The Tumor Vessel Targeting Strategy: A Double-Edged Sword in Tumor Metastasis
by
Xiaobo Li
1,2,†, 
Yong Li
1,2,†,
Weijin Lu
1,2,
Minfeng Chen
1,2,
Wencai Ye
1,2 and
Dongmei Zhang
1,2,* 1
College of Pharmacy, Jinan University, No. 601, Huangpu Road West, Guangzhou 510632, China
2
Guangdong Province Key Laboratory of Pharmacodynamic Constituents of Traditional Chinese Medicine and New Drugs Research, Jinan University, Guangzhou 510632, China
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Cells 2019, 8(12), 1602; https://0-doi-org.brum.beds.ac.uk/10.3390/cells8121602
Submission received: 25 October 2019
/
Revised: 3 December 2019
/
Accepted: 5 December 2019
/
Published: 10 December 2019
(This article belongs to the Special Issue Angiogenesis in Cancer)
Abstract
:Tumor vessels provide essential paths for tumor cells to escape from the primary tumor and form metastatic foci in distant organs. The vessel targeting strategy has been widely used as an important clinical cancer chemotherapeutic strategy for patients with metastatic tumors. Our review introduces the contribution of angiogenesis to tumor metastasis and summarizes the application of Food and Drug Administration (FDA)-approved vessel targeting drugs for metastatic tumors. We recommend the application and mechanisms of vascular targeting drugs for inhibiting tumor metastasis and discuss the risk and corresponding countermeasures after vessel targeting treatment.
1. Introduction
Tumor metastasis is the main cause of cancer-induced death. More than 90% of mortality from cancer is due to metastasis [1]. Tumor vessels are necessary for tumor growth and metastasis. Irregular and tortuous tumor vessels have low pericyte coverage with high permeability, providing favorable conditions for tumor metastasis. Cancer cell dissemination via blood vessels (hematogenous spread) plays a vital role in the metastasis cascade [2], which predominantly involves local invasion, intravasation, circulation, extravasation, micrometastasis formation and metastatic colonization [3].
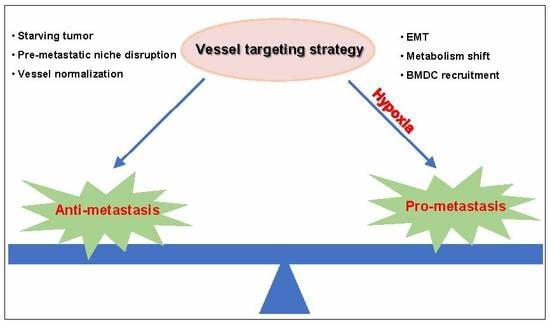
Based on the importance of blood vessels for tumor progression, vessel targeting strategies have been developed. The Food and Drug Administration (FDA) has approved angiogenic inhibitors, including bevacizumab, ramucirumab, sunitinib, sorafenib, regorafenib, vandetanib, cabozantinib, lenvatinib and aflibercept, for metastatic tumors. These agents block angiogenic signaling pathways and blood supply to metastatic tumor cells. The vessel targeting strategy affects the pre-metastatic niche, which suppresses angiogenesis and the inflammatory response, leading to reduced metastatic foci formation [4,5]. The vessel targeting strategy also leads to vessel normalization with decreased interstitial fluid pressure (IFP) and enhanced vessel perfusion, which inhibits disseminated cells’ intravasation [6].
However, it remains controversial in the application of vessel targeting therapy. Many investigations have shown that vessel targeting treatment leads to tumor metastasis. The disrupted endothelial cell barrier facilitates tumor cell extravasation. Hypoxia induced by vessel targeting treatment contributes to tumor cell epithelial to mesenchymal transition (EMT) and metabolism shift, directly leading to tumor cell invasion and metastasis [7,8]. In addition, hypoxia induces bone marrow derived cell (BMDC) recruitment, which indirectly facilitates tumor cell metastasis by inducing angiogenesis and producing chemokines that recruit metastatic cells to distant organs [9].
In this review, we provide an overview of angiogenesis in tumor metastasis and summarize the application of a vessel targeting strategy to metastatic cancer. We introduce the anti-metastatic effects of the vessel targeting strategy in cancer patients and the underlying mechanisms. We also analyze the risk of vessel targeting agents that induce metastasis after treatment and propose therapies to address this problem.
2. Angiogenesis and Metastasis
Tumor vessels provide sufficient oxygen and nutrients for tumor growth and metastasis. Angiogenesis is a critical process for tumor vessel formation. In 1971, Folkman first introduced the tumor angiogenesis theory, indicating that angiogenesis is a consecutive process in which neovessels grow from preexisting vessels [10]. Compared with normal vessels, tumor vessels are arranged chaotically, with discontinuous pericyte coverage and high permeability [11]. For metastatic tumor cells, they detach from the primary tumor and pass through the tumor-blood barrier with the help of extracellular matrix proteinase, and then the cells transmigrate the endothelium and circulate in the blood flow. For survival, circulating tumor cells (CTCs) need to overcome blood flow sheer stress and escape the anti-tumor surveillance of immune cells. Finally, disseminated tumor cells extravasate and form metastatic colonization in a secondary site (Figure 1) [12,13].
2.1. Tumor Cell Intravasation
Tumor cell intravasation depends on various factors, including the vasculature properties and stromal cells in the tumor microenvironment [14]. Vessels more than 100 μm in diameter in the primary tumor may provide favorable conditions for tumor intravasation with respect to the liver metastasis of colorectal tumors [15]. Vessel density is the other factor that determines tumor intravasation. Fibroblast growth factor 1 (FGF1) is a pro-angiogenic factor that promotes MCF-7 breast cancer cell intravasation via increasing vessel density [16]. The blocking of vascular endothelial growth factor (VEGF) with anti-VEGF antibody was observed to decrease vessel density and suppress intravasation of human prostate carcinoma cells [17]. Tumor intravasation is closely related to the vascular barrier consisting of endothelial cells and pericytes. To enhance intravasation, malignant tumor cells release matrix metalloproteinase-1 (MMP-1) and induce high vascular permeability via the activation of protease-activated receptor-1 (PAR1) on endothelial cells. The disrupted endothelial cell barrier provides a conduit for tumor cell intravasation. Deletion of MMP-1 leads to decreased vascular permeability and reduced transendothelial migration [18]. VEGF-B secreted by tumor cells also facilitates tumor intravasation by modulating vascular structure, which increases vascular permeability and leakage, promoting tumor cell metastasis in human and mouse tumor models [19]. Except for tumor cells, endothelial cells automatically produce angiopoietin 2 (Ang2), which interacts with integrin β1 and results in reduced barrier function, leading to enhanced transendothelial migration [20]. Pericytes are another physical barrier for tumor vessels, as they attach to the endothelial tube and participate in the formation and stability of blood vessels [21]. Pericytes have dual effects on tumor intravasation. In the resting state, vessels with high pericyte coverage often display increasing stabilization and normalization compared with normal vessels, which prohibits disseminated tumor cell intravasation into the blood stream [22]. Genetic and pharmaceutical deletion of pericyte results in increased vascular leakage and intratumoral hypoxia, leading to EMT-driven metastasis [23,24]. However, pericytes can be activated under the effect of the tumor microenvironment (TME). Certain subpopulations of pericytes, such as endosialin (CD248) and desmin-overexpressing pericytes, increase tumor vessel permeability; thus, directly promoting tumor cell intravasation [25,26]. Activated pericytes indirectly promote tumor metastasis via transition into fibroblasts upon platelet derived growth factor-BB (PDGF-BB) treatment. Co-injection of pericyte–fibroblast transition (PFT) cells with less-metastatic cells facilitates tumor cell intravasation in the primary tumor [27]. PFT is also mediated by tumor-secreted exosomes through the bone morphogenetic proteins (BMPs), phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) and MAP kinases (MEK)/extracellular regulated protein kinase (ERK) signaling pathways [28]. Fibroblasts increase vascular permeability to promote tumor intravasation via enhancing C–X–C motif chemokine ligand 12 (CXCL12) expression. Fibroblast-derived CXCL12 is associated with poor overall survival (OS) in breast cancer patients [29].
As an important component of the TME, immune cells also play a role in tumor cell intravasation. Tie2-expressing macrophages (TEMs) are enriched in the TME with high expression of VEGF-A, which mediates loosening of vascular junctions and enhances vascular permeability, promoting tumor cell intravasation [30]. Tumor-associated macrophages (TAMs) perform pro-angiogenic effects through their response to colony-stimulating factor-1 (CSF-1) and Ang2 [31,32]; meanwhile, TAMs produce VEGF-A and Wnt7B [33,34]. These effects mediated by TAMs, facilitate tumor cell intravasation by increasing the density of blood vessels [35]. Tumor-associated neutrophils (TANs) contribute to tumor intravasation by inducing neovessel formation via MMP-9. Inhibition of neutrophil infiltration by interleukin 8 (IL-8) neutralization leads to diminished tumor angiogenesis and intravasation [36]. These data strongly support a role of stromal cells in the tumor environment in the promotion of intravasation via vessel formation and stabilization [37].
2.2. Tumor Cell Extravasation
Tumor cells adhere to endothelial cells and then transmigrate into the extravascular stroma through paracellular migration, in which tumor cells migrate between two endothelial cells via cellular rearrangements and disruption of inter-endothelial cell junctions [38]. Multiple factors are involved in the adhesion stage of extravasation, including selectins, integrins, N-cadherin, CD44, MUC1, and intercellular cell adhesion molecule-1 (ICAM-1) [39]. Dynamic imaging shows that integrin β1 contributes to tumor protrusion formation and mediates adhesion to the subendothelial matrix, which protects tumor cells from detachment into the blood flow and facilitates tumor extravasation [40]. In addition, ICAM-1 participates in tumor cell adhesion and transendothelial migration [41]. ICAM-1 is expressed both in endothelial and tumor cells. The binding of ICAM-1 with αLβ2 and β2 integrin on neutrophils facilitates tumor cell adhesion and extravasation [42,43]. During the process of extravasation, tumor cells and other cells, including endothelial cells, platelets and myeloid cells co-contribute to cell extravasation. The endothelial to mesenchymal transition (EndoMT) is a new type of transdifferentiation in which endothelial cells acquire mesenchymal or myofibroblastic markers, such as α-smooth muscle actin (α-SMA) and type I collagen, with the stimulation of TGF-β [44]. EndoMT leads to endothelial cytoskeleton remodeling and increased vessel permeability, which facilitates melanoma extravasation [45]. Platelets promote tumor extravasation by inducing an invasive, mesenchyme-like phenotype in tumor cells by activating the TGF-β/Smad/nuclear factor-κappa B (NF-κB) pathway. Platelets interact with endothelial cells through the ATP/P2Y2 signaling pathway, which facilitates tumor cell extravasation by opening the endothelial barrier [46]. Myeloid progenitor cells are also involved in tumor extravasation by transdifferentiating into metastasis-associated macrophages (MAMs) and secreting VEGF-A to increase vascular permeability; thus, enhancing the extravasation of cancer cells [35]. However, only a minority of disseminated tumor cells form metastasis in the secondary site because most tumor cells are cleared away by immune cells or apoptosis after extravasation. To form a metastatic tumor, tumor cells need to proliferate and colonize in the target organ with the help of neovessels, which provide nutrients and oxygen for secondary tumor growth [1].
3. Application of Vessel Targeting Drugs in Metastatic Cancer
The FDA has approved a series of anti-angiogenic drugs applied for chemotherapy of malignant tumors. Anti-angiogenic drugs are mainly classified into three types: monoclonal antibodies, tyrosine kinase inhibitors (TKIs) and fusion peptides [47]. These anti-angiogenic drugs are widely applied in a range of malignant metastatic cancers, such as colorectal cancer, non-small cell lung cancer, renal cell cancer and medullary thyroid cancer (Table 1).
3.1. Monoclonal Antibody Therapy
A monoclonal antibody is designed to specifically bind with a signaling molecule in angiogenesis and neutralize its function. At present, two kinds of monoclonal antibodies are approved: bevacizumab and ramucirumab.
Bevacizumab is a humanized monoclonal anti-VEGF-A antibody that was approved by the FDA as the first anti-angiogenic agent in 2004 [85]. Bevacizumab is widely applied to the treatment of metastatic colorectal cancer (mCRC), metastatic non-small cell lung cancer (mNSCLC), metastatic renal cell carcinoma (mRCC) and metastatic cervical cancer as the first and second-line agent in combination with standard chemotherapy. For instance, bevacizumab is approved for first-line treatment of mCRC in conjunction with 5-fluorouracil-irinotecan or 5-fluorouracil-oxaliplatin-based chemotherapy [48]. A large amount of clinical experimental data suggest that intravenous injection of bevacizumab prolongs the progression-free survival (PFS) and OS of patients with mCRC [49,50,51]. Bevacizumab prevents brain metastases’ formation of nonsquamous non–small cell lung cancer (nsNSCLC) in mouse models, leading to a survival benefit, which suggests that bevacizumab might benefit patients with stage III nsNSCLC who are at a high risk of developing brain metastases [52]. The combination of bevacizumab with interferon-α (IFN-α) is more effective in patients with mRCC than IFN-α treatment alone, which leads to significant improvement in PFS [53]. Bevacizumab is also effective in the treatment of metastatic cervical cancer in combination with chemotherapy, such as cisplatin and paclitaxel, which increases the OS of patients [54]. Although bevacizumab was removed by the FDA for the treatment of metastatic breast cancer, citing safety concerns, the clinical data show that bevacizumab treatment significantly prolongs PFS of metastatic breast cancer patients with an acceptable toxicity profile and is still advised for application in the clinic [55,56].
Ramucirumab is a humanized monoclonal antibody that selectively targets vascular endothelial growth factor receptor-2 (VEGFR-2) and blocks the interaction of VEGFR-2 with VEGF. Ramucirumab has been approved for application in colorectal cancer, non-small cell lung cancer and stomach adenocarcinoma or an gastroesophageal junction adenocarcinoma that has metastasized [57,58,59]. Clinical experiments have shown that the combination of ramucirumab and docetaxel prolonged PFS in patients with locally advanced or metastatic urothelial carcinoma, which further guarantees investigation in a phase III trial [60].
3.2. Tyrosine Kinase Inhibitor Therapy
TKIs are the second group of anti-angiogenic agents that block the activation signal of the intracellular domain after binding of the cell surface receptors and ligands. TKIs may exclusively target one receptor during angiogenesis; however, in most cases, TKIs are multitarget agents; targets include VEGFR, platelet derived growth factor receptor (PDGFR), fibroblast growth factor receptor (FGFR) and c-Kit. Sunitinib and sorafenib are the most common TKIs in the clinic [86].
Sunitinib targets include VEGFR1-3, PDGFR-α, PDGFR-β, c-Kit, colony-stimulating factor-1 receptor (CSF-1R) and RET. Sunitinib is approved by the FDA for the treatment of metastatic pancreatic cancer. Sunitinib was observed to markedly improve the two year OS of patients with metastatic pancreatic adenocarcinoma [61,62]. Sunitinib is also effective against mRCC and is applied as a first-line treatment of mRCC, since it shows superiority compared with IFN-α in a phase III trial [63,64].
Sorafenib inhibits the tyrosine phosphorylation of VEGFR-1, 2 and 3; PDGFR-β; Flt-3; and c-Kit [87]. Sorafenib is the priority drug used for advanced hepatocellular carcinoma (HCC) [65], as it inhibits the lung metastasis of HCC mediated by CD90+ cancer stem cells (CSCs) [66]. The combination of sorafenib and carfilzomib synergistically suppresses the metastasis of HCC via inducing tumor cell apoptosis [67]. Sorafenib may benefit patients with metastatically differentiated thyroid cancers that are resistant to radioactive iodine [68].
Regorafenib is a multikinase inhibitor that potentially targets angiogenesis-associated kinase and the mutant oncogenic kinases, including VEGFR-1 and 3, PDGFR-β, FGFR1, KIT, RET and B-RAF [69]. Regorafenib was approved in 2012 for the treatment of patients with metastatic colorectal cancer previously treated with other therapy. Later, in 2013, regorafenib was applied to patients with locally advanced, unresectable or metastatic gastrointestinal stromal tumors (GISTs) who had been previously treated with imatinib and sunitinib as third-line treatment [70]. A phase II study reported that regorafenib benefits patients with progressive metastatic osteosarcoma and other bone sarcomas by delaying disease progression and extending the PFS of patients with metastatic osteosarcomas [71].
Vandetanib selectively inhibits VEGFR-2 and VEGFR-3, blocking VEGF-stimulated endothelial cell proliferation and migration to inhibit angiogenesis. Vandetanib was approved by the FDA to treat metastatic medullary thyroid cancer (MTC) in 2011 and in 2012 by the European Medicines Agency (EMA), which was observed to improve PFS (30.5 versus 19.3 months in the placebo group) in patients with MTC [72].
Cabozantinib was also approved by the FDA to treat progressive metastatic MTC in 2012, which is a TKI against MET, RET, AXL, VEGFR-2, FLT3 and c-Kit [73]. Cabozantinib has therapeutic effects on metastatic castration-resistant prostate cancer (mCRPC) by eliminating the bone metastasis of mCRPC with pain relief [74,75]. Cabozantinib was approved as a first and second-line treatment for advanced/metastatic renal cell carcinoma based on two clinical trials [76].
Lenvatinib is a multitargeted tyrosine kinase inhibitor targeting the families of VEGFR, FGFR, PDGFRα, KIT and RET [88]. Lenvatinib was approved by the FDA to treat patients with metastatic, progressive and radioactive iodine–refractory differentiated thyroid cancer in 2015, which, thus, provides a new oral option for differentiated thyroid cancer [77]. Later, in 2018, lenvatinib was approved for the treatment of metastatic HCC patients, which had a favorable clinical activity and tolerable toxicity [78,79]. Clinical data show that lenvatinib alone improves the PFS of patients with mRCC who have disease progression after VEGF-targeted therapy [80].
3.3. Fusion Protein Therapy
Ziv-aflibercept is the representative agent of the third type of angiogenesis inhibitors: fusion protein. Ziv-aflibercept is composed of the extracellular domain of both VEGFR-1 and VEGFR-2 fused to the Fc region of IgG1, which interacts with VEGF-B and placental growth factor (PlGF) and halts the pro-angiogenic effects of the VEGF/VEGFR signaling pathway [81,82]. In 2012, the U.S. FDA approved aflibercept in combination with 5-fluorouracil, leucovorin and irinotecan (FOLFIRI) for the treatment of patients with mCRC that is resistant to or has progressed after oxaliplatin chemotherapy, significantly improving the OS, PFS and partial responses in patients with mCRC [83,84].
4. Mechanism of the Anti-Metastatic Effects Mediated by Vessel Targeting Therapies
Vessel-targeting therapies inhibit tumor metastasis through multiple mechanisms (Figure 2). The relevant mechanism is that cancer cells are starved to death without the supply of vessels [89]. On the one hand, angiogenesis inhibitors suppress neovessel formation with decreased vascular density and functional aberrations. The tumor cells lose their pathway to disseminate to other organs. On the other hand, these agents block the pro-angiogenic signaling pathways such as VEGF, PDGF, FGF, Ang, hematopoietic growth factor (HGF), IL-6 and so on, which regulate vessel permeability, remodeling, endothelial cell survival, proliferation and migration, leading to abnormal angiogenesis and tumor metastasis [90,91]. Here, we discuss some anti-metastatic mechanisms of vessel targeting agents that have recently aroused concern.
4.1. Pre-Metastatic Niche Disruption
The pre-metastatic niche provides a favorable environment for seeding and colonization of disseminated tumor cells in the second organ, which is essential for metastatic foci formation. Angiogenesis and the inflammatory response are the main characteristics of the pre-metastatic niche, which provide the necessary conditions for disseminated tumor cells to form metastatic foci and achieve colonization [92]. Regarding the role of angiogenesis in the pre-metastatic niche, anti-angiogenic agents can suppress metastasis via directly blocking metastatic routes through inhibiting neovessel sprouting. Ang2 is a potential target for anti-angiogenic agents. Anti-Ang2 antibody inhibits vessel sprouting and formation mediated by the angiogenic cytokine bombina variegate 8 (Bv8), which is produced by myeloid cells in metastatic nodules [5]. Vessel targeting agents also suppress metastasis via inflammatory responses, which affect the pre-metastatic niche through various inflammatory cells and chemokines. Ang2 neutralization inhibits the recruitment of CCR2+Tie2− MAMs via blocking C–C motif chemokine ligand 2 (CCL2) and ICAM-1, leading to reduced metastases [5]. The neutrophil-mediated inflammatory response also facilitates tumor metastasis, as neutrophils are recruited to the pre-metastatic niche and produce leukotrienes that contribute to the colonization and metastasis of breast cancer cells in the lung [93]. TSU-68 is an antiangiogenic agent targeting multiple tyrosine kinase receptors, such as VEGFR-2, PDGFR and FGFR [94]. TSU-68 significantly inhibits the expression of inflammatory chemokines, including CXCL1, S100A8 and S100A9, which suppresses the CXCL1/C–X–C motif chemokine receptor 2 (CXCR2)-mediated inflammatory response with decreased CXCR2+neutrophils infiltration in the pre-metastatic niche, leading to a marked inhibition of liver metastasis [4].
4.2. Vessel Normalization
Vessel normalization changes tortuous and leaky vessels into straight and regular vessels, which decreases vessel permeability and IFP. The high perfusion and low IFP halt the intravasation of tumor cells, leading to decreased metastasis [6,95]. Tie1 is expressed in activated endothelial cells, which is a key regulator of angiogenesis and vascular remodeling [96]. Tie1 deficiency leads to reduced angiogenesis and increased mural cell coverage with improved vessel perfusion via activating the Ang1/Tie2 signaling pathway. The normalized tumor vessels inhibit tumor intravasation, extravasation and metastatic foci formation [97]. Directly activating Tie2 with the Ang2-binding and TIE2-activating antibody (ABTAA) complex induces tumor normalization with increased pericyte and collagen type IV basement membrane coverage, which suppresses tumor cell metastasis and extravasation in glioma, Lewis lung carcinoma (LLC) and breast cancer [98]. Vessel normalization is also associated with the hyperglycolytic metabolism of tumor endothelial cell (TEC). The genetic inhibition of the glycolytic activator PFKFB3 improves vessel perfusion and tightens the endothelial cell barrier. The normalized tumor vessels inhibit cancer cell intravasation and metastasis [99]. A number of investigations showed that pharmacologic intervention with vessel targeting drugs induces vessel normalization [100]. Bevacizumab used at a certain dose leads to vessel normalization, which is accompanied by increased pericyte coverage and vessel perfusion [101,102]. The preclinical data reveal that bevacizumab-mediated vessel normalization improves the oxygen supply in tumors and facilitates the delivery of chemotherapy drugs, which benefit patients with mCRC and metastatic breast cancer [103]. Infigratinib is an FGFR kinase inhibitor that suppresses basic FGF (bFGF)-stimulated angiogenesis and mediates vessel normalization with increased perfusion. Infigratinib treatment inhibits tumor hypoxia via vessel normalization, which significantly reduces lung metastasis in HCC models [104]. From the above studies, vessel normalization mediated by antiangiogenic agents is a direct and effective method to recover the normal functions of vessels and then suppress metastasis.
5. Metastasis Risk after Vessel Targeting Therapies
Vessel targeting therapy is not always effective for tumor metastasis [105]. Vessel targeting agents lead to vessel destabilization, including endothelial barrier disruption, which facilitates tumor cell extravasation. Sunitinib disrupts endothelial barrier integrity by decreasing the expression of vascular endothelial cadherin (VE-cadherin) and ZO-1, which maintains the endothelial cell junction. The leaky vessel post-sunitinib treatment contributes to tumor cell extravasation, which increases the number and area of metastatic lung nodules [106]. The poor vasculature with decreased vessel density and stability after vessel targeting treatment often leads to tumor hypoxia compared with neighboring tissues. Hypoxia is associated with poor patient prognosis and has been validated to promote tumor cell invasion and metastasis [107]. Anti-angiogenic agents, including DC101 (antibody against VEGFR-2) and sunitinib, promote local invasiveness and increase distant metastasis in the mouse models of pancreatic neuroendocrine cancer and glioblastoma multiforme. Mice treated with DC101 or sunitinib show severe hypoxia, especially in the micrometastases of distant organs, indicating the crucial role of hypoxia in promoting metastasis after anti-angiogenic therapy [108]. Hypoxia is the most influential factor of hypoxia-inducible factors’ (HIF) activation. The HIF family (HIF-1α, HIF-2α, HIF-3α, HIF-1β and HIF-2β) is widely expressed in solid tumors, including those of non-small cell lung cancer, breast cancer, colon cancer, head and neck cancer, gastric cancer and prostate cancer. Hypoxia/HIF induce metastasis through a series of pathways, including regulation of EMT, metabolism shift and BMDC recruitment (Figure 3) [7,9].
5.1. EMT
EMT is characterized by changes in cell morphology and cell-to-cell and cell-to-matrix adhesions, which endow tumor cells with migratory and invasive capacities. The phenotypic changes in EMT cells include the down-regulation of epithelial cell markers, such as E-cadherin, and high expression of mesenchymal cell markers, including N-cadherin, vimentin and metalloproteases [109,110]. HIF directly regulates EMT transcription factors, including zinc finger E-box binding homeobox (ZEB), snail, slug and twist, which have prognostic significance in tumor metastasis [111]. The HIF pathway also indirectly promotes EMT through a number of cell signaling pathways, including Notch, TGF-β, Sonic Hedgehog and Wnt, which all play key roles in the process of EMT [112,113]. Bevacizumab was reported to induce EMT via activation of Wnt/β catenin signaling in glioblastoma progression, which is accompanied by the increased expression of mesenchymal markers, such as N-cadherin and vimentin [114,115]. Bevacizumab also induces the upregulation of EMT-induced factors including TGFβ1, 2 and 3 in breast cancer patients, which is mediated by acute hypoxia after bevacizumab treatment. Sorafenib treatment also promotes invasiveness and the metastasis of orthotopic HCC tumors in mice by EMT. Sorafenib downregulates the expression of HIV-1 Tat interactive protein 2 (HTATIP2) via the Janus kinase (JAK)-signal transducer and activator of transcription 3 (STAT3) signaling pathway, leading to induced EMT in HCC cells [116].
5.2. Metabolism Shift
A study has demonstrated that metabolism reprogramming induced by anti-angiogenesis drugs facilitates tumor metastasis. Bevacizumab treatment leads to a significant upregulation of glycolytic genes, which results in a metabolism shift towards anaerobic glycolysis with enhanced lactate production, promoting cancer cell invasion and metastasis in glioblastoma [117,118]. Lipid metabolism also plays a crucial role in metastasis mediated by angiogenic inhibitors. Bevacizumab treatment leads to lipid droplet (LD) accumulation by promoting the expression of fatty acid binding protein 3 (FABP3) and FABP7 [119]. Sunitinib and sorafenib both show clinical benefits to cancer patients. However, they are reported to induce tumor regrowth and metastasis after drug withdrawal. Cancer cell metabolism is altered from glycolytic metabolism to lipid metabolism after the withdrawal of angiogenesis inhibitors. Citric acid cycle activity increases accompanied by reduced glycolysis after treatment ends. Fatty acid synthase (FASN) is a critical enzyme in lipogenesis [120], and blocking FASN inhibits tumor regrowth and metastasis after the withdrawal of sunitinib treatment. The result has been validated in a colorectal cancer xenograft and transgenic mouse model of mammary carcinoma [121,122].
5.3. BMDC Recruitment
Several investigations show that vessel targeting agents enhance tumor metastasis by recruiting bone marrow-derived pro-angiogenic cells. The infiltrated BMDCs promote tumor revascularization and metastasis. Hypoxia induced by vessel targeting agents upregulates the expression of stromal-derived factor-1α (SDF-1α), which is a chemoattractant of CD45+myeloid cells and induces recruitment of Tie2, VEGFR-1, CD11b and F4/80-expressing subtypes of myeloid cells; myeloid cells then promote angiogenesis and induce tumor cell intravasation, dissemination and metastasis [123,124]. CD11b+myeloid cells contribute to hepatic metastasis through downregulation of angiopoietin-like 7 (ANGPTL7). Depletion of CD11b+myeloid cells or overexpression of ANGPTL7 significantly inhibits hepatic metastasis formation and angiogenesis [125]. Immune-suppressive cells, such as TAMs, participate in tumor angiogenesis and metastasis, inducing tumor cell invasion, extravasation and survival in the metastatic niche. The recruitment and polarization of TAMs are driven by a series of growth factors, including CSF-1, granulocyte-macrophage colony-stimulating factor (GM-CSF) and CCL2, which are secreted by tumor cells and TAMs, and then produce epidermal growth factor (EGF), MMP-9, MMP-2 and urokinase plasminogen activator (uPA) to promote tumor cell metastasis [126,127]. TAMs play a central role in bevacizumab treatment. High TAM infiltration and variations in genes regulating TAM-related functions such as TBK1, CCL2, CCL18 and IRF3 predict poor outcome in metastatic patients treated with bevacizumab [128,129]. Sorafenib treatment enhances the infiltration of F4/80 and CD11b-positive cells in the peripheral blood of HCC xenograft model via CSF-1, SDF-1α and VEGF, which are key cytokines for macrophage recruitment. The combination of sorafenib with macrophage-targeting drugs including zoledronic acid (ZA) and clodrolip suppresses the recruitment of macrophage and further reduces lung metastasis [130].
6. Discussion
Hematogenous metastasis is the principal pathway for malignant tumor metastasis. Vessel targeting treatment can inhibit metastasis through starving tumor cells, inducing vessel normalization and disrupting the pre-metastatic niche. However, vessel targeting treatment still poses a pro-metastatic risk for patients. Here, we mainly discuss some potential methods to circumvent the problem.
Hypoxia is considered to be the greatest hindrance to vessel targeting treatment. Therefore, a combination medication of a vessel targeting treatment with a hypoxia targeting therapy is a better choice in the clinic. To monitor hypoxia, dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI) and 18F-Fluoromisonidazole (18F-FMISO) are the most effective methods for tumor areas. In addition, multiple HIF inhibitors have been investigated and demonstrated to block the hypoxia pathway and exert antitumor effects [131,132]. These inhibitors suppress the mRNA expression, protein synthesis, protein degradation and dimerization, DNA binding and transcriptional activity of HIF-1 and HIF-2, and some of inhibitors have progressed into clinical trials [133]. Hypoxia-directed gene therapy is another strategy achieved by designing therapeutic genes that are controlled by hypoxia response elements (HREs) or other promoters under HIF-1 activation. A therapeutic gene was used to selectively activate prodrug and increase drug cytotoxicity under hypoxia conditions [134,135]. Bioreductive prodrugs target tumor hypoxia in an oxygen-sensitive manner, which are activated by endogenous oxidoreductases and metabolized to cytotoxins, including nitro compounds, N-oxides, quinones and metal complexes [136].
Both hypoxia and abnormal tumor vasculature induce dysfunction of a tumor’s immune microenvironment, which regulates the functions of the innate and adaptive immune system towards immunosuppression [137,138,139,140]. The expression of programmed cell death 1 ligand 1 (PD-L1) on dendritic cells (DCs), TAMs and tumor ECs is also increased [141,142]. Anti-angiogenic agents normalize abnormal vessels, which facilitate T cell recruitment and decrease the infiltration of pro-tumor immune cells, including regulatory T cells, M2-like TAMs and myeloid-derived suppressor cells (MDSCs) [143,144,145]. Therefore, a potential strategy is to combine anti-angiogenesis agents with immunotherapy, especially T-cell based immunotherapy. Inhibition of VEGFA and Ang-2 normalizes tumor vessels and increases IFNγ+ CD8+ T cells’ extravasation and accumulation, which further enhances the antitumor effects of PD-1 inhibitors [146,147]. Moreover, the combination of VEGFR-2 and PD-L1 antibodies induces high endothelial venules (HEVs) to facilitate IFNγ+ CD4+ and IFNγ+ CD8+ lymphocyte infiltration in breast cancer and pancreatic neuroendocrine tumors, finally leading to tumor cell apoptosis and necrosis [148]. This combination therapy has achieved certain results in the treatment of metastatic cancer. The combination of anti-angiogenic agents with PD-1/PD-L1 inhibitors is safe and tolerable in patients with metastatic, clear cell, renal cell carcinoma [149] and metastatic mucosal melanoma [150]. The combined application of atezolizumab (anti-PD-L1) with bevacizumab, carboplatin and paclitaxel significantly prolongs PFS and OS in patients with metastatic nsNSCLC [151]. These data indicate that the combination of anti-angiogenic therapy with immunotherapy can synergistically benefit patients with metastatic cancer.
Drug resistance is also associated with the failure of anti-angiogenic therapies in clinical applications. Vessel cooption is a key mechanism mediating resistance to anti-angiogenic therapy, in which tumor cells hijack the pre-existing vasculature to support tumor growth without the need for angiogenesis [152]. Vessel cooption is commonly found in human lung, liver and brain metastases [153]. The co-opted vessels facilitate metastatic foci formation and colonization, leading to the failure of treatment with bevacizumab, sunitinib and ZD6474 [154,155,156]. Therefore, combined inhibition of angiogenesis and vessel cooption might be an optimized strategy for the application of vessel targeting drugs in the metastatic tumors.
7. Conclusions
Angiogenesis provides advantageous conditions for tumor metastasis, providing an avenue for the development of antiangiogenic drugs. The vessel targeting strategy is an important strategy for metastatic cancer patients in the clinic, though it creates a risk for tumor metastasis under certain conditions. Strategies for monitoring and decreasing the pro-metastatic risk of vessel targeting agents should be further developed.
Author Contributions
Conceptualization, D.Z.; writing—original draft preparation, X.L. and Y.L.; writing—review and editing, W.L., M.C. and W.Y.
Funding
This review was supported by the National Natural Science Foundation of China (grant numbers: 81803566, 81973340 and 81573455); the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (grant number: 2017BT01Y036); the National Science and Technology Major Project (grant number: 2018ZX09711001-008-008); and the Guangzhou Science and Technology Plan Project (grant number: 201905010003).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paduch, R. The role of lymphangiogenesis and angiogenesis in tumor metastasis. Cell Oncol. (Dordr) 2016, 39, 397–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.Y.; Hynes, R.O. Lymphatic or hematogenous dissemination: How does a metastatic tumor cell decide? Cell Cycle 2006, 5, 812–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, M.; Kikuchi, H.; Ohta, M.; Kawabata, T.; Hiramatsu, Y.; Kondo, K.; Baba, M.; Kamiya, K.; Tanaka, T.; Kitagawa, M.; et al. TSU68 prevents liver metastasis of colon cancer xenografts by modulating the premetastatic niche. Cancer Res. 2008, 68, 9754–9762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, K.; Hu, J.; Korn, C.; Savant, S.; Teichert, M.; Kapel, S.S.; Jugold, M.; Besemfelder, E.; Thomas, M.; Pasparakis, M.; et al. Postsurgical adjuvant tumor therapy by combining anti-angiopoietin-2 and metronomic chemotherapy limits metastatic growth. Cancer Cell 2014, 26, 880–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef]
- De Bock, K.; Mazzone, M.; Carmeliet, P. Antiangiogenic therapy, hypoxia, and metastasis: Risky liaisons, or not? Nat. Rev. Clin. Oncol. 2011, 8, 393–404. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Rankin, E.B.; Nam, J.M.; Giaccia, A.J. Hypoxia: Signaling the Metastatic Cascade. Trends Cancer 2016, 2, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar]
- Siemann, D.W. The unique characteristics of tumor vasculature and preclinical evidence for its selective disruption by Tumor-Vascular Disrupting Agents. Cancer Treat. Rev. 2011, 37, 63–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fidler, I.J. The pathogenesis of cancer metastasis: The ‘seed and soil’ hypothesis revisited. Nat. Rev. Cancer 2003, 3, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Mehlen, P.; Puisieux, A. Metastasis: A question of life or death. Nat. Rev. Cancer 2006, 6, 449–458. [Google Scholar] [CrossRef]
- Chiang, S.P.; Cabrera, R.M.; Segall, J.E. Tumor cell intravasation. Am. J. Physiol. Cell Physiol. 2016, 311, C1–C14. [Google Scholar] [CrossRef] [Green Version]
- Yamamura, T.; Tsukikawa, S.; Yamada, K.; Yamaguchi, S. Morphologic analysis of microvessels in colorectal tumors with respect to the formation of liver metastases. J. Surg. Oncol. 2001, 78, 259–264. [Google Scholar] [CrossRef]
- Zhang, L.; Kharbanda, S.; McLeskey, S.W.; Kern, F.G. Overexpression of fibroblast growth factor 1 in MCF-7 breast cancer cells facilitates tumor cell dissemination but does not support the development of macrometastases in the lungs or lymph nodes. Cancer Res. 1999, 59, 5023–5029. [Google Scholar]
- Conn, E.M.; Botkjaer, K.A.; Kupriyanova, T.A.; Andreasen, P.A.; Deryugina, E.I.; Quigley, J.P. Comparative analysis of metastasis variants derived from human prostate carcinoma cells: Roles in intravasation of VEGF-mediated angiogenesis and uPA-mediated invasion. Am. J. Pathol. 2009, 175, 1638–1652. [Google Scholar] [CrossRef] [Green Version]
- Juncker-Jensen, A.; Deryugina, E.I.; Rimann, I.; Zajac, E.; Kupriyanova, T.A.; Engelholm, L.H.; Quigley, J.P. Tumor MMP-1 activates endothelial PAR1 to facilitate vascular intravasation and metastatic dissemination. Cancer Res. 2013, 73, 4196–4211. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Zhang, Y.; Hosaka, K.; Andersson, P.; Wang, J.; Tholander, F.; Cao, Z.; Morikawa, H.; Tegner, J.; Yang, Y.; et al. VEGF-B promotes cancer metastasis through a VEGF-A-independent mechanism and serves as a marker of poor prognosis for cancer patients. Proc. Natl. Acad. Sci. USA 2015, 112, E2900–E2909. [Google Scholar] [CrossRef] [Green Version]
- Hakanpaa, L.; Sipila, T.; Leppanen, V.M.; Gautam, P.; Nurmi, H.; Jacquemet, G.; Eklund, L.; Ivaska, J.; Alitalo, K.; Saharinen, P. Endothelial destabilization by angiopoietin-2 via integrin beta1 activation. Nat. Commun. 2015, 6, 5962. [Google Scholar] [CrossRef]
- Bergers, G.; Song, S. The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol. 2005, 7, 452–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, G.P.; Nguyen, D.X.; Chiang, A.C.; Bos, P.D.; Kim, J.Y.; Nadal, C.; Gomis, R.R.; Manova-Todorova, K.; Massague, J. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature 2007, 446, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Keskin, D.; Kim, J.; Cooke, V.G.; Wu, C.C.; Sugimoto, H.; Gu, C.; De Palma, M.; Kalluri, R.; LeBleu, V.S. Targeting vascular pericytes in hypoxic tumors increases lung metastasis via angiopoietin-2. Cell Rep. 2015, 10, 1066–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooke, V.G.; LeBleu, V.S.; Keskin, D.; Khan, Z.; O’Connell, J.T.; Teng, Y.; Duncan, M.B.; Xie, L.; Maeda, G.; Vong, S.; et al. Pericyte depletion results in hypoxia-associated epithelial-to-mesenchymal transition and metastasis mediated by met signaling pathway. Cancer Cell 2012, 21, 66–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viski, C.; Konig, C.; Kijewska, M.; Mogler, C.; Isacke, C.M.; Augustin, H.G. Endosialin-Expressing Pericytes Promote Metastatic Dissemination. Cancer Res. 2016, 76, 5313–5325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyle, L.T.; Lockman, P.R.; Adkins, C.E.; Mohammad, A.S.; Sechrest, E.; Hua, E.; Palmieri, D.; Liewehr, D.J.; Steinberg, S.M.; Kloc, W.; et al. Alterations in Pericyte Subpopulations Are Associated with Elevated Blood-Tumor Barrier Permeability in Experimental Brain Metastasis of Breast Cancer. Clin. Cancer Res. 2016, 22, 5287–5299. [Google Scholar] [CrossRef] [Green Version]
- Hosaka, K.; Yang, Y.; Seki, T.; Fischer, C.; Dubey, O.; Fredlund, E.; Hartman, J.; Religa, P.; Morikawa, H.; Ishii, Y.; et al. Pericyte-fibroblast transition promotes tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2016, 113, E5618–E5627. [Google Scholar] [CrossRef] [Green Version]
- Ning, X.; Zhang, H.; Wang, C.; Song, X. Exosomes Released by Gastric Cancer Cells Induce Transition of Pericytes Into Cancer-Associated Fibroblasts. Med. Sci. Monit. 2018, 24, 2350–2359. [Google Scholar] [CrossRef]
- Ahirwar, D.K.; Nasser, M.W.; Ouseph, M.M.; Elbaz, M.; Cuitino, M.C.; Kladney, R.D.; Varikuti, S.; Kaul, K.; Satoskar, A.R.; Ramaswamy, B.; et al. Fibroblast-derived CXCL12 promotes breast cancer metastasis by facilitating tumor cell intravasation. Oncogene 2018, 37, 4428–4442. [Google Scholar] [CrossRef]
- Harney, A.S.; Arwert, E.N.; Entenberg, D.; Wang, Y.; Guo, P.; Qian, B.Z.; Oktay, M.H.; Pollard, J.W.; Jones, J.G.; Condeelis, J.S. Real-Time Imaging Reveals Local, Transient Vascular Permeability, and Tumor Cell Intravasation Stimulated by TIE2hi Macrophage-Derived VEGFA. Cancer Discov. 2015, 5, 932–943. [Google Scholar] [CrossRef] [Green Version]
- Lin, E.Y.; Li, J.F.; Gnatovskiy, L.; Deng, Y.; Zhu, L.; Grzesik, D.A.; Qian, H.; Xue, X.N.; Pollard, J.W. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006, 66, 11238–11246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzieri, R.; Pucci, F.; Moi, D.; Zonari, E.; Ranghetti, A.; Berti, A.; Politi, L.S.; Gentner, B.; Brown, J.L.; Naldini, L.; et al. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell 2011, 19, 512–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, E.Y.; Pollard, J.W. Tumor-associated macrophages press the angiogenic switch in breast cancer. Cancer Res. 2007, 67, 5064–5066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, E.J.; Cassetta, L.; Qian, B.Z.; Lewkowich, I.; Li, J.F.; Stefater, J.A., 3rd; Smith, A.N.; Wiechmann, L.S.; Wang, Y.; Pollard, J.W.; et al. Myeloid WNT7b mediates the angiogenic switch and metastasis in breast cancer. Cancer Res. 2014, 74, 2962–2973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, T.; Qian, B.Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86. [Google Scholar] [CrossRef]
- Bekes, E.M.; Schweighofer, B.; Kupriyanova, T.A.; Zajac, E.; Ardi, V.C.; Quigley, J.P.; Deryugina, E.I. Tumor-recruited neutrophils and neutrophil TIMP-free MMP-9 regulate coordinately the levels of tumor angiogenesis and efficiency of malignant cell intravasation. Am. J. Pathol. 2011, 179, 1455–1470. [Google Scholar] [CrossRef]
- Tien, Y.W.; Chang, K.J.; Jeng, Y.M.; Lee, P.H.; Wu, M.S.; Lin, J.T.; Hsu, S.M. Tumor angiogenesis and its possible role in intravasation of colorectal epithelial cells. Clin. Cancer Res. 2001, 7, 1627–1632. [Google Scholar]
- Strilic, B.; Offermanns, S. Intravascular Survival and Extravasation of Tumor Cells. Cancer Cell. 2017, 32, 282–293. [Google Scholar] [CrossRef]
- Reymond, N.; d’Agua, B.B.; Ridley, A.J. Crossing the endothelial barrier during metastasis. Nat. Rev. Cancer 2013, 13, 858–870. [Google Scholar] [CrossRef]
- Chen, M.B.; Lamar, J.M.; Li, R.; Hynes, R.O.; Kamm, R.D. Elucidation of the Roles of Tumor Integrin beta1 in the Extravasation Stage of the Metastasis Cascade. Cancer Res. 2016, 76, 2513–2524. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Li, N.; Li, Z.; Chang, A.; Chen, Y.; Zhao, T.; Li, Y.; Wang, X.; Zhang, W.; Wang, Z.; et al. Tumour-derived Interleukin 35 promotes pancreatic ductal adenocarcinoma cell extravasation and metastasis by inducing ICAM1 expression. Nat. Commun. 2017, 8, 14035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strell, C.; Lang, K.; Niggemann, B.; Zaenker, K.S.; Entschladen, F. Surface molecules regulating rolling and adhesion to endothelium of neutrophil granulocytes and MDA-MB-468 breast carcinoma cells and their interaction. Cell Mol. Life Sci. 2007, 64, 3306–3316. [Google Scholar] [CrossRef] [PubMed]
- Huh, S.J.; Liang, S.; Sharma, A.; Dong, C.; Robertson, G.P. Transiently entrapped circulating tumor cells interact with neutrophils to facilitate lung metastasis development. Cancer Res. 2010, 70, 6071–6082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piera-Velazquez, S.; Li, Z.; Jimenez, S.A. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am. J. Pathol. 2011, 179, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Krizbai, I.A.; Gasparics, A.; Nagyoszi, P.; Fazakas, C.; Molnar, J.; Wilhelm, I.; Bencs, R.; Rosivall, L.; Sebe, A. Endothelial-mesenchymal transition of brain endothelial cells: Possible role during metastatic extravasation. PLoS ONE 2015, 10, e0123845. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, D.; Strilic, B.; Sivaraj, K.K.; Wettschureck, N.; Offermanns, S. Platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor. Cancer Cell 2013, 24, 130–137. [Google Scholar] [CrossRef] [Green Version]
- Al-Abd, A.M.; Alamoudi, A.J.; Abdel-Naim, A.B.; Neamatallah, T.A.; Ashour, O.M. Anti-angiogenic agents for the treatment of solid tumors: Potential pathways, therapy and current strategies—A review. J. Adv. Res. 2017, 8, 591–605. [Google Scholar] [CrossRef]
- De Falco, S. Antiangiogenesis therapy: An update after the first decade. Korean J. Intern. Med. 2014, 29, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [Green Version]
- Bennouna, J.; Sastre, J.; Arnold, D.; Osterlund, P.; Greil, R.; Van Cutsem, E.; von Moos, R.; Vieitez, J.M.; Bouche, O.; Borg, C.; et al. Continuation of bevacizumab after first progression in metastatic colorectal cancer (ML18147): A randomised phase 3 trial. Lancet Oncol. 2013, 14, 29–37. [Google Scholar] [CrossRef]
- Loupakis, F.; Stein, A.; Ychou, M.; Hermann, F.; Salud, A.; Osterlund, P. A Review of Clinical Studies and Practical Guide for the Administration of Triplet Chemotherapy Regimens with Bevacizumab in First-line Metastatic Colorectal Cancer. Target. Oncol. 2016, 11, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Ilhan-Mutlu, A.; Osswald, M.; Liao, Y.; Gommel, M.; Reck, M.; Miles, D.; Mariani, P.; Gianni, L.; Lutiger, B.; Nendel, V.; et al. Bevacizumab Prevents Brain Metastases Formation in Lung Adenocarcinoma. Mol. Cancer Ther. 2016, 15, 702–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escudier, B.; Pluzanska, A.; Koralewski, P.; Ravaud, A.; Bracarda, S.; Szczylik, C.; Chevreau, C.; Filipek, M.; Melichar, B.; Bajetta, E.; et al. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: A randomised, double-blind phase III trial. Lancet 2007, 370, 2103–2111. [Google Scholar] [CrossRef]
- Tewari, K.S.; Sill, M.W.; Long, H.J., 3rd; Penson, R.T.; Huang, H.; Ramondetta, L.M.; Landrum, L.M.; Oaknin, A.; Reid, T.J.; Leitao, M.M.; et al. Improved survival with bevacizumab in advanced cervical cancer. N. Engl. J. Med. 2014, 370, 734–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manso, L.; Moreno, F.; Marquez, R.; Castelo, B.; Arcediano, A.; Arroyo, M.; Ballesteros, A.I.; Calvo, I.; Echarri, M.J.; Enrech, S.; et al. Use of bevacizumab as a first-line treatment for metastatic breast cancer. Curr Oncol. 2015, 22, e51–e60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montero, A.J.; Escobar, M.; Lopes, G.; Gluck, S.; Vogel, C. Bevacizumab in the treatment of metastatic breast cancer: Friend or foe? Curr. Oncol. Rep. 2012, 14, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguerido, A.; Mulet-Margalef, N.; Matos, I.; Ros, J.; Argiles, G.; Elez, E.; Tabernero, J. The safety of ramucirumab for the treatment of colorectal cancer. Expert Opin. Drug Saf. 2018, 17, 945–951. [Google Scholar] [CrossRef]
- Arrieta, O.; Zatarain-Barron, Z.L.; Cardona, A.F.; Carmona, A.; Lopez-Mejia, M. Ramucirumab in the treatment of non-small cell lung cancer. Expert Opin. Drug Saf. 2017, 16, 637–644. [Google Scholar] [CrossRef]
- Aprile, G.; Rijavec, E.; Fontanella, C.; Rihawi, K.; Grossi, F. Ramucirumab: Preclinical research and clinical development. OncoTargets Ther. 2014, 7, 1997–2006. [Google Scholar] [CrossRef] [Green Version]
- Petrylak, D.P.; Tagawa, S.T.; Kohli, M.; Eisen, A.; Canil, C.; Sridhar, S.S.; Spira, A.; Yu, E.Y.; Burke, J.M.; Shaffer, D.; et al. Docetaxel As Monotherapy or Combined With Ramucirumab or Icrucumab in Second-Line Treatment for Locally Advanced or Metastatic Urothelial Carcinoma: An Open-Label, Three-Arm, Randomized Controlled Phase II Trial. J. Clin. Oncol. 2016, 34, 1500–1509. [Google Scholar] [CrossRef]
- Ribatti, D.; Annese, T.; Ruggieri, S.; Tamma, R.; Crivellato, E. Limitations of Anti-Angiogenic Treatment of Tumors. Transl. Oncol. 2019, 12, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Reni, M.; Cereda, S.; Milella, M.; Novarino, A.; Passardi, A.; Mambrini, A.; Di Lucca, G.; Aprile, G.; Belli, C.; Danova, M.; et al. Maintenance sunitinib or observation in metastatic pancreatic adenocarcinoma: A phase II randomised trial. Eur. J. Cancer 2013, 49, 3609–3615. [Google Scholar] [CrossRef] [PubMed]
- Schmid, T.A.; Gore, M.E. Sunitinib in the treatment of metastatic renal cell carcinoma. Ther. Adv. Urol. 2016, 8, 348–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rini, B.I.; Hutson, T.E.; Figlin, R.A.; Lechuga, M.J.; Valota, O.; Serfass, L.; Rosbrook, B.; Motzer, R.J. Sunitinib in Patients With Metastatic Renal Cell Carcinoma: Clinical Outcome According to International Metastatic Renal Cell Carcinoma Database Consortium Risk Group. Clin. Genitourin Cancer 2018, 16, 298–304. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Yoshida, M.; Yamashita, T.; Okada, H.; Oishi, N.; Nio, K.; Hayashi, T.; Nomura, Y.; Hayashi, T.; Asahina, Y.; Ohwada, M.; et al. Sorafenib suppresses extrahepatic metastasis de novo in hepatocellular carcinoma through inhibition of mesenchymal cancer stem cells characterized by the expression of CD90. Sci. Rep. 2017, 7, 11292. [Google Scholar] [CrossRef]
- Jiang, C.; Xu, R.; Li, X.X.; Zhou, Y.F.; Xu, X.Y.; Yang, Y.; Wang, H.Y.; Zheng, X.F.S. Sorafenib and Carfilzomib Synergistically Inhibit the Proliferation, Survival, and Metastasis of Hepatocellular Carcinoma. Mol. Cancer Ther. 2018, 17, 2610–2621. [Google Scholar] [CrossRef] [Green Version]
- Brose, M.S.; Nutting, C.M.; Jarzab, B.; Elisei, R.; Siena, S.; Bastholt, L.; de la Fouchardiere, C.; Pacini, F.; Paschke, R.; Shong, Y.K.; et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: A randomised, double-blind, phase 3 trial. Lancet 2014, 384, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, S.M.; Dumas, J.; Adnane, L.; Lynch, M.; Carter, C.A.; Schutz, G.; Thierauch, K.H.; Zopf, D. Regorafenib (BAY 73-4506): A new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int. J. Cancer 2011, 129, 245–255. [Google Scholar] [CrossRef]
- Lyseng-Williamson, K.A. Regorafenib: A guide to its use in advanced gastrointestinal stromal tumor (GIST) after failure of imatinib and sunitinib. BioDrugs 2013, 27, 525–531. [Google Scholar] [CrossRef]
- Duffaud, F.; Mir, O.; Boudou-Rouquette, P.; Piperno-Neumann, S.; Penel, N.; Bompas, E.; Delcambre, C.; Kalbacher, E.; Italiano, A.; Collard, O.; et al. Efficacy and safety of regorafenib in adult patients with metastatic osteosarcoma: A non-comparative, randomised, double-blind, placebo-controlled, phase 2 study. Lancet Oncol. 2019, 20, 120–133. [Google Scholar] [CrossRef]
- Fallahi, P.; Ferrari, S.M.; Baldini, E.; Biricotti, M.; Ulisse, S.; Materazzi, G.; Miccoli, P.; Antonelli, A. The safety and efficacy of vandetanib in the treatment of progressive medullary thyroid cancer. Expert Rev. Anticancer Ther. 2016, 16, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Grullich, C. Cabozantinib: Multi-kinase Inhibitor of MET, AXL, RET, and VEGFR2. Recent Results Cancer Res. 2018, 211, 67–75. [Google Scholar] [PubMed]
- Smith, D.C.; Smith, M.R.; Sweeney, C.; Elfiky, A.A.; Logothetis, C.; Corn, P.G.; Vogelzang, N.J.; Small, E.J.; Harzstark, A.L.; Gordon, M.S.; et al. Cabozantinib in patients with advanced prostate cancer: Results of a phase II randomized discontinuation trial. J. Clin. Oncol. 2013, 31, 412–419. [Google Scholar] [CrossRef]
- Smith, M.; De Bono, J.; Sternberg, C.; Le Moulec, S.; Oudard, S.; De Giorgi, U.; Krainer, M.; Bergman, A.; Hoelzer, W.; De Wit, R.; et al. Phase III Study of Cabozantinib in Previously Treated Metastatic Castration-Resistant Prostate Cancer: COMET-1. J. Clin. Oncol. 2016, 34, 3005–3013. [Google Scholar] [CrossRef]
- Vecchio, S.J.D.; Ellis, R.J. Cabozantinib for the Management of Metastatic Clear Cell Renal Cell Carcinoma. J. Kidney Cancer VHL 2018, 5, 1–5. [Google Scholar] [CrossRef]
- Fala, L. Lenvima (Lenvatinib), a Multireceptor Tyrosine Kinase Inhibitor, Approved by the FDA for the Treatment of Patients with Differentiated Thyroid Cancer. Am. Health Drug Benefits 2015, 8, 176–179. [Google Scholar]
- Ikeda, K.; Kudo, M.; Kawazoe, S.; Osaki, Y.; Ikeda, M.; Okusaka, T.; Tamai, T.; Suzuki, T.; Hisai, T.; Hayato, S.; et al. Phase 2 study of lenvatinib in patients with advanced hepatocellular carcinoma. J. Gastroenterol. 2017, 52, 512–519. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, M.; Okusaka, T.; Mitsunaga, S.; Ueno, H.; Tamai, T.; Suzuki, T.; Hayato, S.; Kadowaki, T.; Okita, K.; Kumada, H. Safety and Pharmacokinetics of Lenvatinib in Patients with Advanced Hepatocellular Carcinoma. Clin. Cancer Res. 2016, 22, 1385–1394. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Hutson, T.E.; Glen, H.; Michaelson, M.D.; Molina, A.; Eisen, T.; Jassem, J.; Zolnierek, J.; Maroto, J.P.; Mellado, B.; et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: A randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 2015, 16, 1473–1482. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.; Sun, W. Ziv-aflibercept in metastatic colorectal cancer. Biologics 2014, 8, 13–25. [Google Scholar] [PubMed] [Green Version]
- Singh, S.R.; Dogra, A.; Stewart, M.; Das, T.; Chhablani, J. Intravitreal Ziv-Aflibercept: Clinical Effects and Economic Impact. Asia Pac. J. Ophthalmol (Phila) 2017, 6, 561–568. [Google Scholar]
- Ciombor, K.K.; Berlin, J.; Chan, E. Aflibercept. Clin. Cancer Res. 2013, 19, 1920–1925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, P.A.; Moore, M.J. Aflibercept in the treatment of patients with metastatic colorectal cancer: Latest findings and interpretations. Therap Adv. Gastroenterol. 2013, 6, 459–473. [Google Scholar] [CrossRef] [Green Version]
- Ellis, L.M. Bevacizumab. Nat. Rev. Drug Discov. 2005, 4, S8–S9. [Google Scholar] [CrossRef]
- Bennasroune, A.; Gardin, A.; Aunis, D.; Cremel, G.; Hubert, P. Tyrosine kinase receptors as attractive targets of cancer therapy. Crit. Rev. Oncol. Hematol. 2004, 50, 23–38. [Google Scholar] [CrossRef]
- Iyer, R.; Fetterly, G.; Lugade, A.; Thanavala, Y. Sorafenib: A clinical and pharmacologic review. Expert Opin. Pharmacother. 2010, 11, 1943–1955. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Matsui, J.; Matsushima, T.; Obaishi, H.; Miyazaki, K.; Nakamura, K.; Tohyama, O.; Semba, T.; Yamaguchi, A.; Hoshi, S.S.; et al. Lenvatinib, an angiogenesis inhibitor targeting VEGFR/FGFR, shows broad antitumor activity in human tumor xenograft models associated with microvessel density and pericyte coverage. Vasc. Cell 2014, 6, 18. [Google Scholar] [CrossRef] [Green Version]
- Folkman, J. The role of angiogenesis in tumor growth. Semin. Cancer Biol. 1992, 3, 65–71. [Google Scholar]
- Maj, E.; Papiernik, D.; Wietrzyk, J. Antiangiogenic cancer treatment: The great discovery and greater complexity (Review). Int. J. Oncol. 2016, 49, 1773–1784. [Google Scholar] [CrossRef] [Green Version]
- Marech, I.; Leporini, C.; Ammendola, M.; Porcelli, M.; Gadaleta, C.D.; Russo, E.; De Sarro, G.; Ranieri, G. Classical and non-classical proangiogenic factors as a target of antiangiogenic therapy in tumor microenvironment. Cancer Lett. 2016, 380, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cao, X. Characteristics and Significance of the Pre-metastatic Niche. Cancer Cell 2016, 30, 668–681. [Google Scholar] [CrossRef] [Green Version]
- Wculek, S.K.; Malanchi, I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 2015, 528, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Kanai, F.; Yoshida, H.; Tateishi, R.; Sato, S.; Kawabe, T.; Obi, S.; Kondo, Y.; Taniguchi, M.; Tagawa, K.; Ikeda, M.; et al. A phase I/II trial of the oral antiangiogenic agent TSU-68 in patients with advanced hepatocellular carcinoma. Cancer Chemother. Pharmacol. 2011, 67, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Viallard, C.; Larrivee, B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis 2017, 20, 409–426. [Google Scholar] [CrossRef]
- Korhonen, E.A.; Lampinen, A.; Giri, H.; Anisimov, A.; Kim, M.; Allen, B.; Fang, S.; D’Amico, G.; Sipila, T.J.; Lohela, M.; et al. Tie1 controls angiopoietin function in vascular remodeling and inflammation. J. Clin. Investig. 2016, 126, 3495–3510. [Google Scholar] [CrossRef] [Green Version]
- La Porta, S.; Roth, L.; Singhal, M.; Mogler, C.; Spegg, C.; Schieb, B.; Qu, X.; Adams, R.H.; Baldwin, H.S.; Savant, S.; et al. Endothelial Tie1-mediated angiogenesis and vascular abnormalization promote tumor progression and metastasis. J. Clin. Investig. 2018, 128, 834–845. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Kim, I.K.; Han, S.; Park, I.; Kim, C.; Bae, J.; Oh, S.J.; Lee, S.; Kim, J.H.; Woo, D.C.; et al. Normalization of Tumor Vessels by Tie2 Activation and Ang2 Inhibition Enhances Drug Delivery and Produces a Favorable Tumor Microenvironment. Cancer Cell 2017, 31, 157–158. [Google Scholar] [CrossRef]
- Cantelmo, A.R.; Conradi, L.C.; Brajic, A.; Goveia, J.; Kalucka, J.; Pircher, A.; Chaturvedi, P.; Hol, J.; Thienpont, B.; Teuwen, L.A.; et al. Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell 2016, 30, 968–985. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.B.; Tang, Y.L.; Liang, X.H. Targeting VEGF pathway to normalize the vasculature: An emerging insight in cancer therapy. OncoTargets Ther. 2018, 11, 6901–6909. [Google Scholar] [CrossRef] [Green Version]
- Goel, S.; Wong, A.H.; Jain, R.K. Vascular normalization as a therapeutic strategy for malignant and nonmalignant disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006486. [Google Scholar] [CrossRef] [PubMed]
- Arjaans, M.; Oude Munnink, T.H.; Oosting, S.F.; Terwisscha van Scheltinga, A.G.; Gietema, J.A.; Garbacik, E.T.; Timmer-Bosscha, H.; Lub-de Hooge, M.N.; Schroder, C.P.; de Vries, E.G. Bevacizumab-induced normalization of blood vessels in tumors hampers antibody uptake. Cancer Res. 2013, 73, 3347–3355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, R.K.; Duda, D.G.; Clark, J.W.; Loeffler, J.S. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat. Clin. Pract. Oncol. 2006, 3, 24–40. [Google Scholar] [CrossRef] [PubMed]
- Huynh, H.; Lee, L.Y.; Goh, K.Y.; Ong, R.; Hao, H.X.; Huang, A.; Wang, Y.; Graus Porta, D.; Chow, P.; Chung, A. Infigratinib Mediates Vascular Normalization, Impairs Metastasis, and Improves Chemotherapy in Hepatocellular Carcinoma. Hepatology 2019, 69, 943–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebos, J.M.; Lee, C.R.; Cruz-Munoz, W.; Bjarnason, G.A.; Christensen, J.G.; Kerbel, R.S. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 2009, 15, 232–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, A.S.; Kowanetz, M.; Wu, X.; Zhuang, G.; Ngu, H.; Finkle, D.; Komuves, L.; Peale, F.; Ferrara, N. Differential drug class-specific metastatic effects following treatment with a panel of angiogenesis inhibitors. J. Pathol. 2012, 227, 404–416. [Google Scholar] [CrossRef]
- Lv, X.; Li, J.; Zhang, C.; Hu, T.; Li, S.; He, S.; Yan, H.; Tan, Y.; Lei, M.; Wen, M.; et al. The role of hypoxia-inducible factors in tumor angiogenesis and cell metabolism. Genes Dis. 2017, 4, 19–24. [Google Scholar] [CrossRef]
- Paez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Vinals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009, 15, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Rankin, E.B.; Giaccia, A.J. Hypoxic control of metastasis. Science 2016, 352, 175–180. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Joseph, J.P.; Harishankar, M.K.; Pillai, A.A.; Devi, A. Hypoxia induced EMT: A review on the mechanism of tumor progression and metastasis in OSCC. Oral Oncol. 2018, 80, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Yeo, C.D.; Kang, N.; Choi, S.Y.; Kim, B.N.; Park, C.K.; Kim, J.W.; Kim, Y.K.; Kim, S.J. The role of hypoxia on the acquisition of epithelial-mesenchymal transition and cancer stemness: A possible link to epigenetic regulation. Korean J. Intern. Med. 2017, 32, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tian, X.J.; Xing, J. Signal Transduction Pathways of EMT Induced by TGF-beta, SHH, and WNT and Their Crosstalks. J. Clin. Med. 2016, 5, 41. [Google Scholar] [CrossRef]
- Iwadate, Y. Epithelial-mesenchymal transition in glioblastoma progression. Oncol Lett. 2016, 11, 1615–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Zhang, C.; Cui, M.; Niu, J.; Ding, W. Inhibition of Bevacizumab-induced Epithelial-Mesenchymal Transition by BATF2 Overexpression Involves the Suppression of Wnt/beta-Catenin Signaling in Glioblastoma Cells. Anticancer Res. 2017, 37, 4285–4294. [Google Scholar] [PubMed]
- Zhang, W.; Sun, H.C.; Wang, W.Q.; Zhang, Q.B.; Zhuang, P.Y.; Xiong, Y.Q.; Zhu, X.D.; Xu, H.X.; Kong, L.Q.; Wu, W.Z.; et al. Sorafenib down-regulates expression of HTATIP2 to promote invasiveness and metastasis of orthotopic hepatocellular carcinoma tumors in mice. Gastroenterology 2012, 143, 1641–1649.e5. [Google Scholar] [CrossRef]
- Keunen, O.; Johansson, M.; Oudin, A.; Sanzey, M.; Rahim, S.A.; Fack, F.; Thorsen, F.; Taxt, T.; Bartos, M.; Jirik, R.; et al. Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc. Natl. Acad. Sci. USA 2011, 108, 3749–3754. [Google Scholar] [CrossRef] [Green Version]
- Kumar, K.; Wigfield, S.; Gee, H.E.; Devlin, C.M.; Singleton, D.; Li, J.L.; Buffa, F.; Huffman, M.; Sinn, A.L.; Silver, J.; et al. Dichloroacetate reverses the hypoxic adaptation to bevacizumab and enhances its antitumor effects in mouse xenografts. J. Mol. Med. (Berl) 2013, 91, 749–758. [Google Scholar] [CrossRef] [Green Version]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.L.; et al. Fatty acid uptake and lipid storage induced by HIF-1alpha contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef] [Green Version]
- Papaevangelou, E.; Almeida, G.S.; Box, C.; deSouza, N.M.; Chung, Y.L. The effect of FASN inhibition on the growth and metabolism of a cisplatin-resistant ovarian carcinoma model. Int J. Cancer 2018, 143, 992–1002. [Google Scholar] [CrossRef]
- Sounni, N.E.; Cimino, J.; Blacher, S.; Primac, I.; Truong, A.; Mazzucchelli, G.; Paye, A.; Calligaris, D.; Debois, D.; De Tullio, P.; et al. Blocking lipid synthesis overcomes tumor regrowth and metastasis after antiangiogenic therapy withdrawal. Cell Metab. 2014, 20, 280–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIntyre, A.; Harris, A.L. Metabolic and hypoxic adaptation to anti-angiogenic therapy: A target for induced essentiality. EMBO Mol. Med. 2015, 7, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Achyut, B.R.; Arbab, A.S. Myeloid cell signatures in tumor microenvironment predicts therapeutic response in cancer. OncoTargets Ther. 2016, 9, 1047–1055. [Google Scholar] [PubMed] [Green Version]
- Du, R.; Lu, K.V.; Petritsch, C.; Liu, P.; Ganss, R.; Passegue, E.; Song, H.; Vandenberg, S.; Johnson, R.S.; Werb, Z.; et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 2008, 13, 206–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.Y.; Gordon-Weeks, A.; Allen, D.; Kersemans, V.; Beech, J.; Smart, S.; Muschel, R.J. Cd11b(+) myeloid cells support hepatic metastasis through down-regulation of angiopoietin-like 7 in cancer cells. Hepatology 2015, 62, 521–533. [Google Scholar] [CrossRef]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.Y.; Yuen, V.W.; Wong, C.C. Hypoxia and the Metastatic Niche. Adv. Exp. Med. Biol. 2019, 1136, 97–112. [Google Scholar]
- Dost Gunay, F.S.; Kirmizi, B.A.; Ensari, A.; Icli, F.; Akbulut, H. Tumor-associated Macrophages and Neuroendocrine Differentiation Decrease the Efficacy of Bevacizumab Plus Chemotherapy in Patients With Advanced Colorectal Cancer. Clin. Colorectal Cancer 2019, 18, e244–e250. [Google Scholar] [CrossRef]
- Sunakawa, Y.; Stintzing, S.; Cao, S.; Heinemann, V.; Cremolini, C.; Falcone, A.; Yang, D.; Zhang, W.; Ning, Y.; Stremitzer, S.; et al. Variations in genes regulating tumor-associated macrophages (TAMs) to predict outcomes of bevacizumab-based treatment in patients with metastatic colorectal cancer: Results from TRIBE and FIRE3 trials. Ann. Oncol. 2015, 26, 2450–2456. [Google Scholar]
- Zhang, W.; Zhu, X.D.; Sun, H.C.; Xiong, Y.Q.; Zhuang, P.Y.; Xu, H.X.; Kong, L.Q.; Wang, L.; Wu, W.Z.; Tang, Z.Y. Depletion of tumor-associated macrophages enhances the effect of sorafenib in metastatic liver cancer models by antimetastatic and antiangiogenic effects. Clin. Cancer Res. 2010, 16, 3420–3430. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Yang, Y.; Lu, M.; Lei, Y.; Xu, L.; Jiang, Z.; Xu, X.; Guo, X.; Zhang, X.; Sun, H.; et al. Recent Advances in the Discovery of HIF-1alpha-p300/CBP Inhibitors as Anti-Cancer Agents. Mini Rev. Med. Chem. 2018, 18, 296–309. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Saez, O.; Gajate Borau, P.; Alonso-Gordoa, T.; Molina-Cerrillo, J.; Grande, E. Targeting HIF-2 alpha in clear cell renal cell carcinoma: A promising therapeutic strategy. Crit. Rev. Oncol. Hematol. 2017, 111, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Tang, B.; Sun, X. Development of Inhibitors Targeting Hypoxia-Inducible Factor 1 and 2 for Cancer Therapy. Yonsei Med. J. 2017, 58, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Patterson, A.V.; Williams, K.J.; Cowen, R.L.; Jaffar, M.; Telfer, B.A.; Saunders, M.; Airley, R.; Honess, D.; van der Kogel, A.J.; Wolf, C.R.; et al. Oxygen-sensitive enzyme-prodrug gene therapy for the eradication of radiation-resistant solid tumours. Gene Ther. 2002, 9, 946–954. [Google Scholar] [CrossRef] [Green Version]
- Silva, V.L.; Al-Jamal, W.T. Exploiting the cancer niche: Tumor-associated macrophages and hypoxia as promising synergistic targets for nano-based therapy. J. Control. Release 2017, 253, 82–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guise, C.P.; Mowday, A.M.; Ashoorzadeh, A.; Yuan, R.; Lin, W.H.; Wu, D.H.; Smaill, J.B.; Patterson, A.V.; Ding, K. Bioreductive prodrugs as cancer therapeutics: Targeting tumor hypoxia. Chin. J. Cancer 2014, 33, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Sitkovsky, M.V.; Kjaergaard, J.; Lukashev, D.; Ohta, A. Hypoxia-adenosinergic immunosuppression: Tumor protection by T regulatory cells and cancerous tissue hypoxia. Clin. Cancer Res. 2008, 14, 5947–5952. [Google Scholar] [CrossRef] [Green Version]
- Leone, R.D.; Horton, M.R.; Powell, J.D. Something in the air: Hyperoxic conditioning of the tumor microenvironment for enhanced immunotherapy. Cancer Cell 2015, 27, 435–436. [Google Scholar] [CrossRef] [Green Version]
- Casazza, A.; Laoui, D.; Wenes, M.; Rizzolio, S.; Bassani, N.; Mambretti, M.; Deschoemaeker, S.; Van Ginderachter, J.A.; Tamagnone, L.; Mazzone, M. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell 2013, 24, 695–709. [Google Scholar] [CrossRef] [Green Version]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef]
- Yi, M.; Jiao, D.; Qin, S.; Chu, Q.; Wu, K.; Li, A. Synergistic effect of immune checkpoint blockade and anti-angiogenesis in cancer treatment. Mol. Cancer 2019, 18, 60. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.; Hamzah, J.; Ganss, R. More than a scaffold: Stromal modulation of tumor immunity. Biochim. Biophys. Acta 2016, 1865, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yuan, J.; Righi, E.; Kamoun, W.S.; Ancukiewicz, M.; Nezivar, J.; Santosuosso, M.; Martin, J.D.; Martin, M.R.; Vianello, F.; et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc. Natl. Acad. Sci. USA 2012, 109, 17561–17566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Goel, S.; Duda, D.G.; Fukumura, D.; Jain, R.K. Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res. 2013, 73, 2943–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terme, M.; Pernot, S.; Marcheteau, E.; Sandoval, F.; Benhamouda, N.; Colussi, O.; Dubreuil, O.; Carpentier, A.F.; Tartour, E.; Taieb, J. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res. 2013, 73, 539–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmittnaegel, M.; Rigamonti, N.; Kadioglu, E.; Cassara, A.; Wyser Rmili, C.; Kiialainen, A.; Kienast, Y.; Mueller, H.J.; Ooi, C.H.; Laoui, D.; et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Di Tacchio, M.; Macas, J.; Weissenberger, J.; Sommer, K.; Bahr, O.; Steinbach, J.P.; Senft, C.; Seifert, V.; Glas, M.; Herrlinger, U.; et al. Tumor Vessel Normalization, Immunostimulatory Reprogramming, and Improved Survival in Glioblastoma with Combined Inhibition of PD-1, Angiopoietin-2, and VEGF. Cancer Immunol. Res. 2019. [Google Scholar] [CrossRef] [Green Version]
- Allen, E.; Jabouille, A.; Rivera, L.B.; Lodewijckx, I.; Missiaen, R.; Steri, V.; Feyen, K.; Tawney, J.; Hanahan, D.; Michael, I.P.; et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Nadal, R.; Amin, A.; Geynisman, D.M.; Voss, M.H.; Weinstock, M.; Doyle, J.; Zhang, Z.; Viudez, A.; Plimack, E.R.; McDermott, D.F.; et al. Safety and clinical activity of vascular endothelial growth factor receptor (VEGFR)-tyrosine kinase inhibitors after programmed cell death 1 inhibitor treatment in patients with metastatic clear cell renal cell carcinoma. Ann. Oncol. 2016, 27, 1304–1311. [Google Scholar] [CrossRef]
- Sheng, X.; Yan, X.; Chi, Z.; Si, L.; Cui, C.; Tang, B.; Li, S.; Mao, L.; Lian, B.; Wang, X.; et al. Axitinib in Combination With Toripalimab, a Humanized Immunoglobulin G4 Monoclonal Antibody Against Programmed Cell Death-1, in Patients With Metastatic Mucosal Melanoma: An Open-Label Phase IB Trial. J. Clin. Oncol. 2019, 37, 2987–2999. [Google Scholar] [CrossRef]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodriguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef] [PubMed]
- Donnem, T.; Hu, J.; Ferguson, M.; Adighibe, O.; Snell, C.; Harris, A.L.; Gatter, K.C.; Pezzella, F. Vessel co-option in primary human tumors and metastases: An obstacle to effective anti-angiogenic treatment? Cancer Med. 2013, 2, 427–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayson, G.C.; Kerbel, R.; Ellis, L.M.; Harris, A.L. Antiangiogenic therapy in oncology: Current status and future directions. Lancet 2016, 388, 518–529. [Google Scholar] [CrossRef]
- Frentzas, S.; Simoneau, E.; Bridgeman, V.L.; Vermeulen, P.B.; Foo, S.; Kostaras, E.; Nathan, M.; Wotherspoon, A.; Gao, Z.H.; Shi, Y.; et al. Vessel co-option mediates resistance to anti-angiogenic therapy in liver metastases. Nat. Med. 2016, 22, 1294–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridgeman, V.L.; Vermeulen, P.B.; Foo, S.; Bilecz, A.; Daley, F.; Kostaras, E.; Nathan, M.R.; Wan, E.; Frentzas, S.; Schweiger, T.; et al. Vessel co-option is common in human lung metastases and mediates resistance to anti-angiogenic therapy in preclinical lung metastasis models. J. Pathol. 2017, 241, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Leenders, W.P.; Kusters, B.; Verrijp, K.; Maass, C.; Wesseling, P.; Heerschap, A.; Ruiter, D.; Ryan, A.; de Waal, R. Antiangiogenic therapy of cerebral melanoma metastases results in sustained tumor progression via vessel co-option. Clin. Cancer Res. 2004, 10, 6222–6230. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Schematic representation of the metastatic cascade. Inside the primary tumor, tumor cells produce vascular endothelial growth factor (VEGF), and fibroblast growth factor 1 (FGF1) to increase vessel density and interact with endothelial cells via the VEGF and matrix metalloproteinase-1 (MMP-1) signaling pathways to enhance vessel permeability, which provides favorable conditions for tumor metastasis. Endothelial cells (ECs) and pericytes constitute the physical barrier for tumor vessels. Pericytes, in the resting state, suppress tumor intravasation; however, certain populations of activated pericytes and cells undergone pericyte–fibroblast transition (PFT) lose their role as barriers to tumor vessels and facilitate metastasis. Many other cells in the tumor microenvironment, including tumor-associated neutrophils (TANs), tumor associated macrophages (TAMs) and Tie2-expressing macrophages (TEMs), are involved in the pro-metastatic effects, which contribute to angiogenesis and vascular permeability by secreting pro-angiogenic factors, such as VEGF-A, Wnt7B, MMP-9 and C–X–C motif chemokine ligand 12 (CXCL12). During the process of extravasation, tumor cells first adhere to ECs via various adhesion factors. Neutrophils potentiate tumor cell adhesion and transendothelial migration through the integrins/intercellular cell adhesion molecule-1 (ICAM-1) signaling pathway. Endothelial to mesenchymal transition (EndoMT), under transforming growth factor-β (TGF-β) enhances vessel permeability and contributes to the transmigration of metastatic cells. Platelets and metastasis-associated macrophages (MAMs) also enhance extravasation through opening the endothelial barrier. Finally, only a minority of disseminating cells successfully form metastatic foci and colonize in distant organs with the help of angiogenesis. The neovessels provide the necessary oxygen and nutrients for metastatic tumor growth.
Figure 1.
Schematic representation of the metastatic cascade. Inside the primary tumor, tumor cells produce vascular endothelial growth factor (VEGF), and fibroblast growth factor 1 (FGF1) to increase vessel density and interact with endothelial cells via the VEGF and matrix metalloproteinase-1 (MMP-1) signaling pathways to enhance vessel permeability, which provides favorable conditions for tumor metastasis. Endothelial cells (ECs) and pericytes constitute the physical barrier for tumor vessels. Pericytes, in the resting state, suppress tumor intravasation; however, certain populations of activated pericytes and cells undergone pericyte–fibroblast transition (PFT) lose their role as barriers to tumor vessels and facilitate metastasis. Many other cells in the tumor microenvironment, including tumor-associated neutrophils (TANs), tumor associated macrophages (TAMs) and Tie2-expressing macrophages (TEMs), are involved in the pro-metastatic effects, which contribute to angiogenesis and vascular permeability by secreting pro-angiogenic factors, such as VEGF-A, Wnt7B, MMP-9 and C–X–C motif chemokine ligand 12 (CXCL12). During the process of extravasation, tumor cells first adhere to ECs via various adhesion factors. Neutrophils potentiate tumor cell adhesion and transendothelial migration through the integrins/intercellular cell adhesion molecule-1 (ICAM-1) signaling pathway. Endothelial to mesenchymal transition (EndoMT), under transforming growth factor-β (TGF-β) enhances vessel permeability and contributes to the transmigration of metastatic cells. Platelets and metastasis-associated macrophages (MAMs) also enhance extravasation through opening the endothelial barrier. Finally, only a minority of disseminating cells successfully form metastatic foci and colonize in distant organs with the help of angiogenesis. The neovessels provide the necessary oxygen and nutrients for metastatic tumor growth.

Figure 2.
Anti-metastatic mechanisms of vessel targeting agents. Vessel targeting agents (VTAs) inhibit tumor metastasis through diverse mechanisms. First, VTAs starve tumor cells and block metastatic conduits by suppressing neovessel formation and pro-angiogenic factor production. Second, vessel targeting agents inhibit the bombina variegate 8 (Bv8)-induced vessel sprouting and the inflammation mediated by neutrophils, metastasis-associated macrophages (MAMs), C–C motif chemokine ligand 2 (CCL2) and C–X–C motif chemokine ligand 1(CXCL1) in the pre-metastatic niche; thus, disturbing pre-metastatic niche formation and inhibiting metastasis. Thirdly, vessel targeting agents lead to reduced metastasis through vessel normalization with increased endothelial cell (EC) junction and pericyte (PC) coverage and decreased vessel permeability.
Figure 2.
Anti-metastatic mechanisms of vessel targeting agents. Vessel targeting agents (VTAs) inhibit tumor metastasis through diverse mechanisms. First, VTAs starve tumor cells and block metastatic conduits by suppressing neovessel formation and pro-angiogenic factor production. Second, vessel targeting agents inhibit the bombina variegate 8 (Bv8)-induced vessel sprouting and the inflammation mediated by neutrophils, metastasis-associated macrophages (MAMs), C–C motif chemokine ligand 2 (CCL2) and C–X–C motif chemokine ligand 1(CXCL1) in the pre-metastatic niche; thus, disturbing pre-metastatic niche formation and inhibiting metastasis. Thirdly, vessel targeting agents lead to reduced metastasis through vessel normalization with increased endothelial cell (EC) junction and pericyte (PC) coverage and decreased vessel permeability.

Figure 3.
Hypoxia mediates pro-metastatic risk of vessel targeting agents. Hypoxia mediated by vessel targeting agents facilitates tumor epithelial mesenchymal transition (EMT) with decreased expression of epithelial cell markers and enhanced mesenchymal cell markers via the Wnt/β-catenin, transforming growth factor β (TGFβ) and HIV-1 Tat interactive protein 2 (HTATIP2) signaling pathways. Hypoxia contributes to tumor metabolism shift with increased glycolytic metabolism and lipid metabolism by upregulating fatty acid binding protein (FABP3), FABP7 and fatty acid synthase (FASN). Hypoxia also promotes the infiltration of bone marrow derived cells (BMDCs), including tumor associated macrophages (TAMs) and CD11b+ cells through colony-stimulating factor-1 (CSF-1), stromal-derived factor-1α (SDF-1α) and VEGF.
Figure 3.
Hypoxia mediates pro-metastatic risk of vessel targeting agents. Hypoxia mediated by vessel targeting agents facilitates tumor epithelial mesenchymal transition (EMT) with decreased expression of epithelial cell markers and enhanced mesenchymal cell markers via the Wnt/β-catenin, transforming growth factor β (TGFβ) and HIV-1 Tat interactive protein 2 (HTATIP2) signaling pathways. Hypoxia contributes to tumor metabolism shift with increased glycolytic metabolism and lipid metabolism by upregulating fatty acid binding protein (FABP3), FABP7 and fatty acid synthase (FASN). Hypoxia also promotes the infiltration of bone marrow derived cells (BMDCs), including tumor associated macrophages (TAMs) and CD11b+ cells through colony-stimulating factor-1 (CSF-1), stromal-derived factor-1α (SDF-1α) and VEGF.


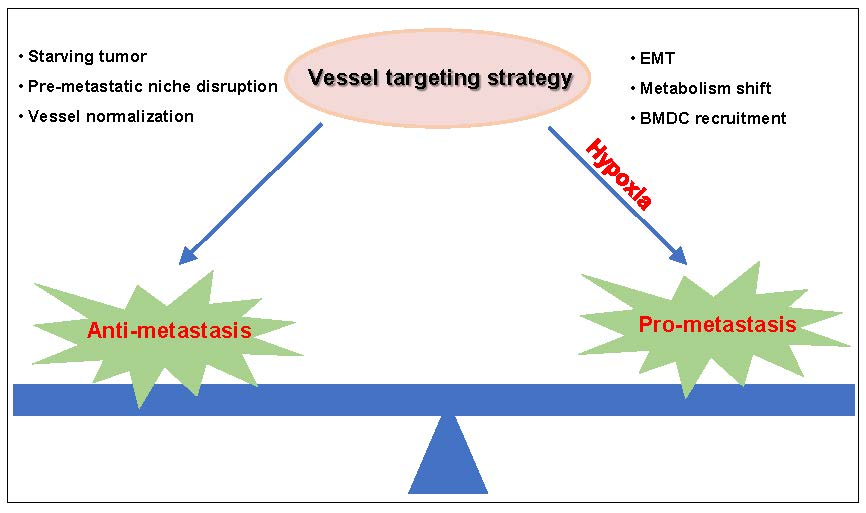{kind=link}
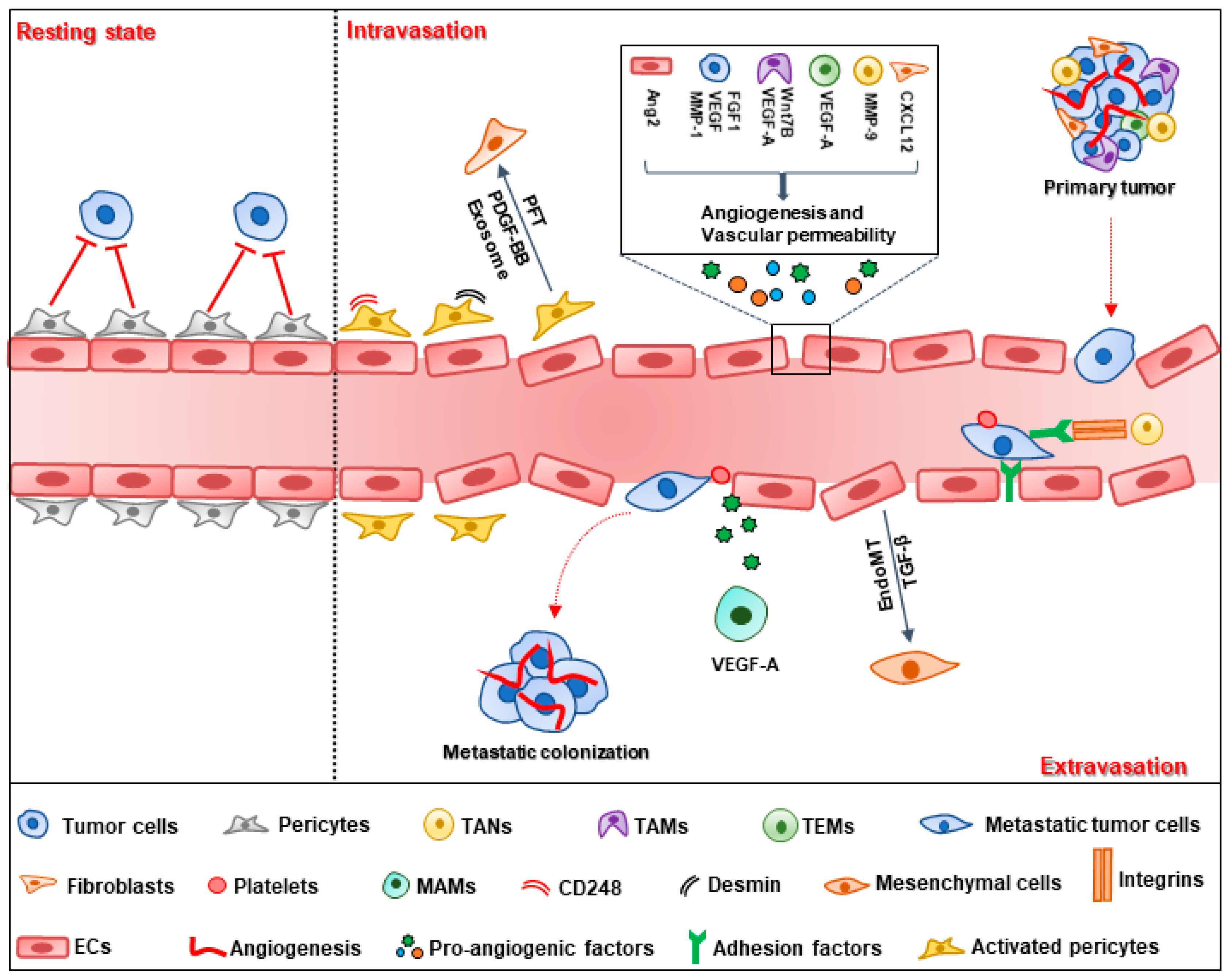{kind=link}
{kind=link}
{kind=link}
Table 1.
Food and Drug Administration (FDA)-approved vessel targeting drugs for metastatic cancer.
| Agent | Type of Inhibitor | Targets | Clinical Application |
|---|---|---|---|
| Bevacizumab | Monoclonal antibody | VEGF-A | Metastatic colorectal cancer [48,49,50,51] Metastatic nonsquamous non-small cell lung cancer [52] Metastatic renal cell carcinoma [53] Metastatic cervical cancer [54] metastatic breast cancer [55,56] |
| Ramucirumab | Monoclonal antibody | VEGFR-2 | Metastatic colorectal cancer [57] Metastatic non-small cell lung cancer [58] Metastatic stomach adenocarcinoma [59] Metastatic gastroesophageal junction adenocarcinoma [59] Metastatic urothelial carcinoma [60] |
| Sunitinib | Tyrosine kinase inhibitor | VEGFR-1, VEGFR -2, VEGFR -3, PDGFR-α, PDGFR-β, c-Kit, CSF-1R, RET | Metastatic pancreatic cancer [61,62] Metastatic renal cell carcinoma [63,64]. |
| Sorafenib | Tyrosine kinase inhibitor | VEGFR-1, VEGFR -2, VEGFR -3, PDGFR-β, Flt-3, c-Kit | Metastatic hepatocellular carcinoma [65,66,67] Metastatic thyroid cancer [68] |
| Regorafenib | Tyrosine kinase inhibitor | VEGFR-1, VEGFR -3, PDGFR-β, FGFR-1 KIT, RET and B-RAF | Metastatic colorectal cancer [69] Metastatic gastrointestinal stromal tumor [70] Metastatic osteosarcoma [71] |
| Vandetanib | Tyrosine kinase inhibitor | VEGFR-2, VEGFR-3, EGFR, RET | Metastatic medullary thyroid cancer [72] |
| Cabozantinib | Tyrosine kinase inhibitor | MET, RET, AXL, VEGFR-2, FLT3, c-Kit | Metastatic medullary thyroid cancer [73] Metastatic castration-resistant prostate cancer [74,75] Metastatic renal cell carcinoma [76] |
| Lenvatinib | Tyrosine kinase inhibitor | VEGFR-1, VEGFR-2, VEGFR-3 FGFR, PDGFRα, KIT and RET | Metastatic differentiated thyroid cancer [77] Metastatic hepatocellular carcinoma [78,79] Metastatic renal cell carcinoma [80] |
| Aflibercept | Fusion protein | VEGF-A, VEGF-B, PlGF | Metastatic colorectal cancer [81,82,83,84] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Li, X.; Li, Y.; Lu, W.; Chen, M.; Ye, W.; Zhang, D. The Tumor Vessel Targeting Strategy: A Double-Edged Sword in Tumor Metastasis. Cells 2019, 8, 1602. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8121602
AMA Style
Li X, Li Y, Lu W, Chen M, Ye W, Zhang D. The Tumor Vessel Targeting Strategy: A Double-Edged Sword in Tumor Metastasis. Cells. 2019; 8(12):1602. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8121602
Chicago/Turabian StyleLi, Xiaobo, Yong Li, Weijin Lu, Minfeng Chen, Wencai Ye, and Dongmei Zhang. 2019. "The Tumor Vessel Targeting Strategy: A Double-Edged Sword in Tumor Metastasis" Cells 8, no. 12: 1602. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8121602
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.