] starting distally at NMJs or “dying-forward” from the CNS contributes to ALS progression remains unsettled and may vary in different patient subsets. Regardless of the direction of communication and relative contribution of different cell types to ALS progression (e.g., glia, neurons, myofibers), NMJ loss and the resulting skeletal muscle denervation is a critical early pathogenic event in both patients and animal models [21,22,23,24]. Because NMJ is the critical site involving bidirectional crosstalk between myofibers and the motor neurons [13,16,18,19,20,25,26,27], sustaining NJM integrity may slow disease progression. The molecular mechanisms accounting for NMJ loss and myofiber degeneration in ALS have attracted more attention in recent years, as strategies to slow these processes and/or enhance intrinsic muscle repair/regeneration could have broad therapeutic significance across different ALS subtypes [28].
Owing to the discovery of multiple genes that constitute the key components of cell membrane repair machinery, including dysferlin, annexin, caveolin, and MG53/TRIM72, etc., multiple review articles have been made that address the membrane repair defects linking to muscular dystrophy and heart failure, etc. [29,30,31,32,33]. However, currently there is none on the topic of membrane repair defects in ALS. This article is to review existing literature supporting the concept that ALS is a disease of oxidative-stress induced disruption of muscle membrane repair that compromise the integrity of the NMJ and hence augmenting membrane repair mechanisms could represent a viable therapeutic strategy for ALS.
2. Mitochondrial Dysfunction and Oxidative Stress in ALS Skeletal Muscle
Skeletal muscle is responsible for voluntary movements of the entire body, thus comprises one of the largest and most metabolically active tissues [34]. Given these energetic demands, it is not surprising that mitochondria account for 4–15% of myofiber volume [35]. Mitochondrial dysfunction is regarded as a major contributor to ALS pathology [36,37]. Both familial and sporadic ALS patients show striking mitochondrial defects and abnormalities of oxidative stress in motor neurons and skeletal muscle [38,39,40,41,42,43]. While mitochondrial dysfunction is a major source of excessive production of reactive oxygen species (ROS) leading oxidative stress, mitochondria themselves are a known target of oxidative stress due to the high sensitivity of their membrane and mtDNA to ROS. It is believed that mitochondrial dysfunction and oxidative stress play key role in neuromuscular degeneration in ALS [44,45,46].
Mouse models expressing human ALS mutations (e.g., SOD1G93A (G93A)) recapitulate many features of human disease [47], and have been widely used to investigate pathogenic mechanisms and preclinical therapies for ALS [37,48,49,50,51]. The G93A mice demonstrate early mitochondrial dysfunction and enhanced reactive oxygen species (ROS) production in skeletal muscle [14,15,16,26,52]. Markedly elevated skeletal muscle ROS production is also an early abnormality in muscles from other ALS mouse models such as mice knockout for TDP-43 and VAPB [4,53,54,55]. The ROS accumulation in G93A skeletal muscle has been revealed by proteomics, biochemical and enzymatic assays [52,53,56]. Our prior studies demonstrated increased cytosolic and mitochondrial ROS levels in G93A myofibers beginning prior to symptom onset [14]. In this study, we generated a double transgenic mouse model (G93A/mt-cpYFP) to visualize dynamic ROS-related “mitoflash” events in G93A myofibers [14,26]. “Mitoflash” events are spontaneous fluorescent transients of the mitochondrial targeted, circularly permuted yellow fluorescent protein (mt-cpYFP), and has been associated with mitochondrial ROS production [57]. Widespread mitoflash activities has been revealed in myofibers derived from G93A/mt-cpYFP mice prior to symptom onset, which was not seen in wild type (WT) mt-cpYFP myofibers. The increased mitoflash activity is associated with increased opening of mitochondrial transition pores (mPTP) [26,57,58,59,60]. mPTP opening can promote mitochondrial ROS production [61,62]. We also detected an elevated level of the mitochondrial protein cyclophilin D (CypD) in G93A skeletal muscle [14], which is accompanied by abnormalities in mitochondrial structure and function [11,13,16]. CypD is a known activator of mPTP opening [63,64,65,66,67] and CypD-related mPTP opening is tightly associated with mitoflash activities [68,69,70]. Enhanced CypD has been associated with more frequent and widespread mitoflash activities in myofibers [58,71,72]. Thus, CypD-mediated mPTP opening may be one of the underlying mechanisms to enhanced ROS production in ALS muscle. However, it is worth to note that increased CypD level may not be a solo cause of excessive ROS generation in ALS skeletal muscle. Other factors, such as mitochondrial Ca2+ overload, oxidizing agents, thiol oxidation, HSP90., etc., also promote high-conductance opening of mPTP in pathological conditions including ALS [73].
3. ALS Mice Exhibit Sarcolemma Fragility and Mitochondrial Dysfunction in Proximity to NMJs Prior to Symptom Onset
Intraperitoneal injection of Evans Blue dye (EB) is widely used as an in vivo marker of abnormal myofiber permeability from sarcolemma damage [74,75,76,77]. We observed consistently elevated intracellular EB penetration in the diaphragm and tibialis anterior muscles of G93A mice after downhill running [78]. Even without the exercise challenge, a significant portion of diaphragm myofibers in 2-month-old G93A mice (pre-symptomatic) showed EB penetration. The phenomenon became more prominent when the G93A mice reached 4-month of age (i.e., after symptom onset). In contrast, EB penetration was not seen in the diaphragm of WT controls, with or without running, at 2- or 4-months of age [78]. These observations indicate that the increase of sarcolemma vulnerability occurs early in the course of the disease in ALS mice.
The enhanced sarcolemma fragility is most striking near NMJs. After downhill running, isolated flexor digitorum brevis (FDB) myofibers from 2-month-old G93A mice exhibited intracellular accumulation of FM 1-43, a cell membrane impermeable dye often used for ex vivo evaluation of sarcolemma integrity [30,77,79], forming a gradient centered around the NMJ. This phenomenon was not observed in myofibers derived from WT mice [78]. These data suggest that NMJs are focally more susceptible to injury than other regions of the sarcolemma, and exercise-induced NMJ injury is exacerbated in G93A mice before ALS symptom onset. Remarkably, mitochondrial lesion appears first at the region harboring NMJ in the G93A muscle with a myofiber segment showing mitochondrial inner membrane depolarization prior to ALS symptom onset, and this myofiber segment at the NMJ with depolarized mitochondria exhibits uncontrolled hyperactive Ca2+ activity, which should never occur in normal WT myofibers [13,16]. This early mitochondrial lesion in proximity to NMJ may be a cause of sarcolemma damage initiated at NMJ.
Nicotinic acetylcholine receptors (nAchR) are abundant in the postsynaptic membrane of NMJ. There is evidence of an elevated Ca2+ permeability of nAchR in adult mammalian muscle [80], and increased nAchR expression was found in G93A muscle [81]. It can be expected that following repetitive stimulations, mitochondria near NMJ could face elevated local intracellular [Ca2+], leading to mitochondrial Ca2+ overload at NMJ [15]. Mitochondrial Ca2+ overload also triggers mPTP opening, which causes mitochondrial depolarization and promotes ROS generation [62]. The depolarized mitochondria have reduced capacity to take up Ca2+, thus, an increased cytosolic Ca2+ release events were observed at region near NMJ of G93A myofibers [13,16]. This abnormal Ca2+ release could further stress neighboring normal mitochondria with Ca2+ overload, causing mPTP opening, mitochondrial depolarization and excessive ROS generation in a vicious cycle started from NMJ. As the cell membrane is a major target of direct ROS attack [46,82], it is possible that early mitochondrial lesion with excessive ROS near NMJ initiates the membrane leakage observed at NMJ in G93A myofibers. Furthermore, those lesions could propagate and affect the entire myofiber during ALS disease progression, although the underlying molecular natures need to be further explored. In addition to damaging the lipid membrane of sarcolemma, elevated ROS level could also affect the normal membrane repair mechanism, which will be further discussed below. Indeed, disorganized T-tubule networks were observed near the NMJ in the myofibers of G93A mice before ALS symptom onset, becoming more striking at later stages of disease. The degree of T-tubule disorganization correlated with the abnormal mitochondrial depolarization at NMJs [78]. As skeletal muscle mitochondrial dysfunction and enhanced oxidative stress are described in different genetic and sporadic ALS subtypes, the resulting sarcolemma fragility may similarly be a common phenomenon.
4. MG53-Mediated Membrane Repair Is Compromised in ALS
The stress to myofiber integrity resulting from repeated muscle contraction-relaxation (such as in the diaphragm muscle during respiration) is offset by intrinsic sarcolemma repair mechanisms. The dynamic balance between potentially injurious stress and membrane repair can be shifted towards myofiber damage in situations where a disease state amplifies the stress beyond the intrinsic capacity for repair, and/or disrupts the repair mechanisms themselves. MG53 is a member of the tri-partite motif (TRIM) E3-ligase family protein (encoded by TRIM72) [83], which is highly expressed in skeletal muscle [84]. Upon acute plasma membrane injury, MG53 acts as a sensor to oxidized intracellular microenvironments, and then facilitates trafficking of intracellular vesicles to form membrane repair patches at focal sites of injured sarcolemma. MG53 is the molecule that first arrives at the injury site (within 2 s following sarcolemma rupture) and plays an essential role in maintaining sarcolemma integrity [77,85,86,87]. Genetic ablation of MG53 results in defective membrane repair and diminished tissue regenerative capacity [77,85,88,89]. Conversely, transgenic mice with sustained elevation of MG53 in the bloodstream (tPA-MG53) live a long, healthy life span with enhanced tissue-regenerative capacity following injury [90].
We have demonstrated that MG53 forms membrane patches on the sarcolemma specifically in proximity to NMJs to preserve NMJ integrity under normal physiological condition [78]. Impaired MG53-related membrane repair function could play an essential role in the progressive degeneration of NMJs in ALS. As discussed above, mitochondrial dysfunction is a major cause of excessive ROS production in ALS muscle [14,38,39,40,41,42,43,52]. Our studies suggest that the prominent and persistently enhanced oxidative stress in ALS muscle limits the movement of intracellular MG53 vesicles, causing abnormal MG53 protein aggregation. The intracellular aggregation of MG53 protein could compromise the MG53-related sarcolemma repair mechanism. We observed pathologic MG53 aggregation in all examined muscle types of the G93A mice including fast and slow twitch muscles, and importantly also in postmortem human diaphragm and psoas muscles samples from both sporadic and familial ALS patients (harboring different ALS mutations), but not in controls. [78]. Therefore, compromised MG53-mediated membrane repair function appears to be a common feature of different familial and sporadic ALS subtypes.
The effects of exercise training on ALS progression remains controversial [91,92]. Several published cohort studies suggest an association between intense physical activities and increased risk of ALS [93,94,95,96]. Furthermore, the effects of exercise training on ALS progression remains controversial, with some studies suggesting modest benefits from mild or moderate exercise while others, especially of more strenuous physical exertion, may be detrimental [91,92]. A trial of diaphragm muscle pacing reduced survival in ALS patients with respiratory insufficiency [97,98,99,100,101]. Similarly, in our ALS mouse studies, even modest exercise worsened diaphragm damage [78].
5. Therapeutic Potential of Exogenously Administered MG53 in ALS
We evaluated the potential efficacy of recombinant human MG53 (rhMG53) using the ALS G93A mouse model [78]. Adding rhMG53 to the extracellular solution significantly attenuated intracellular FM 1-43 accumulation in G93A myofibers in vitro. We treated 3-month-old G93A mice (after symptom onset) with intravenous rhMG53 (2 mg/kg body weight) once daily for 2 weeks. The rhMG53 injected mice exhibited less denervated NMJs in the diaphragm and more surviving motor neuron cell bodies in the spinal cord anterior horns compared with the saline injected control groups [78]. We also produced a PEGylated rhMG53 (PEG-rhMG53) protein with increased half-life of rhMG53 in circulation [102,103]. PEG-rhMG53 was similarly administered to three-month-old G93A mice (2 mg/kg body weight, every other day for one month). The life span of the PEG-rhMG53 injected mice was significantly prolonged for 13 days on average compared with the saline injected group, and the benefits were observed in both male and female mice. The PEG-rhMG53 treatment also slowed weight loss [78].
As MG53 is present at low levels in circulation under normal physiologic conditions in human and rodents [104,105,106,107], administration of exogenous rhMG53 is not likely to produce neutralizing antibodies as peripheral tolerance to this protein has already occurred. Studies in multiple mouse models reported no observable toxic effects with long-term administration of rhMG53 [104,108]. rhMG53 protein has been found to protect various cell types against membrane disruption, and ameliorate the pathology associated with muscular dystrophy [104], acute lung injury [109], myocardial infarction [110], acute kidney injury [111] and ischemic brain damage [112] in rodent and large animal models of these diseases.
MG53 protein is released from skeletal muscle as a myokine [33]. We previously demonstrated elevated serum levels of endogenous MG53 in a mouse model of muscular dystrophy (mdx) compared to WT mice [104]. Similarly, serum MG53 was markedly elevated in the 2-month-old G93A mice after downhill running—likely a reflection of enhanced skeletal muscle membrane injury [78]. Interestingly, serum MG53 levels in later stage ALS mice was reduced to levels lower than in WT mice. This reduced serum MG53 level could be due to muscle atrophy in later stage of disease, perhaps combined with diminished MG53 secretion due to the pathological aggregation we observed with disease progression in the ALS mice.
6. Conclusions and Future Perspectives
Our early studies using the G93A mouse model established a role for mitochondria Ca2+ signaling and ROS in mediating the crosstalk between muscle and neurons at the NMJ during ALS progression [13,14,15,16,26]. As illustrated in Figure 1, during ALS progression, mitochondrial dysfunction in muscle myofibers leads to excessive ROS production, which promotes ectopic aggregation of cytosolic MG53 and loss of function. This worsens sarcolemma disruption, resulting in a vicious cycle of worsening oxidative stress, muscle membrane damage, and NMJ degeneration. Treating ALS mice with exogenous rhMG53 can enhance the formation of membrane sealing patches at the damaged sarcolemma, accounting for the reduced membrane leaking we observed in vitro, preservation of diaphragm muscle NMJs and motor neuron cell bodies in the spinal cord in vivo, as well as prolonging life and slowing weight loss.
NMJs in mice and humans have different anatomical structures: the clefts of the NMJ are deeper in humans compared to mice suggesting a need for amplifying the signal in humans that is greater than in mice. Considering the difference in synaptic transmission, preservation of NMJ integrity could even be more crucial in human. Presumably, rhMG53 can have therapeutic benefits to preserve NMJ integrity in ALS patients.
Our studies in mice with a pathogenic SOD1 mutation and human autopsy muscle samples from decedents who harbored C9orf72 mutations, and “sporadic” ALS (with no known pathogenic mutation)—all showed the same abnormal MG53 aggregation, which was not seen in control mouse or human muscles. This suggests that abnormalities of MG53 function may be seen across different ALS subtypes, and thus treating with exogenous recombinant MG53 could have broad therapeutic applicability to ALS patients. Due to the heterogeneity in etiology, clinical and pathology, the treatment of ALS patients may be best achieved by a multidisciplinary approach with targeting different potential pathological mechanisms. Intriguingly, combining rhMG53 treatment with exercise and/or diaphragm pacing may protect from the worsening of disease seen in prior exercise/pacing trials, while still allowing for the beneficial effects that exercise normally has on muscle physiology and strengthening.
MG53 is present at low levels in blood circulation under normal physiologic conditions in both rodents and humans [104,105,106,107]. Higher circulating levels of MG53 were observed in the G93A mice compared to WT littermates, and correlated with serum CK measurements [78]. If similar elevations of circulating endogenous MG53 are seen in ALS patients, it could be useful as a “prognostic biomarker” to quantify the degree of myofiber degeneration at early disease stages. Furthermore, muscle biopsy is a standardized diagnostic clinical procedure performed at clinical and academic centers around the world. While not generally part of the clinical workup for ALS, biopsies have been used in several ALS clinical trials to look for differences pre- and post-treatment, [https://www.clinicaltrials.gov/ct2/show/NCT04632225 (accessed on 6 September 2022)]. Serum levels of endogenous MG53 and/or muscle biopsy pathology could similarly be promising pharmacodynamic measures to demonstrate therapeutic efficacy for ALS intended to preserve myofiber integrity.
Although MG53 was the first molecules investigated in ALS for its role in sarcolemma repair, there are other membrane repair proteins [29] with undetermined roles in skeletal muscle degeneration in ALS. Future studies should be encouraged to further investigate whether and how those membrane repair proteins are involved in skeletal muscle wasting in ALS. This type of study should provide additional potential therapeutic targets for ALS. We wish this review article could attract more attention of the ALS community and promote research efforts to further explore the possibility of considering preservation of skeletal muscle membrane integrity as a potential therapy for ALS.
Author Contributions
Conceptualization and writing: A.L., J.Z., L.W.O. and J.M.; Review and editing: A.L., J.Y., X.L., L.D., J.M., L.W.O. and J.Z. All authors have read and agreed to the published version of the manuscript.
Funding
Jingsong Zhou was supported by grants from the Department of Defense AL170061(W81XWH1810684), NIH (R01AR057404, R01NS105621 and R01 HL138570), Bank of America Victor E. Speas Foundation, ALS Association (16-IIP-288), and a pilot grant from Kansas City Consortium on Musculoskeletal Diseases. Jianjie Ma was supported by NIH grants (R01AG071676, R01-AG072430, and R01HL157215). Lyle Ostrow was supported by funding from Target ALS Foundation, CDC National ALS Registry and Biorepository, and ALS Association.
Acknowledgments
We would like to apologize in advance to colleagues whose work was not directly discussed in this review article due to space limitations and a focused concept.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiernan, M.C.; Vucic, S.; Talbot, K.; McDermott, C.J.; Hardiman, O.; Shefner, J.M.; Al-Chalabi, A.; Huynh, W.; Cudkowicz, M.; Talman, P. Improving clinical trial outcomes in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2021, 17, 104–118. [Google Scholar] [CrossRef] [PubMed]
- Hardiman, O.; van den Berg, L.H.; Kiernan, M.C. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, J.P.; Picchiarelli, G.; Dupuis, L.; Gonzalez De Aguilar, J.L. The Role of Skeletal Muscle in Amyotrophic Lateral Sclerosis. Brain Pathol. 2016, 26, 227–236. [Google Scholar] [CrossRef]
- de Carvalho, M.; Swash, M.; Pinto, S. Diaphragmatic Neurophysiology and Respiratory Markers in ALS. Front. Neurol. 2019, 10, 143. [Google Scholar] [CrossRef] [Green Version]
- Niedermeyer, S.; Murn, M.; Choi, P.J. Respiratory Failure in Amyotrophic Lateral Sclerosis. Chest 2019, 155, 401–408. [Google Scholar] [CrossRef]
- Nguyen, Q.T.; Son, Y.J.; Sanes, J.R.; Lichtman, J.W. Nerve terminals form but fail to mature when postsynaptic differentiation is blocked: In vivo analysis using mammalian nerve-muscle chimeras. J. Neurosci. 2000, 20, 6077–6086. [Google Scholar] [CrossRef] [Green Version]
- Dobrowolny, G.; Aucello, M.; Rizzuto, E.; Beccafico, S.; Mammucari, C.; Boncompagni, S.; Belia, S.; Wannenes, F.; Nicoletti, C.; Del Prete, Z.; et al. Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab. 2008, 8, 425–436. [Google Scholar] [CrossRef]
- Wong, M.; Martin, L.J. Skeletal muscle-restricted expression of human SOD1 causes motor neuron degeneration in transgenic mice. Hum. Mol. Genet. 2010, 19, 2284–2302. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Ma, C.; Yi, J.; Wu, S.; Luo, G.; Xu, X.; Lin, P.H.; Sun, J.; Zhou, J. Suppressed autophagy flux in skeletal muscle of an amyotrophic lateral sclerosis mouse model during disease progression. Physiol. Rep. 2015, 3, e12271. [Google Scholar] [CrossRef]
- Luo, G.; Yi, J.; Ma, C.; Xiao, Y.; Yi, F.; Yu, T.; Zhou, J. Defective mitochondrial dynamics is an early event in skeletal muscle of an amyotrophic lateral sclerosis mouse model. PLoS ONE 2013, 8, e82112. [Google Scholar] [CrossRef]
- Wang, H.; Yi, J.; Li, X.; Xiao, Y.; Dhakal, K.; Zhou, J. ALS-associated mutation SOD1(G93A) leads to abnormal mitochondrial dynamics in osteocytes. Bone 2018, 106, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Ma, C.; Li, Y.; Weisleder, N.; Rios, E.; Ma, J.; Zhou, J. Mitochondrial calcium uptake regulates rapid calcium transients in skeletal muscle during excitation-contraction (E-C) coupling. J. Biol. Chem. 2011, 286, 32436–32443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.; Karam, C.; Yi, J.; Zhang, L.; Li, X.; Yoon, D.; Wang, H.; Dhakal, K.; Ramlow, P.; Yu, T.; et al. ROS-related mitochondrial dysfunction in skeletal muscle of an ALS mouse model during the disease progression. Pharm. Res. 2018, 138, 25–36. [Google Scholar] [CrossRef]
- Zhou, J.; Li, A.; Li, X.; Yi, J. Dysregulated mitochondrial Ca(2+) and ROS signaling in skeletal muscle of ALS mouse model. Arch. Biochem. Biophys. 2019, 663, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yi, J.; Fu, R.; Liu, E.; Siddique, T.; Rios, E.; Deng, H.X. Hyperactive intracellular calcium signaling associated with localized mitochondrial defects in skeletal muscle of an animal model of amyotrophic lateral sclerosis. J. Biol. Chem. 2010, 285, 705–712. [Google Scholar] [CrossRef] [Green Version]
- Zhu, K.; Yi, J.; Xiao, Y.; Lai, Y.; Song, P.; Zheng, W.; Jiao, H.; Fan, J.; Wu, C.; Chen, D.; et al. Impaired bone homeostasis in amyotrophic lateral sclerosis mice with muscle atrophy. J. Biol. Chem. 2015, 290, 8081–8094. [Google Scholar] [CrossRef] [Green Version]
- Dadon-Nachum, M.; Melamed, E.; Offen, D. The "dying-back" phenomenon of motor neurons in ALS. J. Mol. Neurosci. 2011, 43, 470–477. [Google Scholar] [CrossRef]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 2004, 185, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Frey, D.; Schneider, C.; Xu, L.; Borg, J.; Spooren, W.; Caroni, P. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J. Neurosci. 2000, 20, 2534–2542. [Google Scholar] [CrossRef] [Green Version]
- Clark, J.A.; Southam, K.A.; Blizzard, C.A.; King, A.E.; Dickson, T.C. Axonal degeneration, distal collateral branching and neuromuscular junction architecture alterations occur prior to symptom onset in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. J. Chem. Neuroanat. 2016, 76, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Campanari, M.L.; Bourefis, A.R.; Kabashi, E. Diagnostic Challenge and Neuromuscular Junction Contribution to ALS Pathogenesis. Front. Neurol. 2019, 10, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martineau, E.; Di Polo, A.; Vande Velde, C.; Robitaille, R. Dynamic neuromuscular remodeling precedes motor-unit loss in a mouse model of ALS. Elife 2018, 7, e41973. [Google Scholar] [CrossRef] [PubMed]
- Cappello, V.; Francolini, M. Neuromuscular Junction Dismantling in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2017, 18, 2092. [Google Scholar] [CrossRef]
- Dupuis, L.; Gonzalez de Aguilar, J.L.; Echaniz-Laguna, A.; Eschbach, J.; Rene, F.; Oudart, H.; Halter, B.; Huze, C.; Schaeffer, L.; Bouillaud, F.; et al. Muscle mitochondrial uncoupling dismantles neuromuscular junction and triggers distal degeneration of motor neurons. PLoS ONE 2009, 4, e5390. [Google Scholar] [CrossRef] [Green Version]
- Karam, C.; Yi, J.; Xiao, Y.; Dhakal, K.; Zhang, L.; Li, X.; Manno, C.; Xu, J.; Li, K.; Cheng, H. Absence of physiological Ca 2+ transients is an initial trigger for mitochondrial dysfunction in skeletal muscle following denervation. Skelet. Muscle 2017, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Magrane, J.; Cortez, C.; Gan, W.B.; Manfredi, G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 2014, 23, 1413–1424. [Google Scholar] [CrossRef] [Green Version]
- Scaricamazza, S.; Salvatori, I.; Ferri, A.; Valle, C. Skeletal Muscle in ALS: An Unappreciated Therapeutic Opportunity? Cells 2021, 10, 525. [Google Scholar] [CrossRef]
- Cooper, S.T.; McNeil, P.L. Membrane Repair: Mechanisms and Pathophysiology. Physiol. Rev. 2015, 95, 1205–1240. [Google Scholar] [CrossRef] [Green Version]
- Han, R.; Campbell, K.P. Dysferlin and muscle membrane repair. Curr. Opin. Cell Biol. 2007, 19, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Demonbreun, A.R.; McNally, E.M. Plasma Membrane Repair in Health and Disease. Curr. Top. Membr. 2016, 77, 67–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gazzerro, E.; Bonetto, A.; Minetti, C. Caveolinopathies: Translational implications of caveolin-3 in skeletal and cardiac muscle disorders. Handb. Clin. Neurol. 2011, 101, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, L.; Yue, H.; Whitson, B.A.; Haggard, E.; Xu, X.; Ma, J. MG53, A Tissue Repair Protein with Broad Applications in Regenerative Medicine. Cells 2021, 10, 122. [Google Scholar] [CrossRef] [PubMed]
- Neel, B.A.; Lin, Y.; Pessin, J.E. Skeletal muscle autophagy: A new metabolic regulator. Trends Endocrinol. Metab. TEM 2013, 24, 635–643. [Google Scholar] [CrossRef] [Green Version]
- Larsen, S.; Nielsen, J.; Hansen, C.N.; Nielsen, L.B.; Wibrand, F.; Stride, N.; Schroder, H.D.; Boushel, R.; Helge, J.W.; Dela, F.; et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J. Physiol. 2012, 590, 3349–3360. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Carri, M.T.; D’Ambrosi, N.; Cozzolino, M. Pathways to mitochondrial dysfunction in ALS pathogenesis. Biochem. Biophys. Res. Commun 2017, 483, 1187–1193. [Google Scholar] [CrossRef]
- Echaniz-Laguna, A.; Zoll, J.; Ponsot, E.; N’Guessan, B.; Tranchant, C.; Loeffler, J.P.; Lampert, E. Muscular mitochondrial function in amyotrophic lateral sclerosis is progressively altered as the disease develops: A temporal study in man. Exp. Neurol. 2006, 198, 25–30. [Google Scholar] [CrossRef]
- Napoli, L.; Crugnola, V.; Lamperti, C.; Silani, V.; Di Mauro, S.; Bresolin, N.; Moggio, M. Ultrastructural mitochondrial abnormalities in patients with sporadic amyotrophic lateral sclerosis. Arch. Neurol. 2011, 68, 1612–1613. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, S.; Iwata, M. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2007, 66, 10–16. [Google Scholar] [CrossRef]
- Siciliano, G.; Pastorini, E.; Pasquali, L.; Manca, M.L.; Iudice, A.; Murri, L. Impaired oxidative metabolism in exercising muscle from ALS patients. J. Neurol. Sci. 2001, 191, 61–65. [Google Scholar] [CrossRef]
- Soraru, G.; Vergani, L.; Fedrizzi, L.; D’Ascenzo, C.; Polo, A.; Bernazzi, B.; Angelini, C. Activities of mitochondrial complexes correlate with nNOS amount in muscle from ALS patients. Neuropathol. Appl. Neurobiol. 2007, 33, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, F.R.; Manfredi, G.; Mawrin, C.; Beal, M.F.; Schon, E.A. Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J. Neurochem. 2002, 80, 616–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carri, M.T.; Valle, C.; Bozzo, F.; Cozzolino, M. Oxidative stress and mitochondrial damage: Importance in non-SOD1 ALS. Front. Cell Neurosci. 2015, 9, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amico, E.; Factor-Litvak, P.; Santella, R.M.; Mitsumoto, H. Clinical perspective on oxidative stress in sporadic amyotrophic lateral sclerosis. Free Radic Biol. Med. 2013, 65, 509–527. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Yong, V.W. Oxidized phospholipids as novel mediators of neurodegeneration. Trends Neurosci. 2022, 45, 419–429. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Carri, M.T.; Cozzolino, M. SOD1 and mitochondria in ALS: A dangerous liaison. J. Bioenerg Biomembr. 2011, 43, 593–599. [Google Scholar] [CrossRef]
- Ivanova, M.I.; Sievers, S.A.; Guenther, E.L.; Johnson, L.M.; Winkler, D.D.; Galaleldeen, A.; Sawaya, M.R.; Hart, P.J.; Eisenberg, D.S. Aggregation-triggering segments of SOD1 fibril formation support a common pathway for familial and sporadic ALS. Proc. Natl. Acad. Sci. USA 2014, 111, 197–201. [Google Scholar] [CrossRef] [Green Version]
- Ludolph, A.C.; Bendotti, C.; Blaugrund, E.; Chio, A.; Greensmith, L.; Loeffler, J.P.; Mead, R.; Niessen, H.G.; Petri, S.; Pradat, P.F.; et al. Guidelines for preclinical animal research in ALS/MND: A consensus meeting. Amyotroph. Lateral Scler. 2010, 11, 38–45. [Google Scholar] [CrossRef]
- McGoldrick, P.; Joyce, P.I.; Fisher, E.M.; Greensmith, L. Rodent models of amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2013, 1832, 1421–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, F.L.; Song, W.; Jang, Y.C.; Liu, Y.; Sabia, M.; Richardson, A.; Van Remmen, H. Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am. J. Physiol. Regul Integr. Comp. Physiol. 2007, 293, R1159–R1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halter, B.; Gonzalez de Aguilar, J.L.; Rene, F.; Petri, S.; Fricker, B.; Echaniz-Laguna, A.; Dupuis, L.; Larmet, Y.; Loeffler, J.P. Oxidative stress in skeletal muscle stimulates early expression of Rad in a mouse model of amyotrophic lateral sclerosis. Free Radic Biol. Med. 2010, 48, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Han, S.M.; El Oussini, H.; Scekic-Zahirovic, J.; Vibbert, J.; Cottee, P.; Prasain, J.K.; Bellen, H.J.; Dupuis, L.; Miller, M.A. VAPB/ALS8 MSP ligands regulate striated muscle energy metabolism critical for adult survival in caenorhabditis elegans. PLoS Genet. 2013, 9, e1003738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stallings, N.R.; Puttaparthi, K.; Dowling, K.J.; Luther, C.M.; Burns, D.K.; Davis, K.; Elliott, J.L. TDP-43, an ALS linked protein, regulates fat deposition and glucose homeostasis. PLoS ONE 2013, 8, e71793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capitanio, D.; Vasso, M.; Ratti, A.; Grignaschi, G.; Volta, M.; Moriggi, M.; Daleno, C.; Bendotti, C.; Silani, V.; Gelfi, C. Molecular signatures of amyotrophic lateral sclerosis disease progression in hind and forelimb muscles of an SOD1(G93A) mouse model. Antioxid Redox Signal. 2012, 17, 1333–1350. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Fang, H.; Groom, L.; Cheng, A.; Zhang, W.; Liu, J.; Wang, X.; Li, K.; Han, P.; Zheng, M.; et al. Superoxide flashes in single mitochondria. Cell 2008, 134, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Fang, H.; Chen, M.; Ding, Y.; Shang, W.; Xu, J.; Zhang, X.; Zhang, W.; Li, K.; Xiao, Y.; Gao, F.; et al. Imaging superoxide flash and metabolism-coupled mitochondrial permeability transition in living animals. Cell Res. 2011, 21, 1295–1304. [Google Scholar] [CrossRef]
- Wei, L.; Salahura, G.; Boncompagni, S.; Kasischke, K.A.; Protasi, F.; Sheu, S.S.; Dirksen, R.T. Mitochondrial superoxide flashes: Metabolic biomarkers of skeletal muscle activity and disease. FASEB J. 2011, 25, 3068–3078. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Fang, H.; Shang, W.; Xiao, Y.; Sun, T.; Hou, N.; Pan, L.; Sun, X.; Ma, Q.; Zhou, J.; et al. Mitoflash altered by metabolic stress in insulin-resistant skeletal muscle. J. Mol. Med. (Berl) 2015, 93, 1119–1130. [Google Scholar] [CrossRef]
- Batandier, C.; Leverve, X.; Fontaine, E. Opening of the mitochondrial permeability transition pore induces reactive oxygen species production at the level of the respiratory chain complex I. J. Biol. Chem. 2004, 279, 17197–17204. [Google Scholar] [CrossRef] [Green Version]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Li, A.; Yi, J.; Li, X.; Zhou, J. Physiological Ca2+ Transients Versus Pathological Steady-State Ca2+ Elevation, Who Flips the ROS Coin in Skeletal Muscle Mitochondria. Front. Physiol. 2020, 11, 595800. [Google Scholar] [CrossRef]
- Ott, M.; Robertson, J.D.; Gogvadze, V.; Zhivotovsky, B.; Orrenius, S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc. Natl. Acad. Sci. USA 2002, 99, 1259–1263. [Google Scholar] [CrossRef] [Green Version]
- Turrens, J.F.; Alexandre, A.; Lehninger, A.L. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch. Biochem. Biophys. 1985, 237, 408–414. [Google Scholar] [CrossRef]
- Muller, F.; Crofts, A.R.; Kramer, D.M. Multiple Q-cycle bypass reactions at the Qo site of the cytochrome bc 1 complex. Biochemistry 2002, 41, 7866–7874. [Google Scholar] [CrossRef]
- Muller, F.L.; Roberts, A.G.; Bowman, M.K.; Kramer, D.M. Architecture of the Qo site of the cytochrome bc 1 complex probed by superoxide production. Biochemistry 2003, 42, 6493–6499. [Google Scholar] [CrossRef]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef]
- Basso, E.; Fante, L.; Fowlkes, J.; Petronilli, V.; Forte, M.A.; Bernardi, P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin, D.J. Biol. Chem. 2005, 280, 18558–18561. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, T.; Shimizu, S.; Watanabe, T.; Yamaguchi, O.; Otsu, K.; Yamagata, H.; Inohara, H.; Kubo, T.; Tsujimoto, Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005, 434, 652–658. [Google Scholar] [CrossRef]
- Li, A.; Li, X.; Yi, J.; Ma, J.; Zhou, J. Butyrate Feeding Reverses CypD-Related Mitoflash Phenotypes in Mouse Myofibers. Int. J. Mol. Sci. 2021, 22, 7412. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Dirksen, R.T. Mitochondrial superoxide flashes: From discovery to new controversies. J. Gen. Physiol. 2012, 139, 425–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonora, M.; Giorgi, C.; Pinton, P. Molecular mechanisms and consequences of mitochondrial permeability transition. Nat. Rev. Mol. Cell Biol. 2022, 23, 266–285. [Google Scholar] [CrossRef] [PubMed]
- Hamer, P.W.; McGeachie, J.M.; Davies, M.J.; Grounds, M.D. Evans Blue Dye as an in vivo marker of myofibre damage: Optimising parameters for detecting initial myofibre membrane permeability. J. Anat. 2002, 200, 69–79. [Google Scholar] [CrossRef]
- Wooddell, C.I.; Radley-Crabb, H.G.; Griffin, J.B.; Zhang, G. Myofiber Damage Evaluation by Evans Blue Dye Injection. Curr. Protoc. Mouse Biol. 2011, 1, 463–488. [Google Scholar] [CrossRef]
- Matsuda, R.; Nishikawa, A.; Tanaka, H. Visualization of dystrophic muscle fibers in mdx mouse by vital staining with Evans blue: Evidence of apoptosis in dystrophin-deficient muscle. J. Biochem. 1995, 118, 959–964. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.; Masumiya, H.; Weisleder, N.; Matsuda, N.; Nishi, M.; Hwang, M.; Ko, J.K.; Lin, P.; Thornton, A.; Zhao, X.; et al. MG53 nucleates assembly of cell membrane repair machinery. Nat. Cell Biol. 2009, 11, 56–64. [Google Scholar] [CrossRef] [Green Version]
- Yi, J.; Li, A.; Li, X.; Park, K.; Zhou, X.; Yi, F.; Xiao, Y.; Yoon, D.; Tan, T.; Ostrow, L.W.; et al. MG53 Preserves Neuromuscular Junction Integrity and Alleviates ALS Disease Progression. Antioxidants 2021, 10, 1522. [Google Scholar] [CrossRef]
- McNeil, P.L.; Miyake, K.; Vogel, S.S. The endomembrane requirement for cell surface repair. Proc. Natl. Acad. Sci. USA 2003, 100, 4592–4597. [Google Scholar] [CrossRef] [Green Version]
- Fucile, S. Ca2+ permeability of nicotinic acetylcholine receptors. Cell Calcium 2004, 35, 1–8. [Google Scholar] [CrossRef]
- Dobrowolny, G.; Giacinti, C.; Pelosi, L.; Nicoletti, C.; Winn, N.; Barberi, L.; Molinaro, M.; Rosenthal, N.; Musaro, A. Muscle expression of a local Igf-1 isoform protects motor neurons in an ALS mouse model. J. Cell Biol. 2005, 168, 193–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cejas, P.; Casado, E.; Belda-Iniesta, C.; De Castro, J.; Espinosa, E.; Redondo, A.; Sereno, M.; Garcia-Cabezas, M.A.; Vara, J.A.; Dominguez-Caceres, A.; et al. Implications of oxidative stress and cell membrane lipid peroxidation in human cancer (Spain). Cancer Causes Control 2004, 15, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Ozato, K.; Shin, D.M.; Chang, T.H.; Morse, H.C. TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 2008, 8, 849–860. [Google Scholar] [CrossRef] [Green Version]
- Weisleder, N.; Takeshima, H.; Ma, J. Immuno-proteomic approach to excitation–contraction coupling in skeletal and cardiac muscle: Molecular insights revealed by the mitsugumins. Cell Calcium 2008, 43, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.; Weisleder, N.; Ko, J.K.; Komazaki, S.; Sunada, Y.; Nishi, M.; Takeshima, H.; Ma, J. Membrane repair defects in muscular dystrophy are linked to altered interaction between MG53, caveolin-3, and dysferlin. J. Biol. Chem. 2009, 284, 15894–15902. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.; Zhu, H.; Cai, C.; Wang, X.; Cao, C.; Xiao, R.; Pan, Z.; Weisleder, N.; Takeshima, H.; Ma, J. Nonmuscle myosin IIA facilitates vesicle trafficking for MG53-mediated. cell membrane repair. FASEB J. 2012, 26, 1875–1883. [Google Scholar] [CrossRef]
- Zhu, H.; Lin, P.; De, G.; Choi, K.H.; Takeshima, H.; Weisleder, N.; Ma, J. Polymerase transcriptase release factor (PTRF) anchors MG53 protein to cell injury site for initiation of membrane repair. J. Biol. Chem. 2011, 286, 12820–12824. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.; Masumiya, H.; Weisleder, N.; Pan, Z.; Nishi, M.; Komazaki, S.; Takeshima, H.; Ma, J. MG53 regulates membrane budding and exocytosis in muscle cells. J. Biol. Chem. 2009, 284, 3314–3322. [Google Scholar] [CrossRef] [Green Version]
- Cao, C.M.; Zhang, Y.; Weisleder, N.; Ferrante, C.; Wang, X.; Lv, F.; Zhang, Y.; Song, R.; Hwang, M.; Jin, L.; et al. MG53 constitutes a primary determinant of cardiac ischemic preconditioning. Circulation 2010, 121, 2565–2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, Z.; Wang, Q.; Zhou, X.; Tan, T.; Park, K.H.; Kramer, H.F.; McDougal, A.; Laping, N.J.; Kumar, S.; Adesanya, T.M.A.; et al. Sustained elevation of MG53 in the bloodstream increases tissue regenerative capacity without compromising metabolic function. Nat. Commun. 2019, 10, 4659. [Google Scholar] [CrossRef] [PubMed]
- Shefner, J.M. Effects of Strength Training in Amyotrophic Lateral Sclerosis: How Much Do We Know? Muscle Nerve 2019, 59, 6–7. [Google Scholar] [CrossRef] [Green Version]
- Tsitkanou, S.; Della Gatta, P.; Foletta, V.; Russell, A. The Role of Exercise as a Non-pharmacological Therapeutic Approach for Amyotrophic Lateral Sclerosis: Beneficial or Detrimental? Front. Neurol. 2019, 10, 783. [Google Scholar] [CrossRef] [Green Version]
- Rosenbohm, A.; Peter, R.; Dorst, J.; Kassubek, J.; Rothenbacher, D.; Nagel, G.; Ludolph, A.C.; Group, A.R.S.S. Life Course of Physical Activity and Risk and Prognosis of Amyotrophic Lateral Sclerosis in a German ALS Registry. Neurology 2021, 97, e1955–e1963. [Google Scholar] [CrossRef]
- Julian, T.H.; Glascow, N.; Barry, A.D.F.; Moll, T.; Harvey, C.; Klimentidis, Y.C.; Newell, M.; Zhang, S.; Snyder, M.P.; Cooper-Knock, J. Physical exercise is a risk factor for amyotrophic lateral sclerosis: Convergent evidence from Mendelian randomisation, transcriptomics and risk genotypes. EBioMedicine 2021, 68, 103397. [Google Scholar] [CrossRef]
- Raymond, J.; Mehta, P.; Larson, T.; Factor-Litvak, P.; Davis, B.; Horton, K. History of vigorous leisure-time physical activity and early onset amyotrophic lateral sclerosis (ALS), data from the national ALS registry: 2010–2018. Amyotroph. Lateral Scler. Front. Degener. 2021, 22, 535–544. [Google Scholar] [CrossRef]
- Harwood, C.A.; Westgate, K.; Gunstone, S.; Brage, S.; Wareham, N.J.; McDermott, C.J.; Shaw, P.J. Long-term physical activity: An exogenous risk factor for sporadic amyotrophic lateral sclerosis? Amyotroph. Lateral Scler. Front. Degener. 2016, 17, 377–384. [Google Scholar] [CrossRef] [Green Version]
- Di, P.W.C.; Di, P.S.G.C. Safety and efficacy of diaphragm pacing in patients with respiratory insufficiency due to amyotrophic lateral sclerosis (DiPALS): A multicentre, open-label, randomised controlled trial. Lancet Neurol. 2015, 14, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Bermejo, J.; Morelot-Panzini, C.; Tanguy, M.L.; Meininger, V.; Pradat, P.F.; Lenglet, T.; Bruneteau, G.; Forestier, N.L.; Couratier, P.; Guy, N.; et al. Early diaphragm pacing in patients with amyotrophic lateral sclerosis (RespiStimALS): A randomised controlled triple-blind trial. Lancet Neurol. 2016, 15, 1217–1227. [Google Scholar] [CrossRef]
- McDermott, C.J.; Bradburn, M.J.; Maguire, C.; Cooper, C.L.; Baird, W.O.; Baxter, S.K.; Cohen, J.; Cantrill, H.; Dixon, S.; Ackroyd, R.; et al. DiPALS: Diaphragm Pacing in patients with Amyotrophic Lateral Sclerosis - a randomised controlled trial. Health Technol. Assess 2016, 20, 1–186. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.G.; Lewis, R.A. Diaphragm pacing in patients with amyotrophic lateral sclerosis. Lancet Neurol. 2016, 15, 542. [Google Scholar] [CrossRef]
- Wood, H. Motor neuron disease: Diaphragm pacing is associated with reduced survival in ALS patients with respiratory insufficiency. Nat. Rev. Neurol. 2015, 11, 484. [Google Scholar] [CrossRef]
- Jablonka, S.; Holtmann, B.; Sendtner, M.; Metzger, F. Therapeutic effects of PEGylated insulin-like growth factor I in the pmn mouse model of motoneuron disease. Exp. Neurol. 2011, 232, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Webster, R.; Didier, E.; Harris, P.; Siegel, N.; Stadler, J.; Tilbury, L.; Smith, D. PEGylated proteins: Evaluation of their safety in the absence of definitive metabolism studies. Drug Metab. Dispos 2007, 35, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisleder, N.; Takizawa, N.; Lin, P.; Wang, X.; Cao, C.; Zhang, Y.; Tan, T.; Ferrante, C.; Zhu, H.; Chen, P.J.; et al. Recombinant MG53 protein modulates therapeutic cell membrane repair in treatment of muscular dystrophy. Sci. Transl. Med. 2012, 4, 139ra185. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Hou, J.; Roe, J.L.; Park, K.H.; Tan, T.; Zheng, Y.; Li, L.; Zhang, C.; Liu, J.; Liu, Z.; et al. Amelioration of ischemia-reperfusion-induced muscle injury by the recombinant human MG53 protein. Muscle Nerve 2015, 52, 852–858. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Liu, J.; Bian, Z.; Cui, Y.; Zhou, X.; Zhou, X.; Zhang, B.; Adesanya, T.M.; Yi, F.; Park, K.H.; et al. Effect of metabolic syndrome on mitsugumin 53 expression and function. PLoS ONE 2015, 10, e0124128. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Bian, Z.; Jiang, Q.; Wang, X.; Zhou, X.; Park, K.H.; Hsueh, W.; Whitson, B.A.; Haggard, E.; Li, H.; et al. MG53 does not manifest the development of diabetes in db/db mice. Diabetes 2020, 69, 1052–1064. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, X.; Ong, H.; Tan, T.; Park, K.H.; Bian, Z.; Zou, X.; Haggard, E.; Janssen, P.M.; Merritt, R.E.; et al. MG53 suppresses NF-kappaB activation to mitigate age-related heart failure. JCI Insight 2021, 6, e148375. [Google Scholar] [CrossRef]
- Jia, Y.; Chen, K.; Lin, P.; Lieber, G.; Nishi, M.; Yan, R.; Wang, Z.; Yao, Y.; Li, Y.; Whitson, B.A.; et al. Treatment of acute lung injury by targeting MG53-mediated cell membrane repair. Nat. Commun. 2014, 5, 4387. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Zhu, H.; Zheng, Y.; Xu, Z.; Li, L.; Tan, T.; Park, K.H.; Hou, J.; Zhang, C.; Li, D.; et al. Cardioprotection of recombinant human MG53 protein in a porcine model of ischemia and reperfusion injury. J. Mol. Cell Cardiol. 2015, 80, 10–19. [Google Scholar] [CrossRef]
- Duann, P.; Li, H.; Lin, P.; Tan, T.; Wang, Z.; Chen, K.; Zhou, X.; Gumpper, K.; Zhu, H.; Ludwig, T.; et al. MG53-mediated cell membrane repair protects against acute kidney injury. Sci. Transl. Med. 2015, 7, 279ra236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.; Zhang, B.; Zhu, H.; Li, H.; Han, Y.; Chen, K.; Wang, Z.; Zeng, J.; Liu, Y.; Wang, X.; et al. MG53 permeates through blood-brain barrier to protect ischemic brain injury. Oncotarget 2016, 7, 22474–22485. [Google Scholar] [CrossRef] [PubMed]
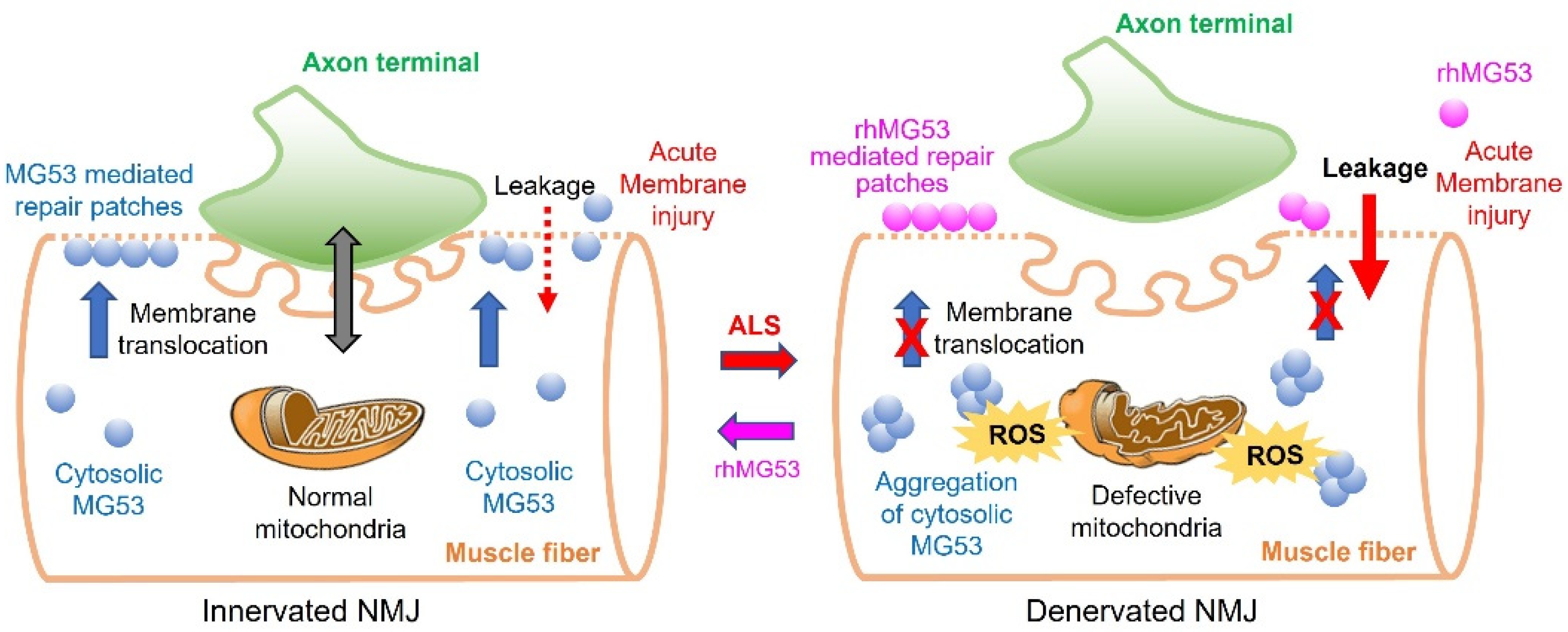
Figure 1.
Proposed mechanisms of membrane repair defects at NMJ in ALS and the restoration by exogenous rhMG53. NMJ is the critical site of neuromuscular interactions. During ALS progression, abnormalities in mitochondrial respiratory activities at NMJ elevates ROS production, leading to ectopic aggregation of cytosolic MG53, undermining its membrane repair function, which exacerbates sarcolemma disruption leading to accumulation of extracellular content in the cytosol. Exogenously applied rhMG53 could be recruited to the sites of membrane injury, forming sealing patches to alleviate the cell membrane leakage.
Figure 1.
Proposed mechanisms of membrane repair defects at NMJ in ALS and the restoration by exogenous rhMG53. NMJ is the critical site of neuromuscular interactions. During ALS progression, abnormalities in mitochondrial respiratory activities at NMJ elevates ROS production, leading to ectopic aggregation of cytosolic MG53, undermining its membrane repair function, which exacerbates sarcolemma disruption leading to accumulation of extracellular content in the cytosol. Exogenously applied rhMG53 could be recruited to the sites of membrane injury, forming sealing patches to alleviate the cell membrane leakage.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Li, A.; Yi, J.; Li, X.; Dong, L.; Ostrow, L.W.; Ma, J.; Zhou, J. Deficient Sarcolemma Repair in ALS: A Novel Mechanism with Therapeutic Potential. Cells 2022, 11, 3263. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11203263
AMA Style
Li A, Yi J, Li X, Dong L, Ostrow LW, Ma J, Zhou J. Deficient Sarcolemma Repair in ALS: A Novel Mechanism with Therapeutic Potential. Cells. 2022; 11(20):3263. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11203263
Chicago/Turabian StyleLi, Ang, Jianxun Yi, Xuejun Li, Li Dong, Lyle W. Ostrow, Jianjie Ma, and Jingsong Zhou. 2022. "Deficient Sarcolemma Repair in ALS: A Novel Mechanism with Therapeutic Potential" Cells 11, no. 20: 3263. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11203263
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.