
Cytochrome c Oxidase Activity as a Metabolic Regulator in Pancreatic Beta-Cells

 , ,
, ,
Abstract
:1. Introduction
2. Materials, Methods and Research Design
2.1. Animals and Diets
2.2. Study Design
2.3. Oral Glucose Tolerance (OGTT) Testing
2.4. Islets Isolation
2.5. PBMCs Isolation
2.6. Assessment of the Mitochondrial Enzymatic Activity
2.7. Data Presentation and Statistical Analysis
3. Results
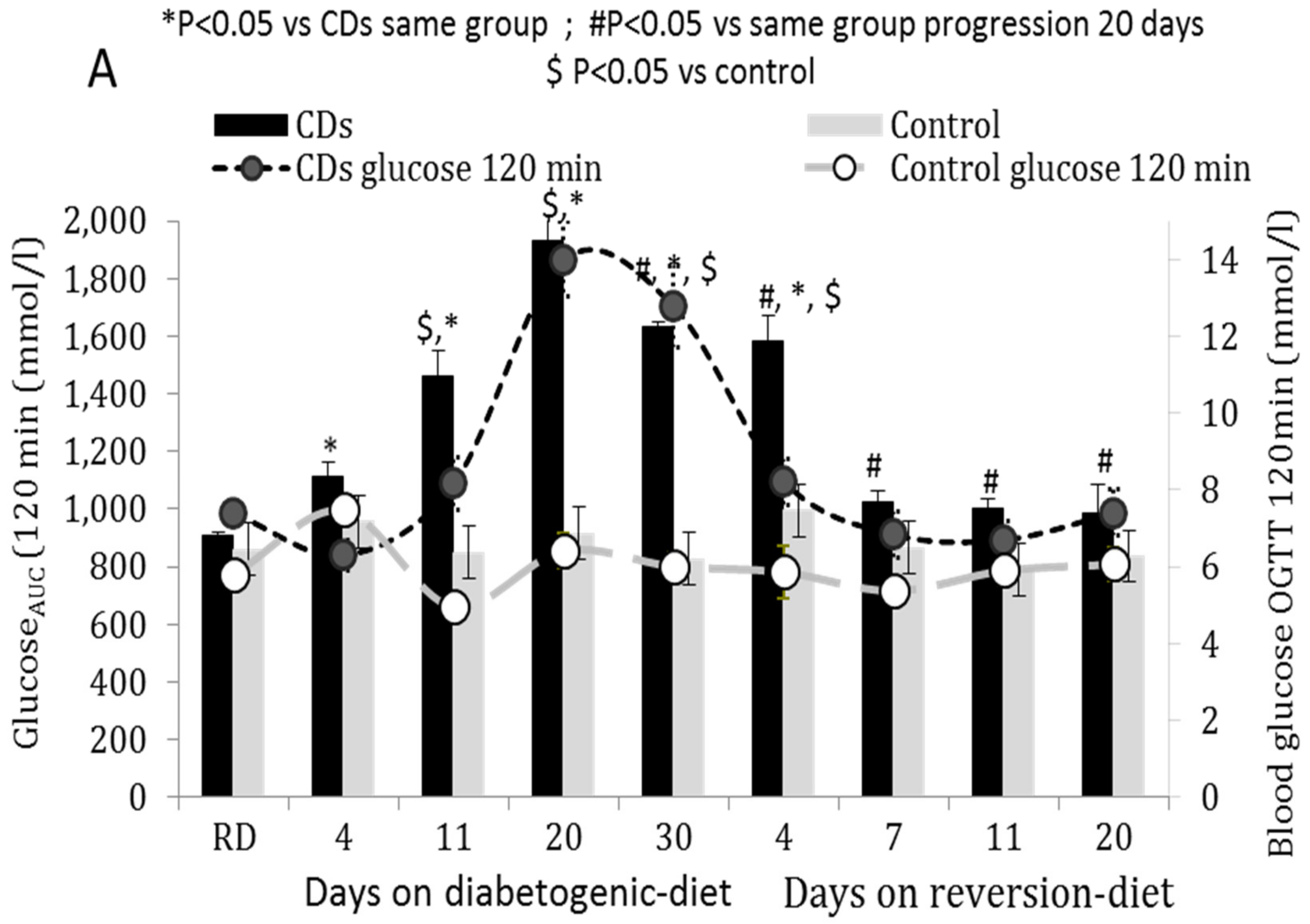
3.1. OGTT of CDs and Control Rats Fed According to the Progression or Reversion Protocols
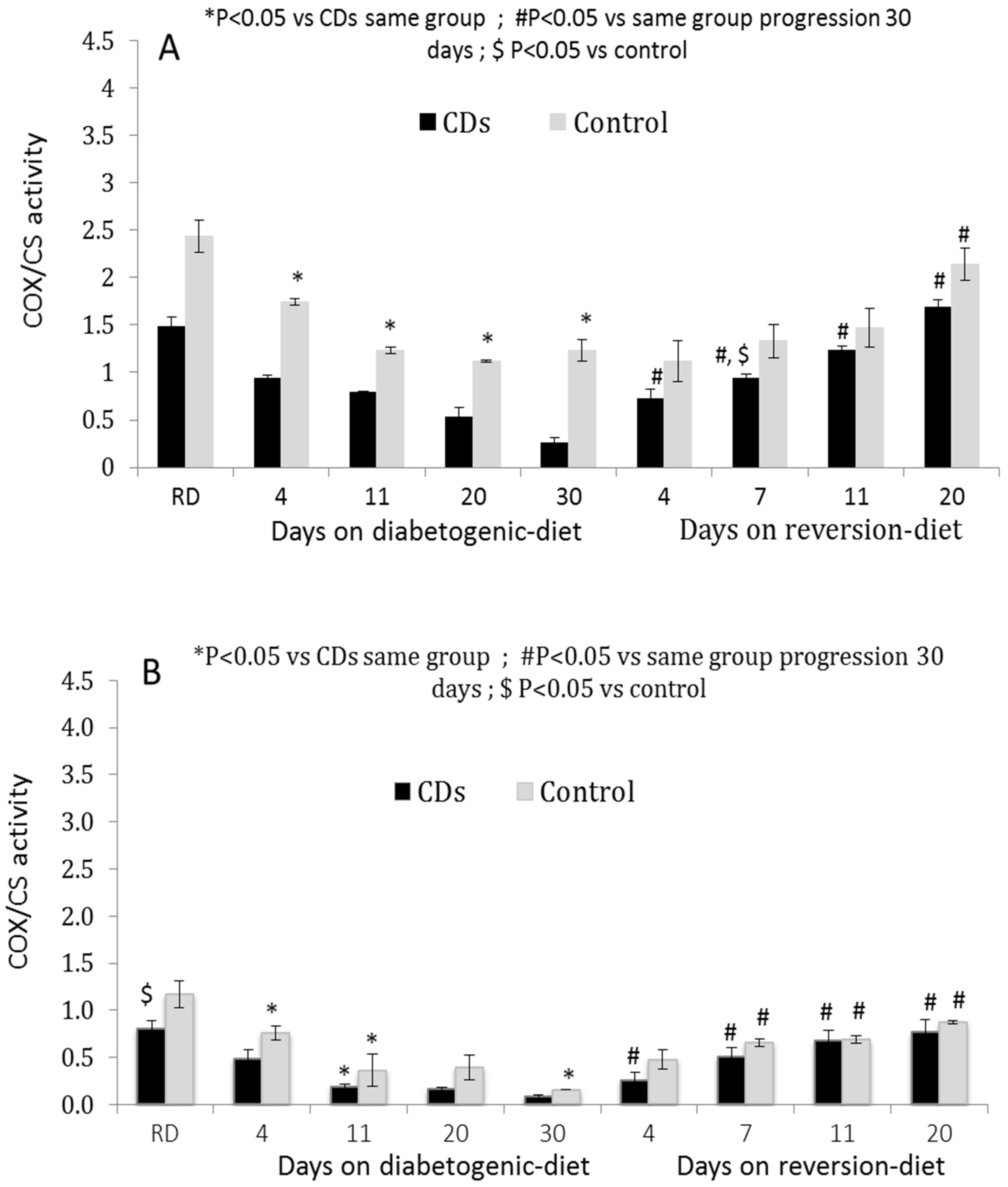
3.2. COX Activity in Isolated Pancreatic Islets and Peripheral PBMCs
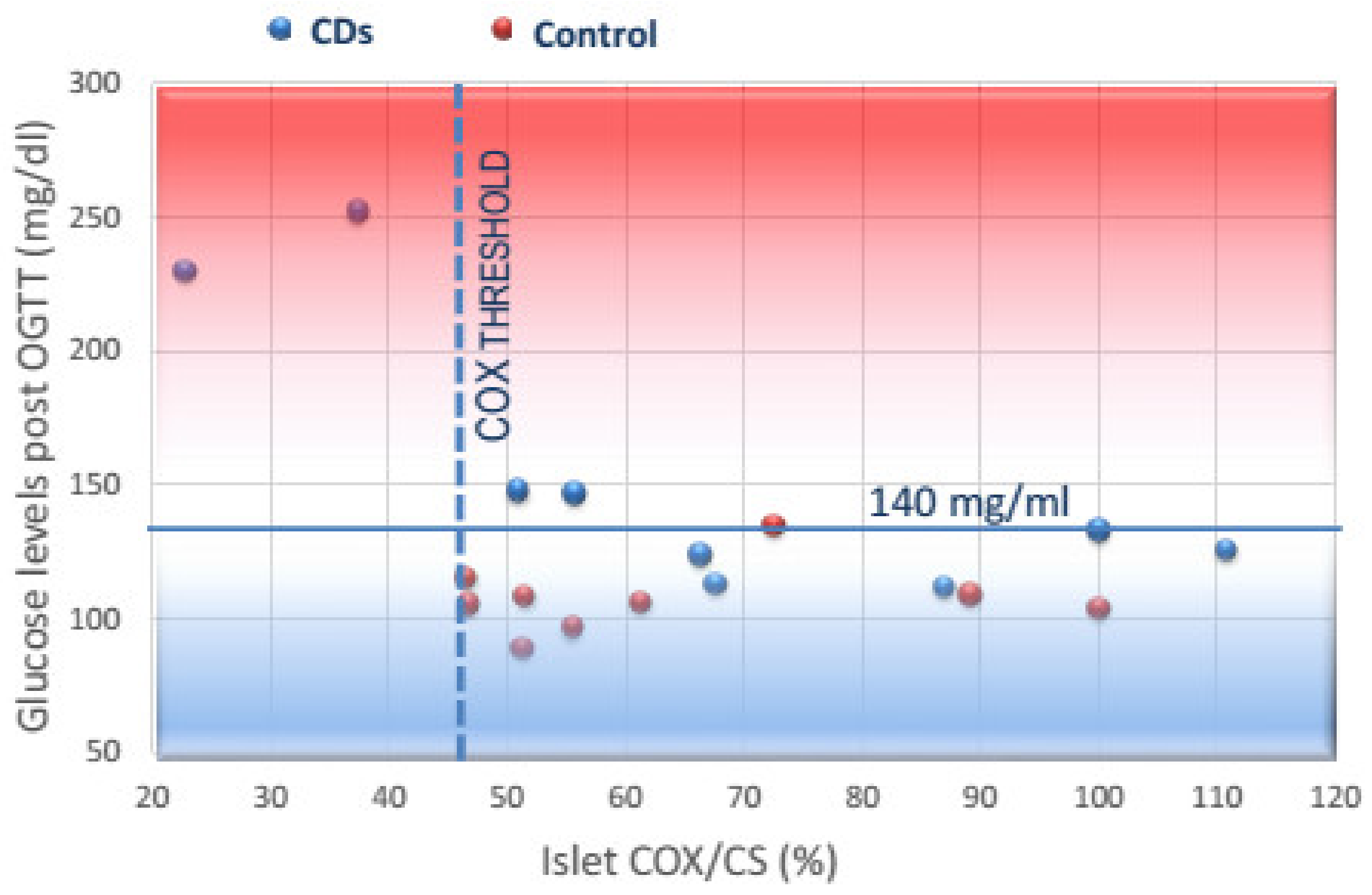
3.3. Correlation of COX Activity in Islets and PBMCs with Post-OGTT Glucose Levels and GSIS
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meier, J.J.; Bonadonna, R.C. Role of reduced β-cell mass versus impaired β-cell function in the pathogenesis of type 2 diabetes. Diabetes Care 2013, 36 (Suppl. 2), S113–S119. [Google Scholar] [CrossRef] [Green Version]
- Parnis, J.; Rutter, G.A. Contributions of Mitochondrial Dysfunction to β Cell Failure in Diabetes Mellitus. In Mitochondria in Obesity and Type 2 Diabetes; Morio, B., Pénicaud, L., Rigoulet, M., Eds.; Elsevier Inc.: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Zhang, E.; Mohammed Al-Amily, I.; Mohammed, S.; Luan, C.; Asplund, O.; Ahmed, M.; Ye, Y.; Ben-Hail, D.; Soni, A.; Vishnu, N.; et al. Preserving Insulin Secretion in Diabetes by Inhibiting VDAC1 Overexpression and Surface Translocation in β Cells. Cell Metab. 2018, 29, 64–77.e6. [Google Scholar] [CrossRef] [PubMed]
- Fex, M.; Nicholas, L.M.; Vishnu, N.; Medina, A.; Sharoyko, V.V.; Nicholls, D.G.; Spegel, P.; Mulder, H. The pathogenetic role of β-cell mitochondria in type 2 diabetes. J. Endocrinol. 2018, 236, R145–R159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholls, D.G. The Pancreatic β-Cell: A Bioenergetic Perspective. Physiol. Rev. 2016, 96, 1385–1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timon-Gomez, A.; Bartley-Dier, E.L.; Fontanesi, F.; Barrientos, A. HIGD-Driven Regulation of Cytochrome c Oxidase Biogenesis and Function. Cells 2020, 9, 2620. [Google Scholar] [CrossRef]
- Timon-Gomez, A.; Nyvltova, E.; Abriata, L.A.; Vila, A.J.; Hosler, J.; Barrientos, A. Mitochondrial cytochrome c oxidase biogenesis: Recent developments. Semin. Cell Dev. Biol. 2018, 76, 163–178. [Google Scholar] [CrossRef]
- Anello, M.; Lupi, R.; Spampinato, D.; Piro, S.; Masini, M.; Boggi, U.; Del Prato, S.; Rabuazzo, A.M.; Purrello, F.; Marchetti, P. Functional and morphological alterations of mitochondria in pancreatic beta cells from type 2 diabetic patients. Diabetologia 2005, 48, 282–289. [Google Scholar] [CrossRef] [Green Version]
- Del Guerra, S.; Lupi, R.; Marselli, L.; Masini, M.; Bugliani, M.; Sbrana, S.; Torri, S.; Pollera, M.; Boggi, U.; Mosca, F.; et al. Functional and molecular defects of pancreatic islets in human type 2 diabetes. Diabetes 2005, 54, 727–735. [Google Scholar] [CrossRef] [Green Version]
- Deng, S.; Vatamaniuk, M.; Huang, X.; Doliba, N.; Lian, M.M.; Frank, A.; Velidedeoglu, E.; Desai, N.M.; Koeberlein, B.; Wolf, B.; et al. Structural and functional abnormalities in the islets isolated from type 2 diabetic subjects. Diabetes 2004, 53, 624–632. [Google Scholar] [CrossRef] [Green Version]
- Pinti, M.V.; Fink, G.K.; Hathaway, Q.A.; Durr, A.J.; Kunovac, A.; Hollander, J.M. Mitochondrial dysfunction in type 2 diabetes mellitus: An organ-based analysis. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E268–E285. [Google Scholar] [CrossRef]
- Prasun, P. Role of mitochondria in pathogenesis of type 2 diabetes mellitus. J. Diabetes Metab. Disord. 2020, 19, 2017–2022. [Google Scholar] [CrossRef]
- Olsson, A.H.; Ronn, T.; Ladenvall, C.; Parikh, H.; Isomaa, B.; Groop, L.; Ling, C. Two common genetic variants near nuclear-encoded OXPHOS genes are associated with insulin secretion in vivo. Eur. J. Endocrinol. 2011, 164, 765–771. [Google Scholar] [CrossRef]
- Olsson, A.H.; Yang, B.T.; Hall, E.; Taneera, J.; Salehi, A.; Nitert, M.D.; Ling, C. Decreased expression of genes involved in oxidative phosphorylation in human pancreatic islets from patients with type 2 diabetes. Eur. J. Endocrinol. 2011, 165, 589–595. [Google Scholar] [CrossRef]
- Silva, J.P.; Kohler, M.; Graff, C.; Oldfors, A.; Magnuson, M.A.; Berggren, P.O.; Larsson, N.G. Impaired insulin secretion and β-cell loss in tissue-specific knockout mice with mitochondrial diabetes. Nat. Genet. 2000, 26, 336–340. [Google Scholar] [CrossRef]
- Antoun, G.; McMurray, F.; Thrush, A.B.; Patten, D.A.; Peixoto, A.C.; Slack, R.S.; McPherson, R.; Dent, R.; Harper, M.E. Impaired mitochondrial oxidative phosphorylation and supercomplex assembly in rectus abdominis muscle of diabetic obese individuals. Diabetologia 2015, 58, 2861–2866. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Liu, Y.; Luo, L.; Lu, Y.; Yew, D.T.; Xu, J.; Guo, K. Platelet mitochondrial dysfunction of DM rats and DM patients. Int. J. Clin. Exp. Med. 2015, 8, 6937–6946. [Google Scholar]
- Khan, S.; Raghuram, G.V.; Bhargava, A.; Pathak, N.; Chandra, D.H.; Jain, S.K.; Mishra, P.K. Role and clinical significance of lymphocyte mitochondrial dysfunction in type 2 diabetes mellitus. Transl. Res. J. Lab. Clin. Med. 2011, 158, 344–359. [Google Scholar] [CrossRef]
- Weksler-Zangen, S.; Aharon-Hananel, G.; Mantzur, C.; Aouizerat, T.; Gurgul-Convey, E.; Raz, I.; Saada, A. IL-1β hampers glucose-stimulated insulin secretion in Cohen diabetic rat islets through mitochondrial cytochrome c oxidase inhibition by nitric oxide. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E648–E657. [Google Scholar] [CrossRef] [Green Version]
- Weksler-Zangen, S.; Jorns, A.; Tarsi-Chen, L.; Vernea, F.; Aharon-Hananel, G.; Saada, A.; Lenzen, S.; Raz, I. Dietary copper supplementation restores β-cell function of Cohen diabetic rats: A link between mitochondrial function and glucose-stimulated insulin secretion. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E1023–E1034. [Google Scholar] [CrossRef] [Green Version]
- Weksler-Zangen, S.; Raz, I.; Lenzen, S.; Jorns, A.; Ehrenfeld, S.; Amir, G.; Oprescu, A.; Yagil, Y.; Yagil, C.; Zangen, D.H.; et al. Impaired glucose-stimulated insulin secretion is coupled with exocrine pancreatic lesions in the Cohen diabetic rat. Diabetes 2008, 57, 279–287. [Google Scholar] [CrossRef] [Green Version]
- Weksler-Zangen, S.; Yagil, C.; Zangen, D.H.; Ornoy, A.; Jacob, H.J.; Yagil, Y. The newly inbred cohen diabetic rat: A nonobese normolipidemic genetic model of diet-induced type 2 diabetes expressing sex differences. Diabetes 2001, 50, 2521–2529. [Google Scholar] [CrossRef] [Green Version]
- Negari, S.B.; Aouizerat, T.; Tenenbaum, A.; Cohen-Cymberknoh, M.; Shoseyov, D.; Kerem, E.; Saada, A. Mitochondrial OXPHOS function is unaffected by chronic azithromycin treatment. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 2013, 12, 682–687. [Google Scholar] [CrossRef] [Green Version]
- Saada, A.; Bar-Meir, M.; Belaiche, C.; Miller, C.; Elpeleg, O. Evaluation of enzymatic assays and compounds affecting ATP production in mitochondrial respiratory chain complex I deficiency. Anal. Biochem. 2004, 335, 66–72. [Google Scholar] [CrossRef]
- Kahn, S.E. The importance of the β-cell in the pathogenesis of type 2 diabetes mellitus. Am. J. Med. 2000, 108 (Suppl. 6), 2S–8S. [Google Scholar] [CrossRef]
- Maechler, P.; Wollheim, C.B. Mitochondrial function in normal and diabetic β-cells. Nature 2001, 414, 807–812. [Google Scholar] [CrossRef]
- Sha, W.; Hu, F.; Bu, S. Mitochondrial dysfunction and pancreatic islet β-cell failure (Review). Exp. Ther. Med. 2020, 20, 266. [Google Scholar] [CrossRef]
- Friday, D.P.; Alleyneb, T.A.; Ignaciob, D.N.; Arrindellb, D.; Raoa, S.R.; Legall, G. The Impact of Diabetes Mellitus on Oxygen Utilization by Complex IV: Preliminary Insights. J. Endocrinol. Metab. 2017, 7, 18–24. [Google Scholar] [CrossRef] [Green Version]
- Van der Schueren, B.; Vangoitsenhoven, R.; Geeraert, B.; De Keyzer, D.; Hulsmans, M.; Lannoo, M.; Huber, H.J.; Mathieu, C.; Holvoet, P. Low cytochrome oxidase 4I1 links mitochondrial dysfunction to obesity and type 2 diabetes in humans and mice. Int. J. Obes. 2015, 39, 1254–1263. [Google Scholar] [CrossRef]
- Dabravolski, S.A.; Orekhova, V.A.; Baig, M.S.; Bezsonov, E.E.; Starodubova, A.V.; Popkova, T.V.; Orekhov, A.N. The Role of Mitochondrial Mutations and Chronic Inflammation in Diabetes. Int. J. Mol. Sci. 2021, 22, 6733. [Google Scholar] [CrossRef]
- Rutter, G.A.; Georgiadou, E.; Martinez-Sanchez, A.; Pullen, T.J. Metabolic and functional specialisations of the pancreatic beta cell: Gene disallowance, mitochondrial metabolism and intercellular connectivity. Diabetologia 2020, 63, 1990–1998. [Google Scholar] [CrossRef]
- Haythorne, E.; Rohm, M.; van de Bunt, M.; Brereton, M.F.; Tarasov, A.I.; Blacker, T.S.; Sachse, G.; Silva Dos Santos, M.; Terron Exposito, R.; Davis, S.; et al. Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic β-cells. Nat. Commun. 2019, 10, 2474. [Google Scholar] [CrossRef]
- Nicholas, L.M.; Valtat, B.; Medina, A.; Andersson, L.; Abels, M.; Mollet, I.G.; Jain, D.; Eliasson, L.; Wierup, N.; Fex, M.; et al. Mitochondrial transcription factor B2 is essential for mitochondrial and cellular function in pancreatic β-cells. Mol. Metab. 2017, 6, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Mulder, H. Transcribing β-cell mitochondria in health and disease. Mol. Metab. 2017, 6, 1040–1051. [Google Scholar] [CrossRef] [PubMed]
- Chow, J.; Rahman, J.; Achermann, J.C.; Dattani, M.T.; Rahman, S. Mitochondrial disease and endocrine dysfunction. Nat. Rev. Endocrinol. 2017, 13, 92–104. [Google Scholar] [CrossRef] [PubMed]
- De Andrade, P.B.; Rubi, B.; Frigerio, F.; van den Ouweland, J.M.; Maassen, J.A.; Maechler, P. Diabetes-associated mitochondrial DNA mutation A3243G impairs cellular metabolic pathways necessary for beta cell function. Diabetologia 2006, 49, 1816–1826. [Google Scholar] [CrossRef] [PubMed]
- Gerbitz, K.D.; van den Ouweland, J.M.; Maassen, J.A.; Jaksch, M. Mitochondrial diabetes mellitus: A review. Biochim. Biophys. Acta 1995, 1271, 253–260. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Nakanishi, K.; Nakase, H.; Kajio, H.; Okubo, M.; Murase, T.; Kosaka, K. In situ characterization of islets in diabetes with a mitochondrial DNA mutation at nucleotide position 3243. Diabetes 1997, 46, 1567–1571. [Google Scholar] [CrossRef]
- Maassen, J.A.; Kadowaki, T. Maternally inherited diabetes and deafness: A new diabetes subtype. Diabetologia 1996, 39, 375–382. [Google Scholar] [CrossRef]
- Velho, G.; Byrne, M.M.; Clement, K.; Sturis, J.; Pueyo, M.E.; Blanche, H.; Vionnet, N.; Fiet, J.; Passa, P.; Robert, J.J.; et al. Clinical phenotypes, insulin secretion, and insulin sensitivity in kindreds with maternally inherited diabetes and deafness due to mitochondrial tRNALeu (UUR) gene mutation. Diabetes 1996, 45, 478–487. [Google Scholar] [CrossRef]
- Lu, H.; Koshkin, V.; Allister, E.M.; Gyulkhandanyan, A.V.; Wheeler, M.B. Molecular and metabolic evidence for mitochondrial defects associated with β-cell dysfunction in a mouse model of type 2 diabetes. Diabetes 2010, 59, 448–459. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.G.; Tassava, T.M.; Romsos, D.R. Threshold for glucose-stimulated insulin secretion in pancreatic islets of genetically obese (ob/ob) mice is abnormally low. J. Nutr. 1993, 123, 1567–1574. [Google Scholar] [CrossRef] [Green Version]
- Leavesley, H.B.; Li, L.; Prabhakaran, K.; Borowitz, J.L.; Isom, G.E. Interaction of cyanide and nitric oxide with cytochrome c oxidase: Implications for acute cyanide toxicity. Toxicol. Sci. Off. J. Soc. Toxicol. 2008, 101, 101–111. [Google Scholar] [CrossRef] [Green Version]
- Letellier, T.; Heinrich, R.; Malgat, M.; Mazat, J.P. The kinetic basis of threshold effects observed in mitochondrial diseases: A systemic approach. Biochem. J. 1994, 302 Pt 1, 171–174. [Google Scholar] [CrossRef]
- Luni, C.; Marth, J.D.; Doyle, F.J., 3rd. Computational modeling of glucose transport in pancreatic β-cells identifies metabolic thresholds and therapeutic targets in diabetes. PLoS ONE 2012, 7, e53130. [Google Scholar] [CrossRef] [Green Version]
- Rossignol, R.; Faustin, B.; Rocher, C.; Malgat, M.; Mazat, J.P.; Letellier, T. Mitochondrial threshold effects. Biochem. J. 2003, 370, 751–762. [Google Scholar] [CrossRef] [Green Version]
- Qin, Q.; Jin, J.; He, F.; Zheng, Y.; Li, T.; Zhang, Y.; He, J. Humanin promotes mitochondrial biogenesis in pancreatic MIN6 β-cells. Biochem. Biophys. Res. Commun. 2018, 497, 292–297. [Google Scholar] [CrossRef]
- Brun, T.; Jimenez-Sanchez, C.; Madsen, J.G.S.; Hadadi, N.; Duhamel, D.; Bartley, C.; Oberhauser, L.; Trajkovski, M.; Mandrup, S.; Maechler, P. AMPK Profiling in Rodent and Human Pancreatic Beta-Cells under Nutrient-Rich Metabolic Stress. Int. J. Mol. Sci. 2020, 21, 3982. [Google Scholar] [CrossRef]
- Viscomi, C.; Bottani, E.; Civiletto, G.; Cerutti, R.; Moggio, M.; Fagiolari, G.; Schon, E.A.; Lamperti, C.; Zeviani, M. In vivo correction of COX deficiency by activation of the AMPK/PGC-1α axis. Cell Metab. 2011, 14, 80–90. [Google Scholar] [CrossRef] [Green Version]
- Alfatni, A.; Riou, M.; Charles, A.L.; Meyer, A.; Barnig, C.; Andres, E.; Lejay, A.; Talha, S.; Geny, B. Peripheral Blood Mononuclear Cells and Platelets Mitochondrial Dysfunction, Oxidative Stress, and Circulating mtDNA in Cardiovascular Diseases. J. Clin. Med. 2020, 9, 311. [Google Scholar] [CrossRef] [Green Version]
- Hargreaves, I.; Mody, N.; Land, J.; Heales, S. Blood Mononuclear Cell Mitochondrial Respiratory Chain Complex IV Activity Is Decreased in Multiple Sclerosis Patients: Effects of β-Interferon Treatment. J. Clin. Med. 2018, 7, 36. [Google Scholar] [CrossRef] [Green Version]
- Arnold, S. The power of life—Cytochrome c oxidase takes center stage in metabolic control, cell signalling and survival. Mitochondrion 2012, 12, 46–56. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Period on the Diet (days) | # of Rats | |
|---|---|---|
| Diets | ||
| Regular diet (RD) | 30 | 40 |
| Diabetogenic High Sucrose diet (HSD) | 4, 11, 20, 30 | 5–8 rats/period on the diet |
| Reversion diet | 4, 7, 11, 20 | 5–8 rats/period on the diet |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aharon-Hananel, G.; Romero-Afrima, L.; Saada, A.; Mantzur, C.; Raz, I.; Weksler-Zangen, S. Cytochrome c Oxidase Activity as a Metabolic Regulator in Pancreatic Beta-Cells. Cells 2022, 11, 929. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11060929
Aharon-Hananel G, Romero-Afrima L, Saada A, Mantzur C, Raz I, Weksler-Zangen S. Cytochrome c Oxidase Activity as a Metabolic Regulator in Pancreatic Beta-Cells. Cells. 2022; 11(6):929. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11060929
Chicago/Turabian StyleAharon-Hananel, Genya, Leonor Romero-Afrima, Ann Saada, Carmit Mantzur, Itamar Raz, and Sarah Weksler-Zangen. 2022. "Cytochrome c Oxidase Activity as a Metabolic Regulator in Pancreatic Beta-Cells" Cells 11, no. 6: 929. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11060929